
A TEM protocol for quality assurance of in vitro cellular barrier models and its application to the assessment of nanoparticle transport mechanisms across barriers†
Dong
Ye
a,
Kenneth A.
Dawson
*a and
Iseult
Lynch
*b
aCentre for BioNano Interactions, School of Chemistry and Chemical Biology, University College Dublin, Belfield, Dublin 4, Ireland. E-mail: kenneth.a.dawson@cbni.ucd.ie
bSchool of Geography, Earth and Environmental Sciences, University of Birmingham, Edgbaston, Birmingham B15 2TT, UK. E-mail: i.lynch@bham.ac.uk
First published on 22nd September 2014
Abstract
We report here a protocol to characterise and monitor the quality of in vitro human cellular barrier models using Transmission Electron Microscopy (TEM), which can be applied for transport assays, mechanistic studies and screening of drug/compound (including nanoparticle) penetration across such biological barriers. Data from two examples of biological barriers are given, namely the hCMEC/D3 endothelial blood–brain barrier model, and the Caco-2 intestinal epithelial barrier model, to show the general applicability of the method. Several aspects of this method are applicable to the quality assurance of in vitro barrier models, e.g., assessment of the multi or mono-layer structure of the endothelial cells; identification of any potential “holes” in the barrier that could confound transport assay results; validation of tight junction expression; and determination of the types and amounts of key cellular organelles present in the barrier to account for any significant changes in phenotype that may occur compared to the in vivo situation. The method described here provides a key advantage in that it prevents loss of the filter membrane during monolayer sectioning, thereby preserving critical details associated with the basal cell membrane. Applicability of the protocol for other in vitro biological barriers, such as the blood–foetus, blood–testes, blood–cerebrospinal fluid (CSF) and lung alveolar–capillary barriers is also discussed. Additionally, we demonstrate the use of the method for assessment of nanoparticle transport across cellular barriers and elucidation of transcytosis mechanisms. Sequential events of cellular endocytosis, localisation and transcytosis can be described in detail by TEM imaging, revealing useful sub-cellular details that provide evidence for the mechanism of nanoparticle transport in the hCMEC/D3 blood–brain barrier model and the Caco-2 intestinal epithelial cell model. Potential artefacts resulting from the nanoparticles interacting with the Transwell membranes can also be assessed.
1. Introduction
There are both ethical (animal welfare) and economic considerations driving the regulatory push towards the development and validation of alternative (in vitro) approaches for safety assessment of chemicals.1,2 Specifically, the use of cells or tissues derived, where possible from humans, which are cultured under controlled conditions in the laboratory (in vitro methods) may provide an important, scientifically valid route for safety assessment. From an economic standpoint, conducting a 2-generation reproductive toxicity study, a 90 day toxicity study, and a developmental toxicity study costs close to one million dollars (in addition to approximately 5000 animals). As such, in vitro methods can be seen (perhaps inappropriately) as a quick and cheap way to register a substance under REACH, based on justification of a waiver to conducting new animal experiments.1,2In light of this regulatory push, a large number of in vitro toxicology assays are being proposed, developed, validated, and approved for regulatory use as alternatives to animal testing.3 Such assays have also been proposed to have additional benefits (besides reducing the number of animals used and the cost of testing), by also reducing the quantity of test materials needed and increasing the predictive ability of testing by focusing more closely on relevant endpoints or adding mechanistic information.3 Validation is the process by which the reliability and relevance of a test method are established for a specific purpose, in order to ensure regulatory acceptance of the method worldwide. Thus, any new method has to exhibit its usefulness and power in describing and predicting possible toxicity per se, in comparison with an established method that reflects the present standard.4
A key challenge for in vitro methods is to reproduce the complexity of the in vivo situation. For example, the cell quantities and types in culture are usually very limited, and often there are simply not enough signalling molecules produced by the low volumes of cells relative to the volume of cell culture medium for signalling to function. Thus, many types of cell signalling may be lost in in vitro systems, such as contact-dependent cell signalling. Even in very well-studied and highly commercial areas such as in vitro fertilisation, the current methods do not succeed in mimicking the in vivo situation completely.5
From a research standpoint, this need for alternative in vitro test methods provides multiple opportunities and challenges. Among the most important tissues to mimic in vitro are the biological barriers that have the dual functions of protecting delicate organs whilst allowing essential nutrients to pass through them. In vivo human biological barriers, such as the lung aveolar–capillary barrier, the intestine, the blood–brain barrier and the blood–placenta barrier, are composed of epithelial and endothelial cells which are associated within junction-anchored networks. All of these biological barriers share several common functions, including: (1) regulation of the permeability of biological fluids and balancing of fluid homeostasis; (2) separation of fluid compartments; and (3) protection of human micro-environments from harmful pathogens.6 However, due to variances in cell type, location and function, different barrier structures have distinct metabolic mechanisms and significance in terms of their function.
A major research and regulatory goal is thus to establish and validate analogous in vitro models for biological barriers and to utilise them as screening tools to assess both exposure (amount of a compound that crosses the barriers) and impacts (effects such as toxicity or drug efficacy) of compounds for risk assessment and regulatory purposes. Key aspects of in vitro barrier models that require validation include barrier morphology and functionality. Epithelial and endothelial cellular barriers can grow into communicating cell–cell networks due to cell polarization and differentiation. Once confluent, they can form morphological and functional cell barriers akin to their “in vivo” counterparts, and impart a degree of restriction for transport of biomolecules via expression of tight junctions.7–11 Utilising a Transwell filter membrane set-up as per previous reports (see Fig. 1),10,11 epithelial and endothelial cells established on Transwells are usually polarised and prompted to communicate (via signalling molecules) between the apical and basolateral chambers, such that they can acquire sufficient differentiation to function as barriers.7–9,12–14 Tight junctions expressed in these in vitro models are usually thought to be a critical standard with which to discriminate a well-differentiated barrier from loosely associated barriers. Due to cell polarisation, endogenous bio-markers, such as junctional proteins, and transport related proteins that are specifically expressed in vivo, are subsequently expressed in these barriers.
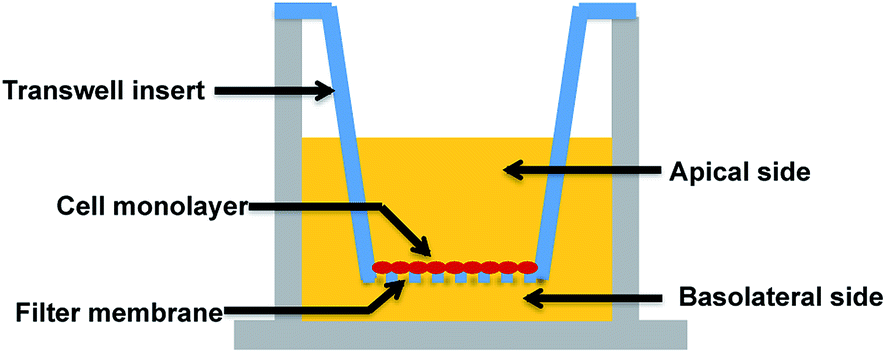 | ||
| Fig. 1 Representation of a Transwell-based approach for in vitro biological barrier models. | ||
The application of such Transwells has been utilised to study a range of end-points such as transport, absorption and secretion of molecules, access into or out of polarised cells, mobility-related microbial pathogenesis, interactions between co-cultured cell types and so on. Despite the Transwell approach being well established and widely used in cell biology since its introduction in 1953,15 there have been persistent concerns that in vitro barriers do not fully mimic the in vivo situation. Potential reasons for this could include incomplete surface coverage, or overgrowth of the cells leading to multiple layers, either of which could lead to significant differences compared to the cell monolayer structure in vivo. However, to date, few studies have imaged the morphology of in vitro cellular barriers in detail as a quality control step to confirm their functionality, relying instead on the older established approaches of transport studies using fluorescently-labelled dextrans of specific molecular weights or electrical resistance studies across the barrier cells.8,13,16 Indeed, recommendations from the ECVAM study of available in vitro alternatives for a Blood–Brain Barrier (BBB) included the need for: (1) further development, characterisation and standardisation of in vitro models; (2) standard protocols for the different models; and (3) methods to characterise (a) the functional expression of transporter mechanisms and (b) the closeness of morphology to that of the in vivo system.17
Here we report a Transmission Electron Microscopy (TEM) based method to monitor the morphological quality of in vitro biological barrier models and to evaluate cell–cell interactions. The method provides direct visualisation of the barrier cells, their organisational structure and format, and allows any defects in the layer to be identified as a quality control tool prior to transport studies. TEM imaging, with its high magnification is capable of scrutinizing ultrastructures from cell level to nano-scale level. The general applicability of the method to in vitro biological barriers is demonstrated using two distinct barrier models: the human capillary microvascular endothelial D3 (hCMEC/D3) cell model of the human brain blood–brain barrier (BBB) and the human colon intestinal Caco-2 model of the human intestinal barrier.
Possible applications of the protocol for TEM imaging of barrier models, could be: (1) to probe any defect or imperfection upon barrier formation (such as multilayer or monolayer discrepancy, holes in the layers and other related issues that confound permeability and absorption tests); and (2) to examine tight junction or other subcellular components. Additionally, we show the application of the method to investigation of the mechanism of uptake and transport of nanoparticles, which have a size that is suitable for detection by TEM, into and across biological barriers. Thus, TEM imaging can also be used to identify and (semi) quantify nanoparticle uptake, localisation and transport across biological barriers,18 both for exposure and impact studies. The present protocol provides an excellent addition to the published protocol for preparation of cells for assessing ultrastructural localisation of nanoparticles with TEM,19 extending it to the study of the morphology of biological barriers and for quality assurance of barrier morphology & function.
Nanomedicine has recently emerged as a hot topic, since nanosized (1–100 nm) materials can interact with endogenous cellular machinery and access sub-cellular locations unavailable to conventional molecular drugs, as a result of their small size and large surface area for protein binding. However, there is also significant scope for nanoparticles not intended to cross biological barriers to do so, and thus, as part of an assessment of the potential health implications of nanotechnologies, detailed understanding of the mechanisms of nanoparticle transport into and across biological membranes, utilising well established in vitro models, is urgently needed. Inclusion of TEM images for quality control of the in vitro barriers as a pre-requisite to uptake and transport studies would go a long way towards facilitating cross-comparison of studies and towards validating the comparability of the in vitro models as alternatives to in vivo biodistribution and biokinetics studies. Coupling these with TEM images of the nanoparticle sub-cellular localisation (including at the basal membrane) can also provide mechanistic insights.
The protocol presented here consists of several techniques including hCMEC/D3 cell and Caco-2 cell culture, microtome sectioning, cell monolayer histology processing and TEM imaging, which are each described in detail. Typical results regarding the morphology of cells grown on filters, tight junction structures and representative subcellular components are demonstrated to illustrate the versatility of the protocol in underpinning subsequent transport and mechanistic studies.
2. Materials and methods
2.1 Materials
hCMEC/D3 cells were obtained under license from INSERM, France and used for experiments between passage 25 and 35.12 Growth medium and assay medium were used for hCMEC/D3 cell propagation and barrier forming respectively (see Reagent setup). Caco-2 cells were obtained from ATCC and passage numbers used were between 53–75, but it is possible to use cells with lower or higher passage numbers (e.g., 30–40).7 Culture medium for Caco-2 was prepared as described in the Reagent setup. 0.01% Trypsin/EDTA solution and foetal bovine serum (FBS, non-heated inactivated) were from Biosciences (Ireland). Hank's balanced salt solution (HBSS) was from Gibco (Ireland). Type I rat tail collagen (see Reagent setup), HEPES buffer, Triton X-100, glutaraldehyde solution, osmium tetroxide solution, absolute ethanol (99.8% purity) (see Reagent setup) and DMSO (99.5% purity) were all purchased from Sigma (Ireland). Sorensen's buffer was prepared as per the reported method20 (see Reagent setup). Epoxy resin preparation kit was purchased from Agar Scientific (Ireland). 12 nm human serum albumin functionalised-gold nanoparticles and 50 nm non-modified SiO2 nanoparticles were prepared as per reported methods.10,21Cells were grown in a 37 °C incubator with CO2 connection (5% CO2 in air) and humidified with a water atmosphere. For hCMEC/D3 cell culture, 25 cm2 collagen pre-coated culture flasks (BD, USA) were used, and for Caco-2 cell culture various non-collagen coated flasks (Cap-vented flasks, with area 25 cm2 or 75 cm2, from Greiner Bio-One, Ireland) were utilised. Filters were purchased from Corning Costar (USA), including polyester membrane (PET) pore size 0.4 μm and 3.0 μm, and PTFE membrane pore size 3.0 μm, all with a growth area of 1.12 cm2. 12-well culture plates with lids were also ordered from Corning Costar. Transmission electron microscope (TEM, TECNAI 12) was from FEI (USA). The microtome used for ultra-thin section preparation was a Leica EM UC6 (Ireland). The cell culture laminar flow fume hood was from Heraeus (Germany). A diamond knife was purchased from Diatome (USA). Copper grids (200 meshes) and other TEM imaging tools were obtained from Agar Scientific (USA). Light Microscopy was accessed via the UCD imaging facility. Glutaraldehyde, osmium tetroxide, ethanol, toluidine blue (tolonium chloride), uranyl acetate and lead citrate were purchased from Laboratory Instruments & Supplies, Ireland.
2.2 Reagent setup
hCMEC/D3 cell growth medium was prepared from EGM-2 basal medium supplemented with VEGF, IGF-1, EGF, bFGF, 2.0% FBS, ascorbate, gentamycin and hydrocortisone, as recommended by the manufacturer (Lonza Bioscience, Ireland) and literature.8,12 For the assay medium, EGM-2 basal medium was supplemented only with bFGF, 2.5% FBS, hydrocortisone, gentamycine and 10 mM HEPES as reported in the literature.9,10,12,22 Type I rat tail collagen was diluted in sterile de-ionized water to a working concentration of 0.1 mg ml−1, according to the manufacturer (Sigma, Ireland).For Caco-2 culture medium, Dulbecco's Modified Eagle Medium (DMEM, 500 ml) was supplemented with high glucose (4500 mg l−1D-glucose), sodium pyruvate (Gibco, Biosciences), 10% FBS, 5 ml penicillin–streptomycin (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 U ml−1) and 5 ml non-essential amino acids (100×) (Gibco Biosciences, Ireland). Sorensen's phosphate buffer was prepared from two separate solutions, A and B (A is 0.2 M Na2HPO4·2H2O; B is 0.2 M NaH2PO4·H2O). At pH 7.4, Sorensen's buffer was freshly prepared by mixing 40.5 ml solution A with 9.5 ml solution B plus 50 ml de-ionized water. 2.5% Glutaraldehyde fixative was prepared with Sorensen's buffer. Osmium tetroxide post-fixative was diluted in de-ionised water to 1%. For specimen dehydration, absolute ethanol was diluted with de-ionised water to 30%, 50%, 70% and 90%. Epoxy resin mixture was prepared as per the manufacturer's instructions. For a better dispersion, human albumin-modified gold nanoparticles were pipetted several times to disperse potential nanoparticle agglomerates before dilution with 2% FBS assay medium to a working concentration of 50 μg ml−1; non-modified silica nanoparticles were dispersed with bath sonication at 25 °C for 2–5 min in order to agitate any agglomerates, and subsequently added to HBSS buffer supplemented with 10% FBS to reach the desired concentration of 100 μg ml−1 and mixed thoroughly. Note that nanoparticle solutions (dilutions in media) should be always made freshly immediately prior to experiments, and their dispersion quality could be monitored via dynamic light scattering or an alternative particle sizing technique to ensure the sample has not agglomerated in the assay medium.
000 U ml−1) and 5 ml non-essential amino acids (100×) (Gibco Biosciences, Ireland). Sorensen's phosphate buffer was prepared from two separate solutions, A and B (A is 0.2 M Na2HPO4·2H2O; B is 0.2 M NaH2PO4·H2O). At pH 7.4, Sorensen's buffer was freshly prepared by mixing 40.5 ml solution A with 9.5 ml solution B plus 50 ml de-ionized water. 2.5% Glutaraldehyde fixative was prepared with Sorensen's buffer. Osmium tetroxide post-fixative was diluted in de-ionised water to 1%. For specimen dehydration, absolute ethanol was diluted with de-ionised water to 30%, 50%, 70% and 90%. Epoxy resin mixture was prepared as per the manufacturer's instructions. For a better dispersion, human albumin-modified gold nanoparticles were pipetted several times to disperse potential nanoparticle agglomerates before dilution with 2% FBS assay medium to a working concentration of 50 μg ml−1; non-modified silica nanoparticles were dispersed with bath sonication at 25 °C for 2–5 min in order to agitate any agglomerates, and subsequently added to HBSS buffer supplemented with 10% FBS to reach the desired concentration of 100 μg ml−1 and mixed thoroughly. Note that nanoparticle solutions (dilutions in media) should be always made freshly immediately prior to experiments, and their dispersion quality could be monitored via dynamic light scattering or an alternative particle sizing technique to ensure the sample has not agglomerated in the assay medium.
2.3 Methods
The detailed information for this TEM protocol has been laid out as described in the following sections, and for associated technical issues, if any, readers are encouraged to consult our troubleshooting list shown in Table 1.| Problem | Solution |
|---|---|
| Defrosted cells are not confluent or reach confluence slowly during cell culture | Ensure removal of DMSO before culturing cells in a culture flask. A smaller-growth area flask is recommended to speed up cell growth. Check cell density and storage time of the cryogenic stock |
| Multilayer formation | Several methods can be used to improve this: reduce cell seeding density on the Transwell and avoid seeding aggregated cells. Improve collagen coating on Transwell membrane |
| A low TEER or a high FD4 value in the barrier model (if applicable) | Improve the cell culture to allow tight junctions to be well-developed |
| Blank filter not permeable | Soak Transwell filter in water before cell seeding. Test equilibration capacity of nanoparticles/test molecules on different types of porous membranes. Optimise collagen coating to minimize blockage of Transwell pores |
| Cell barrier layer becomes completely removed from the Transwell membrane | Cell seeding density is too high or collagen coating is too thick, causing multi-layer formation. Optimise cell seeding density and collagen coating |
| Cells migrate through Transwell pores | Improve collagen coating or change to another Transwell with a smaller pore size |
| Imaging displays holes in the barrier | Improve cell culture procedure and collagen coating. Check cell seeding density and number of growth days |
| Nanoparticles precipitated | Increase sonication time, or if not applicable, improve chemical synthesis |
| Darkened Transwell membranes in specimen | Ensure osmium tetroxide is completely removed and rinsed with washing buffer at the end of the fixation step |
| Bubbles underneath the Transwell in epoxy resin embedding | Avoid vigorous mixing of epoxy resin and take extra care to check bubble formation during embedding |
| Specimen is too hard to cut | Always use manufacturer-recommended recipe for resin preparation |
| Holes in sections | Improve ethanol dehydration, resin embedding and section staining procedures. Alternatively, a vacuum oven may be used to help infiltrate pores of Transwells and remove air bubbles from resin |
| Filter breaks away from epoxy resin | Avoid using liquid nitrogen to separate the plastic surrounding the embedded sample. Use a blade to manually remove plastic and prepare block face |
| Sections are washed away from grids during staining | Dry sections for a longer time (e.g., overnight) prior to staining and always avoid vigorous washing during the staining process |
| Block face always wets during cutting and sections may drag back over knife edge | Water level in the boat of diamond knife is too high and the block face of specimen is too big |
| Heavy metal particle contamination on sections | Improve washing and drying steps during uranyl acetate and lead citrate staining |
| Poor image contrast | Improve the staining with uranyl acetate and lead citrate. Adjust brightness and contrast settings in TEM camera |
For barrier seeding, we used 12-well Transwell plates of different pore sizes. In some cases, Transwells (e.g., Corning polyester or polycarbonate) may require collagen coating. To do so, 0.2–0.5 ml 0.1 mg ml−1 collagen solution is added to the apical chamber of Transwells and incubated for 24 hours at 37 °C in a dry incubator (collagen solution will dry out and deposit on the Transwell membrane). Transwells were washed with PBS before use. It was noted that an optimal density of collagen coating may improve confluent morphology and barrier formation of hCMEC/D3 cells, although this is still subject to using a suitable type of Transwell (i.e. suitable pore size) to avoid potential cell migration through bigger pores (ESI Fig. 2†). The Transwell filters (growth area 1.12 cm2) were soaked in 0.5 ml and 1.5 ml assay medium in the apical and basolateral chambers respectively for at least 10 min at 37 °C after which the medium was removed. 0.5 ml cell solution with an optimal seeding density of 5.0 × 104 cells was added to the apical chamber and 1.5 ml fresh assay medium to the basal chamber. After cells were cultured at 37 °C for 5–6 hours, the apical medium was removed and replaced with pre-warmed fresh medium, in order to avoid any non-attached cells forming a multilayer. Cell medium was then changed every 3 days for 7 days, by aspirating medium from the basolateral side, then from the apical side and adding assay medium to the apical chamber, then to the basolateral chamber (medium volumes as stated before). Note that when pipetting medium, care should be taken to avoid any physical contact with the filter surface which may damage monolayer integrity. On the 7th day, medium should be changed before the experiment (whether quality assurance or transport assay) as starvation may affect protein expression in the endothelial cell monolayer. Tight junction expression is validated using well established assays such as transport of 4 KDa FITC-Dextran (FD4) and Trans-endothelial/epithelial Electrical Resistance (TEER), as per our previous report.11
For barrier seeding, Transwell filters were soaked with fresh medium as stated for hCMEC/D3 cells. With a 12-well Transwell plate (0.4 or 3.0 μm filter), 0.5 ml cell suspension containing 2 × 105 cells was added to the apical side and 1.5 ml DMEM fresh medium added to the basolateral side. Cells were cultured in an incubator for 5–16 hours before the apical medium was removed and replaced with fresh (cell-free) medium, in order to reduce formation of a multilayer. Caco-2 cells were then grown for up to 21 days, with medium changed every two days up to 15 days and then every day until day 21 to obtain well-differentiated monolayers. The medium change was carried out using the same procedure as stated for the hCMEC/D3 cell barrier. Tight junction quality is also evaluated using TEER measurements to allow comparability with established literature.7
An orbital shaker was set up at 37 °C in an incubator with humidified atmosphere and 5% CO2. Apical and basolateral medium were removed from barrier Transwells on which barriers were 90% confluent, followed by rinsing of the barriers with fresh medium or buffer for 30–60 min (assay medium for hCMEC/D3 cell barrier; 10 mM HEPES supplemented HBSS buffer for Caco-2 cell barrier). Next, Transwell filters were moved into another 12-well plate, which already contained fresh assay medium or HBSS (1.5 ml per well). Nanoparticle solutions were loaded into the apical chambers along the inner verge of the filters and placed into an orbital shaker set at 100 rpm. Physical contact of pipette tips with filter membranes should be avoided while adding or emptying solutions as that may damage cell barriers that have formed. Exposure to the nanoparticle solutions was over 4 hours for hCMEC/D3 barriers and over 9 hours for Caco-2 barriers, although exposure times of up to 24 hours for Caco-233 or 48 hours for hCMEC/D39 can be easily achieved. After exposure, nanoparticle solution/media was emptied from both apical and basolateral sides of filters. Fresh PBS was applied and removed in order to wash off any remaining nanoparticles. It should be noted that nanoparticles may cross the monolayer and reach the basolateral compartment during the exposure, thus for enhanced image analysis, it is important to remove nanoparticles/solutions from both chambers of each Transwell prior to performing fixation and embedding. Medium from the basolateral side may be used subsequently for quantification of nanoparticle transport11via particle counting, fluorescence or other approaches.
:1 v/v) for 1 hour, followed by 2 hour embedding in full epoxy resin (100%) at 37 °C, with 250 ml on the top and 600 ml on the bottom. This step is to ensure that the ethanol evaporates completely during the incubation. Later, epoxy resin was removed completely and replaced with fresh resin. Note that resin should be gently added to both the top and bottom space of the Transwell filters and enough used to submerge the Transwell as a whole. Bubbles may form during this step, which should be carefully removed by using a pipette tip; the presence of bubbles below the filter membranes should also be carefully checked: if any are observed the filters should be taken out from the resin and then slowly re-positioned in order to avoid bubble formation underneath. Resin is then polymerised in an oven at 65 °C for 24 hours. Note that the Transwell filter lids should be removed prior to polymerisation.
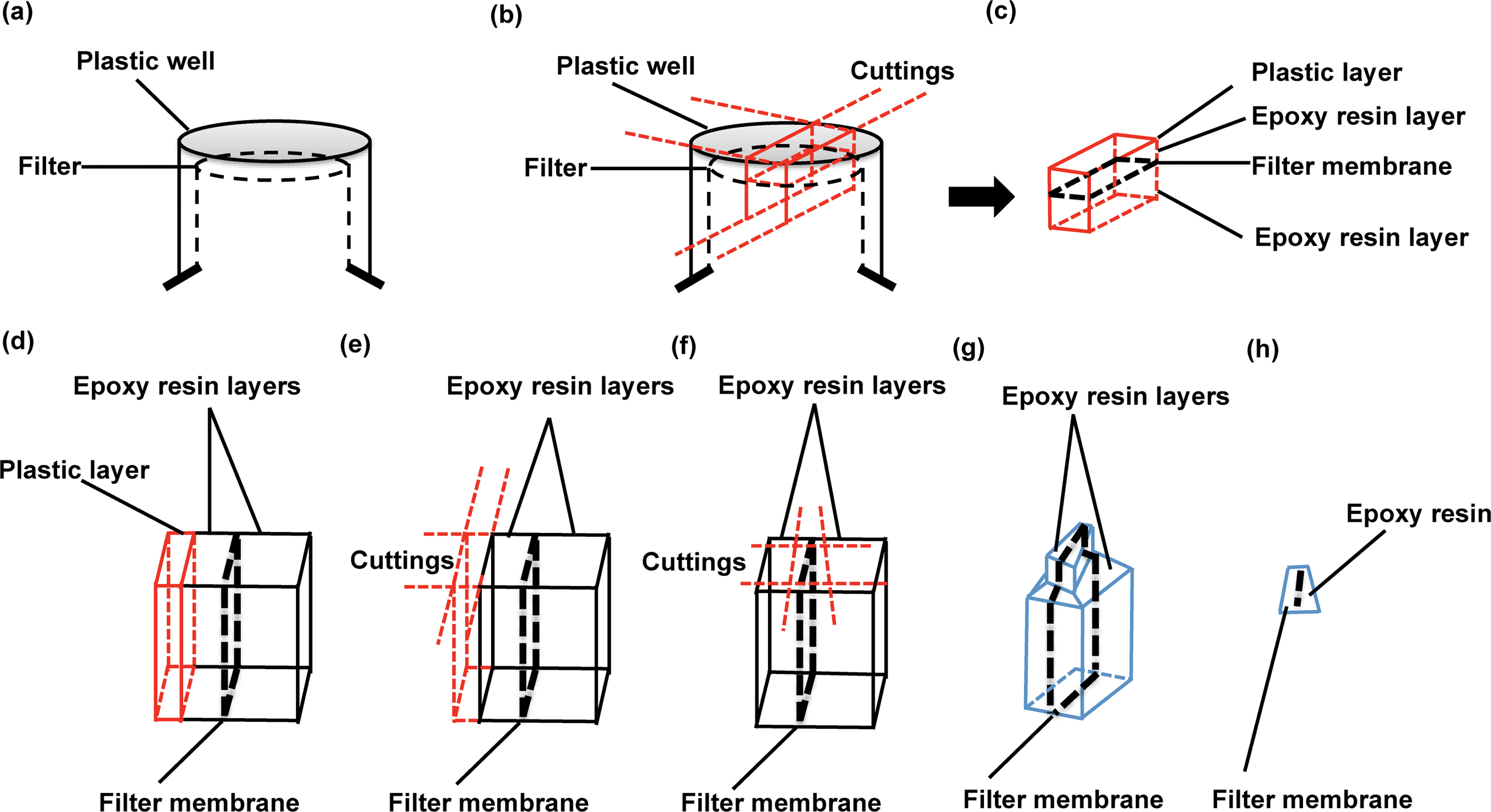 | ||
| Fig. 2 Schematic diagram for separating and sectioning a barrier-seeded Transwell sample. (a) A resin-embedded Transwell filter in an inverted position. (b) The cutting strategy is outlined with red lines to demonstrate the separation of a filter membrane-localised block from the resin-embedded Transwell. (c) An overview (red line) of the block sample after separation. (d) A view across the filter to show the different layers of materials in the sample block. From left to right: plastic (red line), epoxy resin, filter membrane and epoxy resin. (e) The cutting method is outlined with red lines to show how the plastic layer is removed without damaging the filter membrane. (f) After removal of the plastic, serial cuttings (red lines) are applied around the filter membrane. (g) The trimming block surface is revealed by removal of the peripheral resin before microtome sectioning. (h) Overview of an ultrathin section obtained by microtome sectioning. | ||
3. Results
3.1 Barrier morphology quality control
Barriers composed of hCMEC/D3 and Caco-2 cells exhibited confluent and contact-inhibited morphology, as demonstrated in Fig. 3a and c, grown on 0.4 μm and 3.0 μm Transwells (Fig. 3b) respectively. Both barriers completely covered the surface area of the Transwell membranes, as shown in the two longitudinal views for barrier integrity given in Fig. 3a and c, respectively. Following fixation and embedding of the intact cell barriers on the Transwells, cross sections of Transwell membranes were cut using the microtome. Transwell filters were well preserved and closely attached under the layer of cells. | ||
| Fig. 3 Morphology of hCMEC/D3 endothelial (a) and Caco-2 epithelial (b) cell barriers grown on Transwell filters (b, from http://www.eandkscientific.com). Confluent barriers of hCMEC/D3 cells and Caco-2 cells after 7 and 21 days in culture, respectively. Both hCMEC/D3 (a) and Caco-2 (c) cell barriers were stained by toluidine blue and both images were taken with a 5× objective lens using a phase contrast microscope. Cross-sections for hCMEC/D3 monolayers with 0.4 μm PET (d) and 3.0 μm PTFE (e) Transwells, and Caco-2 monolayers with 0.4 μm PET (f) and 3.0 μm PTFE (g) Transwells were obtained and imaged, where the PTFE membranes generally appeared more porous than the PET ones. Caco-2 cells showed relatively bigger nuclei (N, as indicated) than hCMEC/D3 cells. | ||
Using TEM imaging, a much more detailed (and magnified) description of barrier morphology emerged, compared to other methods such as fluorescence microscopic imaging used to characterise and evaluate the same BBB barrier, as reported previously.22 Holes of different sizes in the porous membranes underneath the barrier cell layers could be observed. As expected, 3.0 μm PTFE membranes showed much bigger pores than 0.4 μm PET membranes with both cell types. TEM imaging showed that both hCMEC/D3 cells and Caco-2 cells formed a single layer (of endothelial or epithelial cells respectively) on the apical side of the Transwell. Morphologically, the hCMEC/D3 endothelial monolayer (Fig. 3d and e) was 3–4 times thinner than the Caco-2 epithelial monolayer, which was about 15–20 μm in thickness (Fig. 3f and g).
Caco-2 cells appeared rectangular-shaped with their nuclei staying close to the basal membrane (Fig. 3f and g). The cell boundaries between adjacent epithelial cells were merged without clear division. In comparison, hCMEC/D3 cell barriers consisted of thin, elongated cells. Although different in morphology, both endothelial and epithelial cell monolayers were formed uniformly on the various Transwell filters (as shown in Fig. 3d–g).
In some cases, however, TEM imaging found that hCMEC/D3 cells were able to form sandwich-like bi-layers on both sides of the 3.0 μm PET membranes (even where prelonged coating with collagen resulted in a relatively higher density), as shown in Fig. 4a and ESI Fig. 2.† Presumably the cells may have migrated through the pores of the Transwells and grown underneath by attaching to the bottom of the Transwell membrane, as described in Fig. 4b and c. By comparing with a barrier monolayer formed on 3.0 μm PTFE membrane (collagen coated by the manufacturer) in Fig. 3e and 5b, it is postulated that collagen coating density and/or porosity of the chosen Transwell type may contribute to the migration of cells through the filter. For a given Transwell type, collagen coating density and porosity of Transwells played important roles in formation of homogenous barriers. Barrier homogeneity was also influenced by Transwell composition, even where the pore sizes are nominally the same according to the manufacturer, for example, 3.0 μm PET versus 3.0 PTFE (ESI Fig. 2†).
 | ||
| Fig. 4 Double layers of hCMEC/D3 cells growing on both sides of a 3.0 μm PET Transwell membrane, due to apical-to-basolateral migration of the cells through the pores of the filter. (a) The microtomic section of double cell layers was stained with toluidine blue and visualised with light microscopy. Regional cell attachments on the apical (b) and basolateral (c) sides of the Transwell membrane were examined under TEM. | ||
 | ||
| Fig. 5 An example of multilayer (a) or monolayer (b) formation determined using TEM imaging in order to monitor hCMEC/D3 overgrowth on 3.0 μm PTFE Transwells during the protocol optimisation procedure. The highly porous Transwell membrane without any cells seeded was also imaged as a reference (c). It should be noted that studies inadvertently performed on a multilayer would give false results, thereby emphasizing the need for careful quality control of barriers prior to studies. | ||
Choice of Transwell composition, pore size and collagen coating are important, but optimisation of the growth conditions is also necessary to avoid the formation of multilayers. Thus, multilayer formation (Fig. 5a) was also observed via TEM on 3.0 μm PTFE Transwell membranes, for example (Fig. 5c), using the hCMEC/D3 barrier model, and can be compared to the hCMEC/D3 monolayer formed on the same Transwells (Fig. 5b), as previously reported.10 The hCMEC/D3 cells seemed to undergo partial overgrowth on the Transwell, potentially caused by cellular compatibility of Transwells as well as sub-optimal growth conditions including barrier growth time or cell seeding density. Fig. 5b shows cells grown using the optimised the seeding density of hCMECD3 cells (50000 cells), with growth time (7 days) monitored in relation to tight junction formation. As can be seen from Fig. 5b, optimal seeding density results in a good confluent morphology where cells were elongated and showed contact-inhibited features and a homogenous monolayer, compared to the barriers that were acquired from excess or insufficient seeded cell numbers (ESI Fig. 3b and c†). In addition, TEER values showed that the optimal seeding density generated a similar tight junction quality regardless of variance of pore sizes of the chosen Transwells (ESI Fig. 3a†), although according to the literature, the hCMEC/D3 model always gives low TEER values compared to the in vivo values.8,11,13 Overall, all these distinct characteristics suggested that TEM imaging is a necessary tool to monitor the growth characteristics and ensure formation of a homogenous monolayer as part of barrier morphology quality assurance prior to transport studies.
The paracellular space between adjacent cells in both hCMEC/D3 and Caco-2 cell barriers was observed to be sealed by tight junctions, which were visible as strong electron-dense structures (Fig. 6a and c). Additionally, to complement the information regarding quality of tight junctions from TEM, Triton X-100 was used as a chemical control for disruption of tight junctions. Tight junction disruption was observed in both barrier models using 4 KDa FITC-Dextran and TEER measurements in the presence of Triton X-100 (ESI Fig. 4a and b† respectively).
 | ||
| Fig. 6 Sub-cellular characteristics of hCMEC/D3 endothelial (a and b) and Caco-2 epithelial (b and d) cell barriers. Tight junctions were found to seal the paracellular space between adjacent cells in the endothelial (a) and the epithelial (c) cell monolayers. Different cellular compartments displayed in (b) and (d) included a variety of organelles. Abbreviations: BMV, basal membrane vesicle; EP, endocytic pit; L, lysosome; M, mitochondria; MV, multivesicular body; N, nucleus; R, ribosome; TJs, tight junctions; V, vesicle. | ||
The sub-cellular compartments in the two barrier models are quite distinctive. In hCMEC/D3 monolayers, the cells normally have a big nucleus which occupies a large volume of the cytosolic space (Fig. 6b). Other major subcellular components, such as mitochondria, multivesicular bodies and lysosomes, are also observed (Fig. 6a and b). In Caco-2 monolayers, the apical cell membranes are covered by intensive microvilli (Fig. 6c and d), a series of cellular membrane protrusions, which are involved in adsorption of nutrients. Within the intracellular compartment, in addition to mitochondria, multivesicular bodies, and lysosomes seen in TEM, Caco-2 cells also showed a large amount of ribosomes surrounding the nucleus (Fig. 6d). Endocytotic pits or vesicles were found in both hCMEC/D3 and Caco-2 cells (Fig. 6a and d), presumably undertaking a similar function of cellular uptake processes. Overall, as presented in Fig. 6, it is clear that the degree of sub-cellular ultrastructure visualised by TEM imaging offers an important tool to study morphological changes, barrier integrity and transport mechanisms across the cell barrier models.
3.2 Nanoparticle uptake and transport
To characterise transport of nanoparticles across the hCMEC/D3 cell monolayer, 12 nm gold nanoparticles conjugated with human serum albumin were applied. After a 4 hour incubation, albumin–gold nanoparticles demonstrated a series of active uptake events, which involved both endocytic and exocytic processes. As demonstrated in Fig. 7, vesicle sorting was observed to transport single albumin–gold nanoparticles within the cytoplasm of endothelial cells (Fig. 7a and b), presumably following cell membrane endocytosis (a related event for this, the membrane invagination in Fig. 7a that might lead to subsequent vesicle sorting). The endothelial paracellular space was tightly sealed by tight junctions (seen in Fig. 7b) thus paracellular transport was restricted. The conjugation of albumin to 12 nm gold nanoparticles was potentially responsible for initiating the observed vesicle sorting. Colocalisation of albumin–gold nanoparticles within lysosomal pathway-related organelles such as endosomes (Fig. 7c) and lysosomes (Fig. 7d) was observed. The transport of albumin–gold nanoparticles was dominated by the recycling process of lysosomes, although the attached albumin was expected to lead to active transcytosis across endothelial cells. In all cases, the endo-lysosomal pathway played a major role in the internalisation of albumin-coated gold nanoparticles in the human brain endothelial cells, as the nanoparticles were linked with endogenous proteins thought to promote endothelial receptor-mediated transcytosis.24,34,35 Although it was only rarely observed from previous reports,10,22 a transcytosis event was demonstrated in Fig. 7e, where an exocytotic vesicle docking the basal membrane containing a single gold nanoparticle inside could be observed, which seemed to discharge the nanoparticle into the basal compartment (Fig. 7f). | ||
| Fig. 7 Translocation of 12 nm human serum albumin–gold nanoparticles (Au-NPs) (50 μg ml−1) across the hCMEC/D3 endothelial cell monolayer post 4 hour incubation. (a) Clathrin-coated pit cavity was invaginated on the apical membrane and Au-NPs were found in sorting endocytic vesicles within cytosolic space (the arrows). (b) Au-NPs were trafficked to different cytoplasmic regions as indicated by the arrows. Au-NPs were found co-localised within the endosome (c) and lysosome (d) respectively. A rare event, whereby a single Au-NP exited the basolateral cell membrane from a vesicle, is shown in (e) and the Au-NP present in the basal compartment is observed (f). Abbreviations: EP, endocytic pit; ER, endoplasmic reticulum; EV, exocytic vesicle; E, endosome; L, lysosome; M, mitochondria; N, nucleus; TJ, tight junction. | ||
In the Caco-2 cell monolayer, nanoparticle internalisation was demonstrated using 50 nm SiO2 nanoparticles, following a 9 hour incubation. TEM revealed a shortening and disorganisation of the microvilli on the apical cell membrane (compared to Fig. 3f), suggesting potential disruption resulting from nanoparticle exposure (Fig. 8a and b). The integrity of the Caco-2 epithelial monolayer was not altered as tight junctions were still observable (Fig. 8b). Similar to the hCMEC/D3 cell barrier, 50 nm SiO2 nanoparticles were mainly internalised in endo-lysosomal pathway-related organelles, such as endosomes (Fig. 8c), multivesicular bodies (Fig. 8d) and lysosomes (Fig. 8a, b, d and e). A single nanoparticle seemed to be trafficked within a sorting vesicle and transported close to the basal membrane of the monolayer (Fig. 8f), although little evidence of transcytosis of SiO2 nanoparticles across the Caco-2 cell barrier was found by TEM.
 | ||
| Fig. 8 Internalisation of 50 nm SiO2 nanoparticles (SiO2-NPs) (100 μg ml−1) by Caco-2 cell monolayer post 9 hour exposure. (a) Microvilli appeared to lose integrity and SiO2-NPs were found co-localised within a lysosome (indicated by the arrow). (b) Tight junctions remained associated with neighbouring cells, maintaining the integrity of the Caco-2 barrier, and one single SiO2-NP was also found in a lysosome (the arrow). More nanoparticles were found co-localised with late endosome (c), microvesicular body (d) and lysosomes (d and e). Another single SiO2-NP was seen in a sorting vesicle (f). Abbreviations: CCV, clathrin-coated vesicle; E, endosome; LE, late endosome; L, lysosome; M, mitochondria; MV, multivesicular body; N, nucleus. | ||
4. Discussion
4.1 Barrier models – cell types
The human blood–brain barrier is a structure in brain capillary vessels, where a single layer of endothelial cells lines the wall of blood vessels and forms an impermeable barrier regulating molecule access to the central nerves system (CNS). The functions of the blood–brain barrier are to maintain the extracellular environment of the central nervous system (CNS), balance brain fluid homeostasis via the blood stream and protect the CNS from injury and disease. One well-known model involves human capillary microvascular endothelial cells (hCMEC/D3) grown on collagen-coated permeable filters.8–13,22 Once polarised in vitro, these cells express typical endothelial markers such as CD31, VE-cadherin, maintain contact-inhibited monolayers, and demonstrate excellent tight junction formation and a high capacity to exclude drugs.8,12,13 The hCMEC/D3 cell monolayer has been used to investigate compound transport mechanisms, active transport of proteins such as ABCB1 and ABCC1 that mediate the active efflux machinery for drugs,8,36 as well as to investigate the transport of nanoparticles across the BBB.10,11,22The intestinal barrier is part of the human gastrointestinal (GI) tract and functions as an absorption interface between the external environment and the body.37 The intestinal epithelia are mainly responsible for absorption of nutrients in the GI tract. Human intestine is covered by a single layer of epithelial cells that form an associated network and regulate the intake of biomolecules and drug compounds from intestinal brush-border membranes to the blood.7 The Caco-2 cell barrier is well-characterised and known for its compatibility with various permeable filters used to predict drug absorption and permeability. In culture, Caco-2 cells can slowly seal the Transwell membrane and spontaneously differentiate into a monolayer with many analogous functions to the in vivo small intestinal villus epithelium.7,14,23 Some of the most representative subcellular structures, such as tight junctions, drug transport resistant proteins (e.g., P-gp, BCRP or MRP2) and microvilli consistently appear following polarisation of Caco-2 cell monolayers. Using such a confluent layer of Caco-2 cells, a small piece of human gastrointestinal epithelia can be easily assembled for experimental purposes.
4.2 Transwell filters – material and pore size
Transwell permeable supports are commercially available in three membrane materials: polycarbonate (PC), polyester (PET), and collagen-coated polytetrafluoroethylene (PTFE), depending on the application and the measurement modality to be utilised. Selecting the correct pore size (typically 0.4, 1.0, 3.0, 8.0 microns) for experiments using Transwell® permeable supports is very important, both to ensure good cell growth and viability and to ensure optimal barrier morphology. Additionally, the membranes can be coated with a variety of extra-cellular matrix components, such as collagen and fibrinogen. Transwell manufacturers such as Corning provide excellent technical guidance on the selection of appropriate Transwells for different applications. The TEM protocol outlined here enables detailed assessment of the morphology of the resultant barrier monolayers and can be used to determine whether further optimisation of the growth conditions (including the Transwell filter composition, coating or pore size) is required, for example if cells are observed to be growing into the pores, or if significant gaps remain in the surface coverage of the Transwell filter.For studies involving nanoparticle uptake or transport, an additional set of considerations emerge regarding the potential for interaction of the nanoparticles with the pores of the Transwell membranes. We found that nanoparticles can potentially adhere to the pores in various types of filters, remaining within the filter without moving through to the basolateral chamber and resulting in false negative results for transport studies based on determination of particles in the basolateral chamber.10 Therefore, it is necessary to perform quality control experiments whereby interactions between the nanoparticles and the selected Transwells are performed in the absence of cells. TEM imaging is an excellent tool to assess whether these issues apply with the selected in vitro Transwell system.
4.3 Electron microscopy preparation and imaging considerations
The protocol from 2010 for preparation of cells for assessing ultrastructural localisation of nanoparticles with TEM,19 gives an excellent summary of many of the critical aspects of TEM imaging considerations, which are also relevant to this protocol. Thus, we focus here only on those aspects of relevance specifically for the in vitro barrier models, both for quality assurance purposes and for nanoparticle transport studies across biological barriers.In particular, it is valuable to mention that the filter membrane plays an important role in TEM imaging of biological barriers, not only because it supports barrier formation, but also because it acts as a reference object for orientation of the slides during image analysis. The protocol given in detail above describes a technique that protects the monolayer-filter cross sections from falling apart and enables determination of the orientation of cell monolayer polarity.
TEM was used as the primary tool to control the quality of biological barriers and to visualize their cellular structures, including those related to barrier formation such as tight junctions. TEM also enabled visualisation of the localisation of nanoparticles in cellular organelles such as endosomes, lysosomes and at the basolateral membrane (if transcytosis was occurring). More importantly, an important goal of using TEM imaging was to capture events of nanoparticle interaction with cell monolayers and acquire conclusive evidence to reveal transport mechanisms for nanoparticles.
Because Transwell filters may physically trap nanoparticles and hinder their passage through the pores, resulting in quantitative loss of nanoparticles and deviation of experimental data despite their being transcytosed by endothelial or epithelial cell monolayers,10 it is essential to assess the interaction of nanoparticles with the Transwells in the absence of cells also.
Understanding the direction of nanoparticles travel from one side of the barrier to the other is vital for the determination of transport routes, particularly when investigating endocytosis, or exocytosis versus transcytosis processes in which the directions of movement are opposed. Although TEM users are presumably capable of identifying the apical or basal sides of polarized endothelial monolayer from experience, preserving the filters which exist underneath the BBB layer provides an additional orientation guide. Additionally, it is likely that filter membrane removal from cell monolayer sections may cause loss of critical details under the basal cell membrane and lead to light overexposure during object imaging, especially when studying nanoparticle trafficking processes, exocytosis or transcytosis. The protocol reported here preserves the filter intact with the barrier cells throughout sectioning, ensuring no loss of vital information, and providing an orientation guide for imaging and data interpretation (as well as information regarding particle interaction with the membrane pores).
Another potential problem with transport studies across barriers utilising fluorescently-tagged nanoparticles is that the quantification of nanoparticles in the basolateral chamber is often performed via measurement of fluorescence from the nanoparticles. However, it is becoming quite well known that the dyes incorporated into nanoparticles may leak from the nanoparticles, e.g., silica10,38 or polystyrene39 nanoparticles, potentially resulting from the influence of the biological conditions, such as temperature, pH and cell culture medium.21 Dye leakage may cause false signals during nanoparticle quantification, especially in cases where the nanoparticles themselves remain trapped in the barrier cells or the filters, and only TEM imaging can demonstrate the precise location of the particles visually.
4.4 Confirmation of barrier functionality via TEER and FD transport
Prior to transport studies it is standard practice to perform some well-established tests to confirm that the barrier functionality has been attained, such as the FITC-Dextran (FD) transport assay to test permeability and transepithelial/transendothelial electrical resistance (TEER) measurement. The principles of these two techniques are related to the restrictive effect of a functional barrier on paracellular transport of molecules or ions. FD is a paracellular marker which can passively diffuse through the intercellular space of biological barriers.40,41 When a cell monolayer is established in a Transwell system, FD is applied to the upper compartment, and is able to travel through the barrier and reach the lower compartment. Using a time-elapsed sampling method, transported FD is quantified on the basis of the fluorescence in the basolateral side, whereby a fluorescence versus concentration flux curve is obtained. According to the previously reported equation,7,42 the permeability of FD can be calculated via the slope of flux curve and the initial FD dosing concentration. On the other hand, small ions passing through a cell barrier can be accessed via TEER which measures the impediment introduced by tight junctions formed in a confluent cell barrier.43 Depending on the type of barrier (endothelial or epithelial), TEER is either trans-endothelial or trans-epithelial electrical resistance. By applying an electrical current over the two sides of a Transwell system, a voltammeter reading can be acquired which is the resistance provided by the barrier. 4 KDa FITC-Dextran (FD4) transport and TEER measurements have been widely used as parameters to evaluate the status of barrier integrity.8,11,13,16,41 In a barrier model, their results provide information about tight junction expression quality and allow comparison with other reported results obtained and with in vivo values (although in vitro values are consistently lower).4.5 Nanoparticle types
The selection of nanoparticles used for TEM imaging analysis is dependent on their electron density. Many nanoparticles, including silica, titania, iron, gold, silver and other heavy metals, retain a rather high density under electron beam compared to biological samples (e.g., cells), which therefore provides a strong imaging contrast and excellent visibility for imaging studies. By contrast, other nanomaterials, such as polystyrene and liposomes, have an electron density that is closer to that of cells or tissues, therefore it is difficult to visualize and distinguish their structures in a complex biological environment by TEM. However, using a larger diameter of nanoparticles can help improve their visibility as well as improve image contrast in TEM imaging. It was reported that low-density polystyrene nanoparticles (100–160 nm) could be observed within cells using TEM,9,22,44 although their imaging relied on an improved contrast of cells provided by an optimised osmium tetroxide staining protocol. Visualization of small objects, for example, 1 nm gold nanoparticles within a cell, would require high resolution TEM imaging,45 since their contrast compared to background cellular components would be negligible. In our work, the sizes of our selected nanoparticles ranged from 10 to 50 nm, and both particle types (gold and silica) provided good contrast and could be appropriately visualized under TEM.Efficient nanoparticle internalisation and translocation within a cell is dependent on having a good quality starting dispersion (among other factors). Although nanoparticle agglomerates have a strong electron density under TEM, they limit effective interaction with cells and hinder access to cellular uptake pathways, such as endocytosis and transcytosis. Exocytotic vesicles seem to have a size limit of ∼100 nm from our results, and thus have a limited capacity to transport agglomerated nanoparticles.
A last factor that needs to be taken into account when preparing nanoparticle dispersions for cellular uptake studies is the cell medium composition, as serum proteins used to supplement the medium can absorb to nanoparticles and form a protein corona,46–48 which is now known to mitigate targeting abilities of functionalised nanoparticles.49 A differential uptake efficiency of nanoparticles in the absence (serum-free) or presence of protein coronas of different composition has been illustrated with TEM imaging for cells.10,38,50 However, serum-free uptake generally results from significant membrane damage and is not considered physiologically relevant, as particles would be coated with biomolecules immediately upon contact with the body (e.g. in the gut or lungs the relevant mucus/surfactant proteins bind to the particle surfaces).
Acknowledgements
This research has been supported by the QualityNano Research Infrastructure INFRA-2010-1.1.31-262163 (I.L.). Part of this work was conducted under the framework of the INSPIRE programme, funded by the Irish Government's Programme for Research in Third Level Institutions, Cycle 4, National Development Plan 2007–2013 (D.Y.). Albumin-coated gold nanoparticles and silica nanoparticles from Dr Eugene Mahon and Mr Delyan R. Hristov respectively (funded via the INSPIRE programme) and assistance from Dr Michelle Nic Ragnaill (funded via EU FP7 Small Collaborative project NeuroNano, NMP4-SL-2008-214547), Centre for BioNano Interactions are gratefully acknowledged. Immortalized human brain capillary endothelial cells (hCMEC/D3) were obtained from Drs Pierre-Olivier Couraud (INSERM, France), Ignacio A. Romero (Open University, UK) and Babette Weksler (Weill Cornell Medical College, New York). Use of the UCD Electron Microscopy Core facility is also acknowledged.References
- H. Spielmann, U. G. Sauer and O. Mekenyan, Altern. Lab. Anim., 2011, 39, 481–493 CAS.
- N. Ball, The Way Forward in AltTox Website, 2012 Search PubMed.
- A. P. Worth and M. Balls, Alternative (non-animal) methods for chemicals testing: current status and future prospects, Fund for the Replacement of Animals in Medical Experiments, Atla, 2002 Search PubMed.
- L. Edler and C. Ittrich, ATLA, Altern. Lab. Anim., 2003, 31, 5–42 CAS.
- N. Pfeifer, D. M. Baston-Büst, J. Hirchenhain, U. Friebe-Hoffmann, D. T. Rein, J. S. Krüssel and A. P. Hess, Sci. World J., 2012, 2012, 479315–479323 CrossRef CAS PubMed.
- A. Barragan and L. David Sibley, Trends Microbiol., 2003, 11, 426–430 CrossRef CAS PubMed.
- I. Hubatsch, E. G. Ragnarsson and P. Artursson, Nat. Protoc., 2007, 2, 2111–2119 CrossRef CAS PubMed.
- B. B. Weksler, E. A. Subileau, N. Perriere, P. Charneau, K. Holloway, M. Leveque, H. Tricoire-Leignel, A. Nicotra, S. Bourdoulous, P. Turowski, D. K. Male, F. Roux, J. Greenwood, I. A. Romero and P. O. Couraud, FASEB J., 2005, 19, 1872–1874 CAS.
- M. Nic Raghnaill, M. Bramini, D. Ye, P.-O. Couraud, I. A. Romero, B. Weksler, C. Åberg, A. Salvati, I. Lynch and K. A. Dawson, Analyst, 2014, 139, 923–930 RSC.
- D. Ye, M. N. Raghnaill, M. Bramini, E. Mahon, C. Åberg, A. Salvati and K. A. Dawson, Nanoscale, 2013, 5, 11153–11165 RSC.
- M. Nic Raghnaill, M. Brown, D. Ye, M. Bramini, S. Callanan, I. Lynch and K. A. Dawson, Eur. J. Pharm. Biopharm., 2011, 77, 360–367 CrossRef PubMed.
- B. Poller, H. Gutmann, S. Krahenbuhl, B. Weksler, I. Romero, P. O. Couraud, G. Tuffin, J. Drewe and J. Huwyler, J. Neurochem., 2008, 107, 1358–1368 CrossRef CAS PubMed.
- C. Forster, M. Burek, I. A. Romero, B. B. Weksler, P. O. Couraud and D. Drenckhahn, J. Physiol., 2008, 586, 1937–1949 CrossRef PubMed.
- S. Tavelin, J. Gråsjö, J. Taipalensuu, G. Ocklind and P. Artursson, in Methods Mol Biol, ed. C. Wise, Humana Press, Totowa, New Jersey, 2002, vol. 188, pp. 233–272 Search PubMed.
- C. Grobstein, Nature, 1953, 172, 869–870 CrossRef CAS PubMed.
- M. Sakai, T. Imai, H. Ohtake, H. Azuma and M. Otagiri, J. Pharm. Sci., 1997, 86, 779–785 CrossRef CAS PubMed.
- P. Prieto, B. J. Blaauboer, A. G. De Boer, M. Boveri, R. Cecchelli, C. Clemedson, S. Coecke, A. Forsby, H.-J. Galla and P. Garberg, ATLA, Altern. Lab. Anim., 2004, 37–50 CAS.
- A. Elsaesser, C. A. Barnes, G. McKerr, A. Salvati, I. Lynch, K. A. Dawson and C. V. Howard, Nanomedicine, 2011, 6, 1189–1198 CrossRef CAS PubMed.
- A. M. Schrand, J. J. Schlager, L. Dai and S. M. Hussain, Nat. Protoc., 2010, 5, 744–757 CrossRef CAS PubMed.
- R. M. C. Dawson, D. C. Elliott, W. H. Elliott and K. M. Jones, Data for biochemical research, Clarendon Press, Oxford, 2nd edn, 1969 Search PubMed.
- E. Mahon, D. R. Hristov and K. A. Dawson, Chem. Commun., 2012, 48, 7970–7972 RSC.
- M. Bramini, D. Ye, A. Hallerbach, M. N. Raghnaill, A. Salvati, C. Åberg and K. A. Dawson, ACS Nano, 2014, 8, 4304–4312 CrossRef CAS PubMed.
- J. Karlsson and P. Artursson, Biochim. Biophys. Acta, Biomembr., 1992, 1111, 204–210 CrossRef CAS.
- L. Ghitescu, A. Fixman, M. Simionescu and N. Simionescu, J. Cell Biol., 1986, 102, 1304–1311 CrossRef CAS PubMed.
- N. J. Abbott, L. Ronnback and E. Hansson, Nat. Rev. Neurosci., 2006, 7, 41–53 CrossRef CAS PubMed.
- D. Liu, L. M. Bimbo, E. Makila, F. Villanova, M. Kaasalainen, B. Herranz-Blanco, C. M. Caramella, V. P. Lehto, J. Salonen, K. H. Herzig, J. Hirvonen and H. A. Santos, J. Controlled Release, 2013, 170, 268–278 CrossRef CAS PubMed.
- L. M. Bimbo, M. Sarparanta, E. Makila, T. Laaksonen, P. Laaksonen, J. Salonen, M. B. Linder, J. Hirvonen, A. J. Airaksinen and H. A. Santos, Nanoscale, 2012, 4, 3184–3192 RSC.
- M. P. Sarparanta, L. M. Bimbo, E. M. Mäkilä, J. J. Salonen, P. H. Laaksonen, A. M. K. Helariutta, M. B. Linder, J. T. Hirvonen, T. J. Laaksonen, H. A. Santos and A. J. Airaksinen, Biomaterials, 2012, 33, 3353–3362 CrossRef CAS PubMed.
- L. M. Bimbo, E. Mäkilä, T. Laaksonen, V.-P. Lehto, J. Salonen, J. Hirvonen and H. A. Santos, Biomaterials, 2011, 32, 2625–2633 CrossRef CAS PubMed.
- L. M. Bimbo, M. Sarparanta, H. A. Santos, A. J. Airaksinen, E. Mäkilä, T. Laaksonen, L. Peltonen, V.-P. Lehto, J. Hirvonen and J. Salonen, ACS Nano, 2010, 4, 3023–3032 CrossRef CAS PubMed.
- J. A. Loureiro, B. Gomes, M. A. Coelho, M. d. Carmo Pereira and S. Rocha, Nanomedicine, 2014, 9, 709–722 CrossRef CAS PubMed.
- A. Yildirim, E. Ozgur and M. Bayindir, J. Mater. Chem. B, 2013, 1, 1909–1920 RSC.
- Y. Loo, C. L. Grigsby, Y. J. Yamanaka, M. K. Chellappan, X. Jiang, H. Q. Mao and K. W. Leong, J. Controlled Release, 2012, 160, 48–56 CrossRef CAS PubMed.
- H. H. Li, J. Li, K. J. Wasserloos, C. Wallace, M. G. Sullivan, P. M. Bauer, D. B. Stolz, J. S. Lee, S. C. Watkins, C. M. St Croix, B. R. Pitt and L. M. Zhang, PLoS One, 2013, 8, e81903 Search PubMed.
- V. Tenten, S. Menzel, U. Kunter, E. M. Sicking, C. R. van Roeyen, S. K. Sanden, M. Kaldenbach, P. Boor, A. Fuss, S. Uhlig, R. Lanzmich, B. Willemsen, H. Dijkman, M. Grepl, K. Wild, W. Kriz, B. Smeets, J. Floege and M. J. Moeller, J. Am. Soc. Nephrol., 2013, 24, 1966–1980 CrossRef CAS PubMed.
- L. M. Tai, P. S. Reddy, M. A. Lopez-Ramirez, H. A. Davies, A. D. K. Male, A. J. Loughlin and I. A. Romero, Brain Res., 2009, 1292, 14–24 CrossRef CAS PubMed.
- A. Farhadi, A. L. I. Banan, J. Fields and A. L. I. Keshavarzian, J. Gastroenterol. Hepatol., 2003, 18, 479–497 CrossRef PubMed.
- K. Shapero, F. Fenaroli, I. Lynch, D. C. Cottell, A. Salvati and K. A. Dawson, Mol. BioSyst., 2011, 7, 371–378 RSC.
- A. Salvati, C. Åberg, T. dos Santos, J. Varela, P. Pinto, I. Lynch and K. A. Dawson, Nanomedicine, 2011, 7, 818–826 CrossRef CAS PubMed.
- Z. G. Zhang, L. Zhang, W. Tsang, H. Soltanian-Zadeh, D. Morris, R. Zhang, A. Goussev, C. Powers, T. Yeich and M. Chopp, J. Cereb. Blood Flow Metab., 2002, 22, 379–392 CrossRef CAS PubMed.
- G. Borchard, H. L. Lueβen, A. G. de Boer, J. Verhoef, C.-M. Lehr and H. E. Junginger, J. Controlled Release, 1996, 39, 131–138 CrossRef CAS.
- P. Artursson, J. Pharm. Sci., 1990, 79, 476–482 CrossRef CAS PubMed.
- N. F. Fletcher, D. J. Brayden, B. Brankin, S. Worrall and J. J. Callanan, Vet. Immunol. Immunopathol., 2006, 109, 233–244 CrossRef CAS PubMed.
- A. Musyanovych, J. Dausend, M. Dass, P. Walther, V. Mailander and K. Landfester, Acta Biomater., 2011, 7, 4160–4168 CrossRef CAS PubMed.
- Y.-G. Kim, S.-K. Oh and R. M. Crooks, Chem. Mater., 2004, 16, 167–172 CrossRef CAS.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggård, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- M. P. Monopoli, D. Walczyk, A. Campbell, G. Elia, I. Lynch, F. B. Bombelli and K. A. Dawson, J. Am. Chem. Soc., 2011, 133, 2525–2534 CrossRef CAS PubMed.
- M. P. Monopoli, C. Åberg, A. Salvati and K. A. Dawson, Nat. Nanotechnol., 2012, 7, 779–786 CrossRef CAS PubMed.
- A. Salvati, A. S. Pitek, M. P. Monopoli, K. Prapainop, F. B. Bombelli, D. R. Hristov, P. M. Kelly, C. Åberg, E. Mahon and K. A. Dawson, Nat. Nanotechnol., 2013, 8, 137–143 CrossRef CAS PubMed.
- A. Lesniak, F. Fenaroli, M. P. Monopoli, C. Åberg, K. A. Dawson and A. Salvati, ACS Nano, 2012, 6, 5845–5847 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Uptake and viability of Caco-2 barrier, effects of collagen coating and seeding density on hCMEC/D3 barrier, FD4 and TEER measurements. See DOI: 10.1039/c4an01276c |
| This journal is © The Royal Society of Chemistry 2015 |