DOI:
10.1039/C4TB01401D
(Communication)
J. Mater. Chem. B, 2015,
3, 25-29
Dual secured nano-melittin for the safe and effective eradication of cancer cells†
Received
22nd August 2014
, Accepted 28th October 2014
First published on 28th October 2014
Abstract
The clinical application of natural and synthetic amphipathic peptides (e.g., melittin) for cancer therapy is hindered by their notorious side effect, lysing red blood cells. To safely deliver a therapeutic peptide to the tumor tissue and kill cancer cells, we developed an environment-sensitive peptide delivery system, dual secured nano-sting (DSNS), through the combination of a zwitterionic glycol chitosan and disulfide bonds. Melittin loaded DSNS could kill almost 100% of MCF-7, HCT-116, SKOV-3, and NCI/ADR-RES (multidrug resistant) cancer cells at the concentration of 5 μM, while not showing any hemolytic effect.
Introduction
The host defense amphipathic peptides found in eukaryotic cells have diverse activities in human and other species, originating from their antibiotic, anticancer and anti-inflammatory activities.1 These peptides oligomerize with phospholipids in the cell membrane, result in pore formation, and subsequently cause cell death. In addition, they act in a similar way on the membranes of internal organelles after intracellular transport, and induce cell apoptosis.2 Amphipathic peptides have been explored for cancer chemotherapy because of their wide-spectrum lytic properties. Melittin is one of the most promising amphipathic water-soluble α-helical cationic polypeptides and is derived from toxin of the honey bee Apis melllifera.3 Melittin partitions into and moves laterally in the cell membranes as monomers, followed by oligomerization into toroidal structures, forming pores, which results in cell death.3,4 Furthermore, the most recent research showed that melittin can induce cancer cell apoptosis through the inhibition of the JAK2/STAT3 pathway.5 It is worth mentioning that melittin also suppresses the constitutively activated NF-κB, which is partially responsible for the development of drug resistance in cancer cells.6 It is a very attractive cancer therapeutic agent, because cancer cells are less likely to develop resistance to cytolytic peptides.1a,7
Despite all of these advantages, its non-specific cytolytic activity could lead to off-target effects such as hemolysis (lysis of red blood cells) when administrated intravenously. Besides that, positively charged peptides could be cleared from blood circulation rapidly by the reticuloendothelial system (RES) system.8 Several groups developed melittin delivery systems either by covalently fusing melittin with receptor-targeted peptide motifs or through physically encapsulating it into liposomes or polymer nanoparticles to attenuate its hemolytic effect while achieving therapeutic efficiency comparable to free melittin.9 Compared with free melittin, their anticancer efficacies were significantly decreased for the encapsulated form. Until recently, Soman et al. developed a liposome based melittin nanocarrier (“nanobee”), which showed promising results in inhibiting the growth of melanoma tumors.10 Despite the encouraging outcome of the “nanobee”, they also found that the “nanobee” was about five-fold less effective than that of melittin for the tested cancer cells.
An ideal melittin carrier should be able to completely quench its hemolytic activity while fully retaining its advantages, including a wide-spectrum and potent anticancer ability. To solve this dilemma, we rationally designed a melittin delivery system by integrating a zwitterionic glycol chitosan and disulfide bonds. Due to its zwitterionic property, succinic anhydride modified glycol chitosan (SA-GCS) shows negative surface charges at the physiological pH. Positively charged melittin can form complexes with SA-GCS through the electrostatic effect. The complex is further stabilized through disulfide crosslinking to yield the dual secured nano-sting (DSNS) by aerial oxidation (Scheme 1).
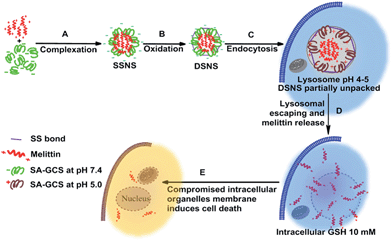 |
| | Scheme 1 Schematic illustration of the formation and intracellular pathway of DSNS. | |
Results and discussion
Synthesis of thiolated zwitterionic glycol chitosan
The zwitterionic glycol chitosan was synthesized from glycol chitosan by amidation with succinic anhydride. First, glycol chitosan was depolymerized by potassium persulfate according to the literature and purified by dialysis against DI water.11 The resulting polymer had a molecular weight of 28 kDa and PDI of 1.38 (Fig. S1†). After that, glycol chitosan was amidized according to our previously published method with succinic anhydride (Fig. S2†).8,12 SA-GCS showed a negative surface charge at pH 7.4, and positive surface charge at pH below its isoelectric point (IEP) (Fig. 1B). Furthermore, the IEP of the amidized glycol chitosan can be tuned by adjusting the feeding ratio of succinic anhydride and glycol chitosan. To introduce free thiol groups, SA-GCS was reacted with N-succinimidyl 3-[2-pyridyldithio]-propionate (SPDP) and subsequently cleaved with tris(2-carboxyethyl)phosphine (TCEP) to achieve the thiolated amidized glycol chitosan (SA-GCS-SH) (Fig. 1A). The DTNB assay showed that each polymer chain contains 8.7 free SH groups. The IEP of the SA-GCS slightly decreased after the thiolation (Fig. 1B).
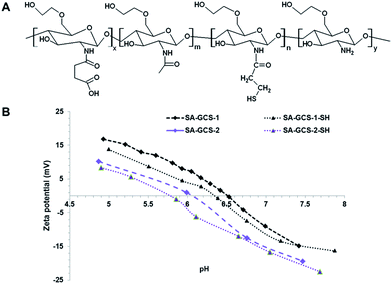 |
| | Fig. 1 The structure of SA-GCS-SH (A) and the surface charges of SA-GCS and SA-GCS-SH at different pHs (B). | |
Fabrication and characterization of melittin–polymer complexes
To verify that zwitterionic glycol chitosan can form complexes with the positively charged melittin, we fabricated the single secure nano-sting (SSNS) by mixing SA-GCS with melittin at pH 7.4 for 2 h at room temperature. The binding efficiency for SA-GCS was determined by measuring the fluorescence intensity of the tryptophan residue of melittin at λEX: 280 nm, λEM: 350 nm. The fluorescence measurement showed that with the increase of the SA-GCS polymer, the detectable free melittin gradually decreased and achieved 100% encapsulation at the polymer to melittin ratio (w/w) of 40 (Fig. S3†).
To further stabilize the complex, inhibit its premature release of melittin, and eliminate its potential side effect, we substituted the SA-GCS with SC-GCS-SH and aerially oxidized the complex to promote the formation of disulfide bonds among the SA-GCS-SH polymers to achieve so called dual secured nano-sting (DSNS). Since safety is an essential requirement for melittin related delivery, a polymer to melittin ratio (w/w) of 200 was selected to ensure that no free melittin remained after the formation of the complexes. The formation of DSNS was confirmed by dynamic light scattering (DLS) (Fig. S4†) and transmission electron microscopy (TEM) (Fig. 2). The hydrodynamic size of the SSNS (220.2 nm, PDI: 0.191) was slightly increased to 223.4 nm after oxidation (PDI: 0.161). The size determined by DLS was larger than that obtained by TEM. This is because the TEM measured the size of solid particles while DLS measured the hydrodynamic size of the particles, which includes the water layer surrounding the particle. This slight size difference between the SSNS and DSNS reflected the size decrease and increase due to the formation of intra-particle and inter-particle crosslinking, respectively. The surface charge of both nano-complexes at pH 7.4 was slightly negative (Fig. S5†), which helps the nano-complexes escape from the detection of the reticuloendothelial system and take advantage of the enhanced permeability and retention effect (EPR) of tumor tissue.13 HPLC confirmed that no free melittin existed in the particle suspensions of SSNS and DSNS (Fig. S6†).
 |
| | Fig. 2 Transmission electron microscopy images of SSNS (A) and DSNS (B). Scale bars are 100 nm. | |
Investigate the pH responsiveness of nano-complexes by FRET
To evaluate the stability of the SSNS and DSNS, Förster resonance energy transfer (FRET) technology was employed.14 Before the fabrication of SSNS and DSNS, melittin and a zwitterionic polymer were conjugated with sulfo-Cy5-NHS and Cy3-NHS, respectively. Cy5-melittin was mixed with Cy3-SA-GCS and Cy3-SA-GCS-SH to achieve the SSNS and DSNS, respectively (Fig. 3A). The DSNS exhibited a higher FRET signal than the SSNS (Fig. 3B) at pH 7.4, indicating that the DSNS was tighter than the SSNS. To evaluate the nano-sting stability at different pH environments, the FRET signal was recorded in the pH range from 7.4 to 3.7. As the pH shifted from 7.4 to the IEPs of the polymers, the FRET intensities of both the SSNS and DSNS increased and reached the maximum at the pH close to the IEPs of the polymers, indicating the formation of more condensed nanoparticles. Similar to other zwitterionic macromolecules, SA-GCS showed lowest solubility at its IEP. The formation of a water insoluble polymer caused the condensation of the SSNS and DSNS, and resulted in the highest FRET signal. SA-GCS displayed a positive surface charge at pH lower than its IEP (Fig. 1B), which would induce the repulsion between SA-GCS and the positively charged melittin, similar to the scenario of a nano-complex inside a lysosome (Scheme 1). As expected, both the SSNS and DSNS displayed reduced FRET signals when environment pH was further decreased. At the pH of 3.7, the SSNS showed a FRET intensity far less than that at pH 7.4, indicating the dissociation of the nanoparticles. By contrast, the lowest FRET intensity that the DSNS reached at pH 3.7 was still higher than that of the SSNS at pH 7.4, suggesting that the formed disulfide bonds restricted melittin from premature release upon the fluctuation of pH. There was one pH unit left shift of the FRET curve from their corresponding IEPs, which we think was due to the lag response of the nano-complexes to the change in the environmental pH. The dual secured effect was also evidenced by the slower melittin release from the DSNS than SSNS, as well as more melittin released at pH 5.0 than at pH 7.4 (Fig. S7†).
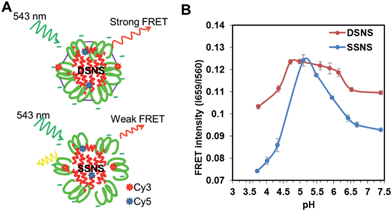 |
| | Fig. 3 The Schematic of FRET produced by DSNS and SSNS (A) and the measured FRET intensities of DSNS and SSNS at different pHs (B). | |
Investigating the hemolytic activity of the nano-complexes
To validate that the combination of the zwitterionic polymer coating and disulfide crosslinking can effectively quench the hemolytic activity of melittin in the DSNS, a hemolytic assay was carried out. The SSNS and DSNS were incubated with red blood cells (RBCs) in PBS (pH 7.4) first, followed by centrifugation to separate the intact RBCs from the released hemoglobin. As shown in Fig. 4A, melittin lysed almost all the RBCs at the concentration of 1 μM. The formation of the SSNS partially inhibited the hemolytic activity of melittin. In contrast, there was no detectable red color in the supernatant of the RBCs incubated with the DSNS at the melittin concentration of 5 μM. The hemolytic activities of the SSNS and DSNS were further quantified by a UV spectrophotometer. Fig. 4B shows that free melittin was highly lytic to RBCs, and it lysed almost 100% the RBCs at 2 μM, which is the major obstacle for its clinical application. The hemolytic activity of melittin in the SSNS was significantly quenched after its complexation with the zwitterionic glycol chitosan. The residual hemolytic activity indicated that some melittin was released when it was incubated with the RBCs. Further stabilized through the formation of disulfide bonds, DSNS did not show any hemolytic activity at 2 μM and only caused very few RBCs to lyse at the concentration of 5 μM. Therefore, we proved that the SSNS was safer than free melittin, while the DSNS was almost non-toxic to RBCs up to the melittin concentration of 5 μM in pH 7.4 buffer.
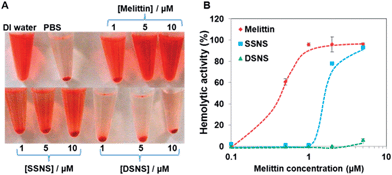 |
| | Fig. 4 Images of RBCs after hemolytic assay (A) and the hemolytic activity of melittin, SSNS, and DSNS (B). | |
To investigate the intracellular membrane lytic activity of the SSNS and DSNS, RBCs were co-incubated with melittin, SSNS, and DSNS in PBS (pH 5.0) buffer and PBS (pH 7.4) supplemented with 10 mM glutathione (GSH) to mimic the environments in an acidic lysosome and reducing cytosol, respectively. The acidic pH and reducing environment quenched the hemolytic activity of melittin (Fig. 5), which is consistent with the observation of others.15 SSNS at the concentration of 0.5 and 1.0 μM displayed considerably higher hemolytic activities in acidic pH than that in pH 7.4, suggesting the release of free melittin at low pH, which was consistent with our FRET observation in Fig. 3B. In contrast, because of the restraint of disulfide bonds, the acidic stimulus couldn't trigger the release of melittin from the DSNS (Fig. 3B), and induced only slightly more RBCs lysis (Fig. 5). As expected, the addition of 10 mM GSH to the pH 7.4 buffer significantly enhanced the DSNS's hemolytic activity, and it reached a similar level as that of the SSNS at the concentration of 2 and 5 μM (Fig. 5). Furthermore, the hemolytic activity of the DSNS was investigated in a 50% serum containing buffer to mimic blood. Fig. S8† reveals that the DSNS was also stable in blood simulating buffer, and did not cause RBC lysis. Based on these observations, we validated that the DSNS should be safe during circulation in the blood stream while effectively lysing intracellular organelles as illustrated in Scheme 1E.
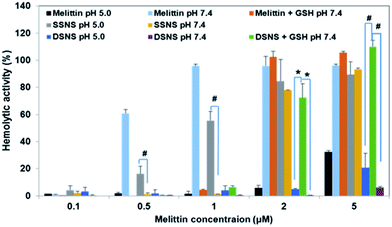 |
| | Fig. 5 Hemolytic activity of melittin, SSNS, and DSNS at different pHs and redox potential conditions. *p < 0.05 and #p < 0.01 (unpaired Student's t-test) of RBCs after hemolytic assay (A) and the hemolytic activity of melittin, SSNS, and DSNS (B). | |
Cellular uptake of nano-complexes
To investigate how the stability of the nano-complexes affects their cellular uptake, confocal microscopy was employed. The SSNS and DSNS were fabricated as described above except that Cy3-SA-GCS was used instead of SA-GCS. More red spots were detected in cells treated with the DSNS than SSNS (Fig. S9†), which suggested that more DSNS nano-complexes entered cancer cells intact than their SSNS counterparts, while some SSNS had dissociated before endocytosis, as evidenced by a lower Cy3 labeled SA-GCS uptake. Since the DSNS was more stable than the SSNS, as shown in Fig. 3 and further proven by Fig. 4, the DSNS won't prematurely release melittin when in contact with serum proteins and red blood cells (Fig. S9†). In contrast, the SSNS was only stabilized by electrostatic effects, which can dissociate by the competing effect of serum proteins. Therefore, more DSNS entered cancer intact than SSNS.
Cell killing effect of the nano-complexes
Due to the limitation of the SSNS associated unwanted hemolytic toxicity, further anticancer efficacy evaluations only included the DSNS. NCI/ADR-RES (OVCAR-8 adriamycin-resistant ovarian) cancer cells were co-cultured with free melittin and DSNS (melittin concentration of 5 μM) for 24 h.16 MTT reagent (3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide) was added after that. Living cells could convert the MTT reagent into water insoluble purple crystals (Fig. 6A). The absence of crystals in both the melittin and DSNS treated cells indicated that the cells in both treatments were dead. To investigate the possible mechanism of cell death, we examined the cell morphology after treatment. Cells collapsed after co-incubating with free melittin and lost its original shape (Fig. 6B). In contrast to its free melittin treated counterpart, the cells in Fig. 6C kept their intact shape after the DSNS treatment. Since melittin can attack cancer cells by forming pore structures on cell membranes,17 we postulate the cell death in the melittin treatment group was mainly due to the loss of cell membrane integrity. DSNS, due to the dual-secured mechanism, could effectively enter cancer cells (Fig. S9†) and release melittin intracellularlly (Scheme 1 and Fig. 5). Therefore, we postulate that the DSNS treated cells were killed mainly due to the compromised membranes of the internal organelles (e.g., mitochondria). After co-incubation with DSNS followed by JC-1 staining, the emerging green fluorescence signals in the DSNS treated cell (Fig. S10†) confirmed that the cancer cells were killed due to mitochondria damage.
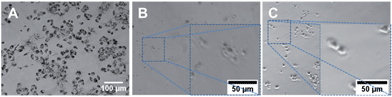 |
| | Fig. 6 The morphology of NCI/ADR-RES cells treated with 5 μM melittin. (A) Control, (B) free melittin, (C) DSNS. | |
The anticancer efficacy of the DSNS was further quantitatively evaluated in four types of cancer cells, HCT-116 colon cancer cells, MCF-7 breast cancer cells, SKOV-3 ovarian cancer cells, and NCI/ADR-RES/OVCAR-8 ovarian (adriamycin-resistant) cancer cells by the MTT assay. As expected, both free melittin and the DSNS showed dose-dependent cytotoxicity and could kill 100% of the cancer cells at a high dose (Fig. 7). It is worthwhile to note that the DSNS was more effective in killing the HCT-116 cells. The DSNS killed 100% of the HCT-116 cells at the melittin concentration of 5 μM, at which free melittin could only kill 76% of the cancer cells (Fig. 7A). Most importantly, the DSNS only showed negligible hemolytic activity at the same concentration (Fig. 4B). A similar anticancer effect was observed for the MCF-7 breast cancer cells and SKOV-3 ovarian cancer cells (Fig. 7B and C). Furthermore, we also found that the DSNS killed 100% of the adriamycin-resistant ovarian cancer cells at the melittin concentration of 5 μM (Fig. 7D), which developed multidrug resistance. Altogether, we proved that the anticancer capacity of the melittin of DSNS, in contrast to other melittin carrier systems,7,9a was fully retained. In addition, the polymer carrier itself was not toxic for all four tested cell lines (Fig. S11†).
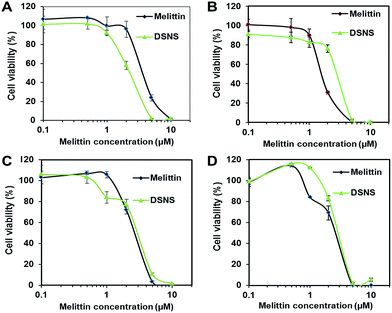 |
| | Fig. 7 Cytotoxicity of melittin and DSNS for (A) HCT-116 colon cancer cells, (B) MCF-7 breast cancer cells, (C) SKOV-3 ovarian cancer cells, and (D) NCI/ADR-RES (OVCAR-8 ovarian adriamycin-resistant) cancer cells. Cells were incubated with melittin and DSNS at a melittin concentration from 0.1 to 10 μM for 24 h. Data represent mean ± SD, n = 3. | |
Conclusions
In summary, we have fabricated DSNS nano-complexes through the electrostatic absorption of zwitterionic glycol chitosan and disulfide crosslinking to deliver melittin for cancer therapy. The hemolytic activity of melittin in DSNS could be completely quenched by our unique dual secured design. Due to the pH and redox potential dual responsiveness of DSNS, the wide-spectrum anticancer activity of melittin was fully retained, thus eradicating 100% of four types of tested cancer cell lines, including a drug resistant cell line. These studies demonstrated that the combination of a zwitterionic polymer and redox sensitive bonds offers a new strategy for a safe and effective therapeutic peptide delivery. The next step of the research would be adding cancer cell targeting ligands, such as folic acid, anisamide, and disaccharide moiety of bleomycin,18 to the DSNS to further enhance its tumor specificity.
Acknowledgements
The authors want to thank the American Cancer Society Institutional Research Grant (ACS-IRG), the ASPIRE award from the Office of the Vice President for Research of The University of South Carolina, the Center for Targeted Therapeutics (1P20 GM109091), and the Center for Colon Cancer Research (5P30 GM103336-02) for financial support.
Notes and references
-
(a) D. W. Hoskin and A. Ramamoorthy, Biochim. Biophys. Acta, 2008, 1778, 357–375 CrossRef CAS PubMed;
(b) N. Papo and Y. Shai, Cell. Mol. Life Sci., 2005, 62, 784–790 CrossRef CAS PubMed.
-
(a) L. Yang, T. A. Harroun, T. M. Weiss, L. Ding and H. W. Huang, Biophys. J., 2001, 81, 1475–1485 CrossRef CAS;
(b) M. T. Lee, W. C. Hung, F. Y. Chen and H. W. Huang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 5087–5092 CrossRef CAS PubMed.
- M. T. Tosteson and D. C. Tosteson, Biophys. J., 1981, 36, 109–116 CrossRef CAS.
-
(a) W. F. DeGrado, G. F. Musso, M. Lieber, E. T. Kaiser and F. J. Kezdy, Biophys. J., 1982, 37, 329–338 CrossRef CAS;
(b) K. A. Brogden, Nat. Rev. Microbiol., 2005, 3, 238–250 CrossRef CAS PubMed;
(c) J. F. Popplewell, M. J. Swann, N. J. Freeman, C. McDonnell and R. C. Ford, Biochim. Biophys. Acta, 2007, 1768, 13–20 CrossRef CAS PubMed.
- M. Jo, M. H. Park, P. S. Kollipara, B. J. An, H. S. Song, S. B. Han, J. H. Kim, M. J. Song and J. T. Hong, Toxicol. Appl. Pharmacol., 2012, 258, 72–81 CrossRef CAS PubMed.
- M. H. Park, M. S. Choi, D. H. Kwak, K.-W. Oh, D. Y. Yoon, S. B. Han, H. S. Song, M. J. Song and J. T. Hong, Prostate, 2011, 71, 801–812 CrossRef CAS PubMed.
- H. Pan, N. R. Soman, P. H. Schlesinger, G. M. Lanza and S. A. Wickline, WIREs Nanomed. Nanobiotechnol., 2011, 3, 318–327 CrossRef CAS PubMed.
- P. Xu, E. A. Van Kirk, Y. Zhan, W. J. Murdoch, M. Radosz and Y. Shen, Angew. Chem., Int. Ed., 2007, 46, 4999–5002 CrossRef CAS PubMed.
-
(a) C. Huang, H. Jin, Y. Qian, S. Qi, H. Luo, Q. Luo and Z. Zhang, ACS Nano, 2013, 7, 5791–5800 CrossRef CAS PubMed;
(b) C. Leuschner and W. Hansel, Curr. Pharm. Des., 2004, 10, 2299–2310 CrossRef CAS;
(c) W. Hansel, C. Leuschner and F. Enright, Mol. Cell. Endocrinol., 2007, 269, 26–33 CrossRef CAS PubMed;
(d) C. S. Kumar, C. Leuschner, E. E. Doomes, L. Henry, M. Juban and J. Hormes, J. Nanosci. Nanotechnol., 2004, 4, 245–249 CrossRef CAS PubMed.
- N. R. Soman, S. L. Baldwin, G. Hu, J. N. Marsh, G. M. Lanza, J. E. Heuser, J. M. Arbeit, S. A. Wickline and P. H. Schlesinger, J. Clin. Invest., 2009, 119, 2830–2842 CrossRef CAS PubMed.
- D. K. Knight, S. N. Shapka and B. G. Amsden, J. Biomed. Mater. Res., Part A, 2007, 83A, 787–798 CrossRef CAS PubMed.
- P. Xu, G. Bajaj, T. Shugg, W. G. Van Alstine and Y. Yeo, Biomacromolecules, 2010, 11, 2352–2358 CrossRef CAS PubMed.
-
(a) K. Xiao, Y. Li, J. Luo, J. S. Lee, W. Xiao, A. M. Gonik, R. G. Agarwal and K. S. Lam, Biomaterials, 2011, 32, 3435–3446 CrossRef CAS PubMed;
(b) K. C. R. Bahadur, B. Thapa and P. Xu, Macromol. Biosci., 2012, 12, 637–646 CrossRef PubMed.
-
(a) Y. P. Ho, H. H. Chen, K. W. Leong and T. H. Wang, J. Controlled Release, 2006, 116, 83–89 CrossRef CAS PubMed;
(b) C. A. Alabi, K. T. Love, G. Sahay, T. Stutzman, W. T. Young, R. Langer and D. G. Anderson, ACS Nano, 2012, 6, 6133–6141 CrossRef CAS PubMed.
-
(a) Y.-X. Tan, C. Chen, Y.-L. Wang, S. Lin, Y. Wang, S.-B. Li, X.-P. Jin, H.-W. Gao, F.-S. Du, F. Gong and S.-P. Ji, J. Gene Med., 2012, 14, 241–250 CrossRef CAS PubMed;
(b) N. J. Baumhover, K. Anderson, C. A. Fernandez and K. G. Rice, Bioconjugate Chem., 2010, 21, 74–83 CrossRef CAS PubMed.
-
(a) D. A. Scudiero, A. Monks and E. A. Sausville, J. Natl. Cancer Inst., 1998, 90, 862 CrossRef CAS PubMed;
(b) L. A. Garraway, H. R. Widlund, M. A. Rubin, G. Getz, A. J. Berger, S. Ramaswamy, R. Beroukhim, D. A. Milner, S. R. Granter, J. Y. Du, C. Lee, S. N. Wagner, C. Li, T. R. Golub, D. L. Rimm, M. L. Meyerson, D. E. Fisher and W. R. Sellers, Nature, 2005, 436, 117–122 CrossRef CAS PubMed.
- M. Lee, T. Sun, W. Hung and H. Huang, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 14243–14248 CrossRef CAS PubMed.
-
(a) H. S. Yoo and T. G. Park, J. Controlled Release, 2004, 100, 247–256 CrossRef CAS PubMed;
(b) H. He, A. Cattran, T. Nguyen, A. Nieminen and P. Xu, Biomaterials, 2014, 35, 9546–9553 CrossRef CAS PubMed;
(c) Z. Yu, R. M. Schmaltz, T. C. Bozeman, R. Paul, M. J. Rishel, K. S. Tsosie and S. M. Hecht, J. Am. Chem. Soc., 2013, 135, 2883–2886 CrossRef CAS PubMed;
(d) C. Bhattacharya, Z. Yu, M. J. Rishel and S. M. Hecht, Biochemistry, 2014, 53, 3264–3266 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures for the synthesis of thiolated amidized glycol chitosan, nano-complexes preparation and characterization, FRET measurement, release kinetics, hemolytic assay, confocal microscopy, and cytotoxicity assay. See DOI: 10.1039/c4tb01401d |
| ‡ B.C. and B.T. contributed equally. |
|
| This journal is © The Royal Society of Chemistry 2015 |








