
Biological materials and molecular biomimetics – filling up the empty soft materials space for tissue engineering applications
Ali
Miserez
*ab,
James C.
Weaver
c and
Ovijit
Chaudhuri
d
aSchool of Materials Science and Engineering, Nanyang Technological University, Singapore. E-mail: ali.miserez@ntu.edu.sg
bSchool of Biological Sciences, Nanyang Technological University, Singapore
cWyss Institute for Biologically Inspired Engineering, Harvard University, Cambridge, MA, USA
dDepartment of Mechanical Engineering, Stanford University, Stanford, CA 94305, USA
First published on 15th October 2014
Abstract
An extensive range of biomaterials, frequently derived from extracellular matrix (ECM) proteins or other natural biopolymers, have been developed for biomedical applications. Their mechanical response, a key requirement for regenerative medicine, is often stiff but exhibits limited extensibility (e.g. silk), or inversely, is compliant with higher strain to failure (e.g. elastin). While synthetic biocompatible materials exhibiting a mechanical response between these boundaries are rare, several biological materials demonstrate unexpected combinations of these properties. In order to replicate these performance metrics in synthetic systems, a central requirement is to first reveal the molecular design of their constituent building blocks, which has traditionally been an extremely time-consuming task. Here, we highlight the recent application of Next Gen sequencing technologies for the characterization of several protein-based natural biopolymers, a technique which circumvents this research bottleneck. Successful molecular biomimicry of these model protein systems could thus have the potential to significantly expand the range of intrinsic material properties available for biomedical applications.
 Ali Miserez | Ali Miserez is a Faculty member in the Schools of Materials Science and Biological Sciences at Nanyang Technological University in Singapore, and a recipient of the Singapore National Research Foundation Fellowship. He obtained his PhD in Materials Sciences from EPFL, and was a post-doctoral researcher at UC Santa Barbara working on biomimetic materials. His research aims at revealing the molecular, physico-chemical, and structural principles from biological materials, and on translating these designs into biomimetic materials. His research group is strongly cross-disciplinary and tackles bio-inspired engineering from various angles, including protein biochemistry, high-throughput sequencing, biomimetic polymer and peptide chemistry, and nanomechanics. |
 James C. Weaver | James C. Weaver is a senior research scientist at the Wyss Institute for Biologically inspired Engineering at Harvard University. He received his Bachelor's degree in Aquatic Biology and PhD in Marine Science from the University of California, Santa Barbara, and went on to pursue postdoctoral studies in Molecular Biology, Chemical Engineering, Physics, and Earth Sciences. Working at the interface between zoology, materials science, and multi-material additive manufacturing, his main research interests focus on investigating structure function relationships in hierarchically ordered biological composites and the fabrication of their synthetic analogs. |
 Ovijit Chaudhuri | Ovijit Chaudhuri is an Assistant Professor in the Department of Mechanical Engineering at Stanford University. He received his Bachelor of Science degree in Engineering Physics and PhD in Bioengineering from the University of California, Berkeley. He conducted postdoctoral research focusing on the areas of Biomaterials, Cancer Biology, and Tissue Engineering at Harvard University. His research focuses on elucidating the mechanics of cells and the extracellular matrix as well as understanding how cells sense and respond to the extracellular matrix, utilizing engineered biomaterials and high-resolution force measurement instrumentation. |
1. Introduction
The last decade has witnessed an increasing interest by researchers from the physical sciences in the exploration of biodiversity as inspiration for materials design strategies. For materials scientists and chemists working in biomimetic and bio-inspired materials synthesis, this attention has largely focused on a multitude of extracellular materials from a diverse assemblage of biological systems used for locomotion, protection, or predation.1–3 The appeal of these biological systems as design inspiration is rooted in the ingenious methods by which these species are specifically adapted to survive and thrive under a wide range of hostile environmental conditions. This interest in biological materials design, however, has led to some misconceptions, principally, that nature produces superior materials when compared to their anthropogenic counterparts, a notion likely encouraged by their intricate structural designs across multiple hierarchical length scales. It is perhaps more accurate to state that Nature makes things differently, with a more efficient use of energetic resources, and that biological materials are inherently multi-functional,4 with evolutionary pressures guiding the selection of multiple simultaneous local design maxima rather than a single global maximum. From a biomedical perspective, structural or load-bearing materials have received a large share of this research interest since fibrous structures, for example, are critical in applications such as tendon or ligament repair, for sutures, or in wound healing for emergency care.5,6 Tissue engineering scaffolds also largely rely on fibrillar materials including, among others, collagen,7,8 silk,9 fibrin,10 and elastin.11 In this report, we will provide an overview of the current trends and needs for the use of synthetic and natural biomaterials in biomedical applications, followed by a discussion of some recent discoveries investigating structure–function relationships in non-mineralized structural biological materials (whelk egg capsules, caddisfly silks, and squid sucker ring teeth) and the engineering design lessors that have been learned from their analysis.2. A brief overview of soft materials for biomedical applications
Polymeric scaffolds have found increasing use in biomedical research, with major applications including drug delivery, tissue engineering, and more recently, in situ cell programming (for extensive reviews on these topics, see Peppas et al.,12 Lutolf and Hubbell,13 and Kearney and Mooney14). In drug delivery, these biomaterials are commonly used for the targeted release of therapeutic agents such as small molecule drugs or protein growth factors. One of the major advantages provided by biomaterial-based delivery is that drug release is highly localized and can directly target a specific region of interest adjacent to the site of implantation, thus permitting lower doses relative to systemic delivery. Additionally, the temporal dynamics of drug release can be precisely tuned through modulation of the physicochemical properties of the scaffold material, so as to optimize drug efficacy. In tissue engineering applications, 3D hydrogels have been utilized as cell-laden carriers to promote tissue regeneration in vivo. In practice, a specific cell type of interest can be highly enriched ex vivo and delivered via the scaffold carrier directly to the site of injury. The physical and biochemical properties of the scaffold can also be engineered to maximally promote tissue regeneration. Finally, a more recent biomedical application of polymeric scaffolds has been to program cells for a specific targeted activity. For example, scaffolds containing the appropriate biochemical cues can be designed to recruit a specific population of cells into the material, and activate these cells towards a specific set of behaviors.15Biocompatible polymeric scaffolds for medical applications can be typically grouped into one of three main classes. One class consists of scaffolds formed from Extracellular Matrix (ECM) proteins such as fibrin and collagen. These biopolymers are semi-flexible, resulting in gel formation at low concentrations (0.1–1% weight/volume is typical) and the elasticity of these gels is highly nonlinear, with significant stiffening at low strains.16 Fibrin also has the specific advantage of being highly extensible, due to unfolding of coiled-coil domains within the protein under tensile loads17 and/or via the unraveling of flexible end domains of the fibrinogen constitutive proteins.18 A clinical application for this class of scaffolds is in the INFUSE bone graft device, in which a collagen sponge is used for sustained delivery of recombinant human bone morphogenetic protein-2, thereby promoting site-specific bone formation.19 A second class of scaffold-forming materials includes naturally occurring biopolymers such as alginate, hyaluronic acid, agarose, or chitosan. These flexible polymers form high water content hydrogels through either chemical or physical crosslinking at much higher concentrations (typically greater than 1% weight/volume) relative to the semi-flexible protein biopolymer-based gels. In one example application, bone progenitor cells transplanted via degradable alginate hydrogels along with dual growth factors have been demonstrated to induce bone formation in vivo.20 Finally, biomedical scaffolds can be constructed from synthetic polymers, with common examples including polyethylene glycol and polyesters such as polylactide or polyglycolide.21 In the next section, a comparative overview of the mechanical responses of these various classes of biocompatible polymers is presented.
3. Empty spaces in the “soft” materials landscape
The mechanical performance of the chosen material is a critical parameter for regenerative medical applications. Hence, together with their assessment of physico-chemical and biological compatibility, knowledge of their stress–strain characteristics is equally important. When plotting the main types of ECM-derived and natural biopolymeric materials used in tissue repair and regeneration on a single tensile stress–strain plot (Fig. 1a, linear scale) a notable feature emerges, specifically that these fibrous materials are mostly segregated into two broad classes. They are either elastically stretchable with limited ultimate tensile strength (<1 MPa), as is exhibited by materials such as elastin- and fibrin-based hydrogels, or as in the case of silks, they are stiff and strong, but feature limited extensibility. While native collagen is one exception, appearing somewhere in the middle,22 such properties have not been recapitulated with synthetically processed collagens. Although there has been recent progress in this area of research,8,23 synthetic collagen fibril synthesis remains a challenging task because it consists of a multi-step and hierarchical assembly process, spanning from the classical triple-helix secondary structure to the nano-staggered arrangement, all of which are important requisites governing the mechanical response of this material.24,25 Chitosan biopolymers are mid-range between these upper and lower boundaries, with Young's modulus values in the 1–10 MPa range under hydrated conditions26,27 and exhibit a rather limited extensibility of 10–15%. This contrast in mechanical performance is perhaps better perceived on the log stress–strain representation shown in Fig. 1b. Here it appears rather clearly that between the stiff silks (and even native collagens) and the highly elastic materials and hydrogels, there exists ample room to tailor the structural properties of biomaterials. This material tailorability has clear advantages in cases where such intermediate properties would be required. For example, a central requirement of connective tissues is to bond different tissues that often exhibit significant differences in mechanical properties,28 and this task is best achieved by using materials with stress–strain characteristics ranging between those of the tissues to which they attach. Even more suitable are materials that feature mechanically-graded properties, because smooth gradients can shield the interfaces against stress concentration that are often the sites of mechanical failure.29 Achieving these macrostructural gradients is easier if the intrinsic properties of the building blocks can be controlled over a few orders of magnitude, since the mechanical gradient can be obtained by spatially varying the volume fraction of the starting materials30,31 through a simple composite effect or by manipulating the cross-linking density.32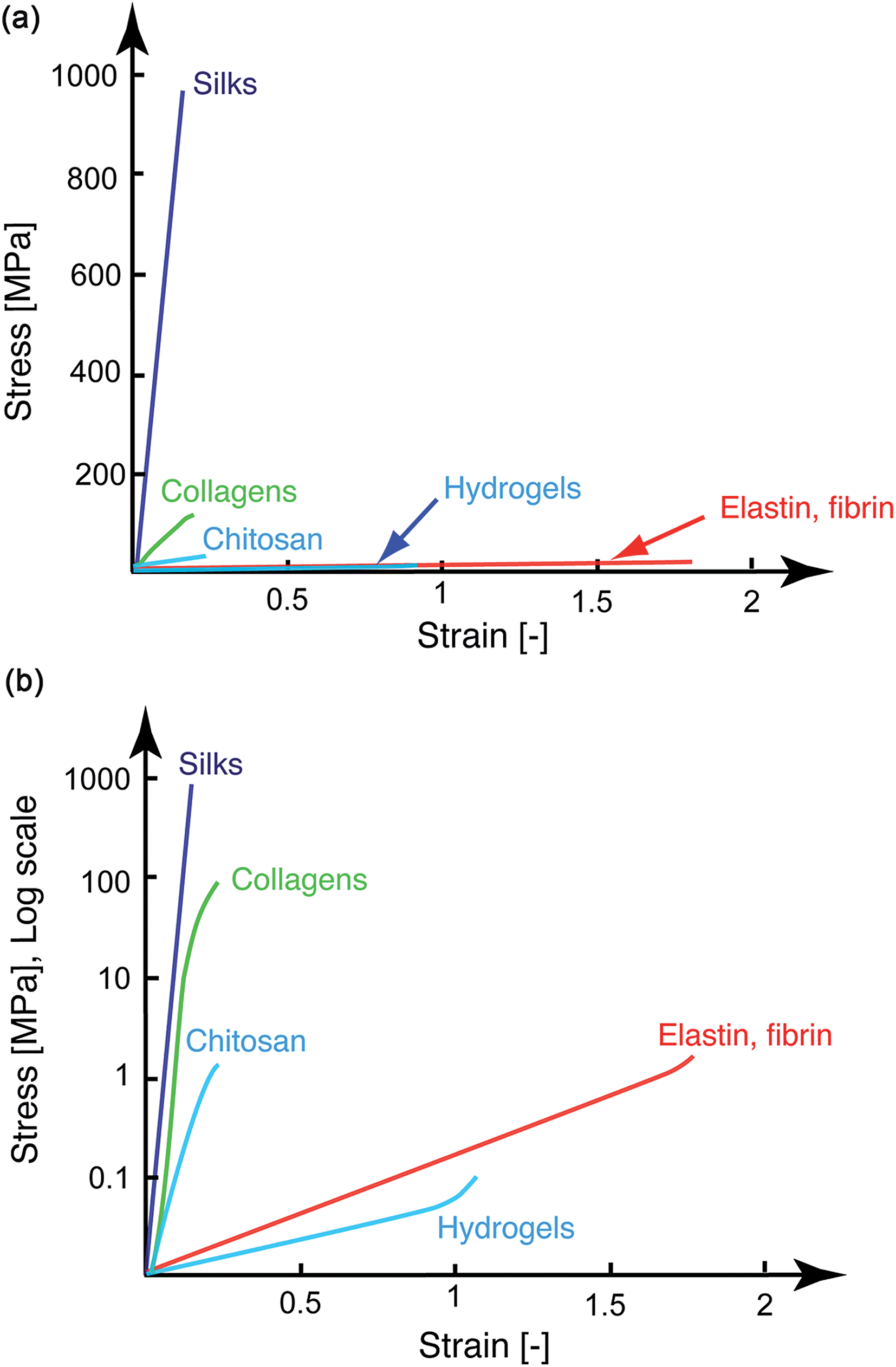 | ||
| Fig. 1 Tensile stress–strain curves of representative soft biomaterials used in medical applications (top: linear stress scale; bottom log stress scale). Between the stiff silks and (native and cross-linked) collagens and the more compliant bioelastomers and hydrogels (which exhibit a higher strain to failure), there are limited materials with properties that fall between these boundaries. | ||
Let us consider the simplest case of linear elastic fibers, where the failure energy or work of fracture wf is given by the relation:
 | (1) |
 | ||
| Fig. 2 Materials selection charts of representative soft biomaterials. (a) Strain to failure vs. elastic modulus, with blue dotted lines denoting the guidelines along which materials exhibit an equivalent strain energy to failure. (b) Characteristic tensile response of materials featuring a mechanical hysteresis during load cycles. The total elastic energy stored at peak stress corresponds to the full area under the curves, whereas the internally-absorbed mechanical energy during a load cycle corresponds to the colored areas. (c) Ashby plot of total hysteresis vs. elastic modulus. Yellow areas in (a) and (c) correspond to the “empty soft materials space”, in which limited biocompatible soft materials are found. Silks refer to spider dragline silks and Bombyx mori silks. Marine snail egg capsules and caddisfly silks are found within these areas. | ||
There also exist other, more subtle characteristics, which would be useful in applications involving cell and tissue encapsulation. For example, the ability to dissipate mechanical loads can be critical for the protection of delicate cells,35 for artificial fibers used for connective tissue replacement, or in cartilaginous tissue engineering. This property manifests itself on the stress–strain curve as hysteresis upon unloading (schematically illustrated in Fig. 2b); the larger the hysteresis during a loading/unloading curve, the more efficiently the material can dissipate elastic energy. When commonly used soft biomaterials are placed on a hysteresis vs. elastic modulus plot, only a few examples exhibit more than a 10% of absorption capacity upon unloading. Notable exceptions to this trend are native dragline silks36 and genetically-engineered titin–resilin hybrid materials.37 For the titin–resin synthetic constructs, however, the macro-scale performance has not been evaluated and their mechanical characterization has so far been limited to single-molecule pulling experiments by AFM. More recently, there have been significant advances in the synthesis of high performance hydrogels38 that exhibit significantly greater toughness compared to traditional formulations. Such toughened gels (generally referred as “physically cross-linked gels” in the materials selection chart of Fig. 2a) appear to bridge the mechanical property gap between soft tissues and synthetic gels.39 Interestingly, the underlying nano- and micro-scale toughening mechanisms behind these properties are conceptually similar to those responsible for the unique mechanical response of natural biological materials described in the following section. Based on these observations, there thus exists a large empty soft material parameter space (highlighted in yellow in Fig. 2a) where new materials could be developed.
4. Looking to nature to fill the void in soft material property space
Over the past several years, we have identified and characterized, across multiple length scales, several intriguing biological materials systems from various species of marine invertebrates that exhibit a wide range of unique properties for potential biomedical applications,30,40–42 a selection of which are described below. Other research groups have focused their attention on fibrous structures from insects, which are also briefly described.The oviparous marine snails from the family Melongenidae (the whelks), for example, deposit their eggs within tough and stretchable egg capsules. These capsules are exclusively proteinaceous and exhibit unusual mechanical tensile responses.43,44 After an initial linear elastic regime, with an elastic modulus of ca. 50 to 70 MPa, they display a pseudo-yield behavior up to 70% strain. Upon further stretching, the capsular materials significantly strain-harden and can be deformed up to 170%. When the external load is released, the material recovers its initial length. In comparison to most other elastomeric materials, however, a high amount of elastic energy is internally absorbed as witnessed by the high hysteresis during a full load/unloading cycle. In other words, the egg capsules are able to absorb a large fraction of the total elastic energy stored during loading. If we place the egg capsules on the εfvs. E materials selection chart (Fig. 2a, purple box), we see that they are ideally located within the empty materials space described in the previous sections. In addition, on an energy absorption vs. modulus chart, the egg capsules again feature a combination of properties unmatched by any synthetic polymer or soft material (Fig. 2c). Clearly, our ability to duplicate such behaviors would expand the range of existing material properties for a wide range of regenerative medical applications.
What is the source of these unusual properties? At the micron-scale, the egg capsules are composed of fibers, which in a species-specific manner, are arranged either in an orthotropic or random fashion.41,45 These fibers are made of smaller, nano-scale units which feature an axial staggered arrangement of 100 nm,45,46 which is reminiscent of the 67 nm staggered organization of collagen fibrils. In situ tensile testing performed in conjunction with Wide-Angle X-ray diffraction41 and confocal Raman imaging46 have shown that the key features governing the egg capsule tensile behavior are conformational changes of the protein backbone under mechanical stress. The egg capsule micro-fibers are made of coiled-coil proteins in the unstretched state and undergo a conformation transformation into axially-aligned extended β-sheet-like structures during stretching. This solid-state phase transition behavior has recently emerged as a common theme in a variety of biomacromolecules,47 including fibrin,48 intermediate filaments,49 and myosin II.50
Another class of biological fibers recently reported by Stewart and co-workers that exhibit a similar mechanical response to the whelk egg capsules are the larval caddisfly silks,51,52 which are used to glue together solid particles into a protective casing. The tensile response of caddisfly silk is characterized by an initial linear regime up to ∼5% strain, with a Young's modulus of ca. 80 MPa. Like the whelk egg capsules, a yield plateau is then observed up to ca. 20% strain, which is followed by strain hardening until failure at 100 to 170% strain. Upon unloading, a high hysteresis is observed and is accompanied by a large irreversible strain, which is, however, restored after 15 minutes at rest. Plotting the caddisfly silks on the same Ashby plots (Fig. 2, red box) reveals that their performance metrics position is near the marine snail egg capsules, but with an even higher mechanical hysteresis (Fig. 2c). However, in comparison to the whelk egg capsule, the reversible elongation is not instantaneous. Structure–property relationship studies of caddisfly silk with solid-state Nuclear Magnetic Resonance (NMR) and X-ray diffraction (XRD) have indicated that anti-parallel β-sheets are a key contributor to the stiffness and strength of the fibers,51 which is reminiscent of spider silks. An additional striking feature of caddisfly silk is their substantial post-translational phosphorylation as well as the presence of Ca2+ ions, which were shown by Fourier Transform Infra-Red (FTIR) spectroscopy to form Ca2+–phosphate complexes. Upon chelation of Ca2+ with EDTA, the mechanical properties (stiffness, yield, and ultimate strength) are dramatically reduced along with a concomitant reduction in β-sheet content. These characteristics, however, can be largely restored following re-incubation with Ca2+, thus strongly suggesting that phosphate–Ca2+ complexation plays a key role in enhancing their mechanical response52 and in stabilizing the β-sheets.
Another promising model biopolymer we have recently characterized comes from the arms and tentacles of decapodiform cephalopods (squids and cuttlefish) such as the Humboldt (or Jumbo) squid, Dosidicus gigas. Jumbo squid are swift, agile, and aggressive predators that use their muscular tentacles and arms during prey capture and handling. The tentacles and arms are lined with hundreds of suckers, each of which contains a rigid ring-like structure from which robust triangular teeth protrude. Despite being entirely proteinaceous, these sucker ring teeth (SRT) are as hard and stiff as some of the strongest synthetic polymers53 with a Young's modulus ranging from 6 to 8 GPa under dry conditions and 2 to 4 GPa when in a hydrated state. The cross-sectional variation in modulus is correlated to the presence of nanotubules oriented parallel to the long axis of the teeth. Perhaps even more remarkable is the fact that there are no inter-chain covalent interactions within the teeth. Instead, SRT are assembled into large supramolecular networks containing a high volume fraction of nano-confined β-sheets and held together primarily through hydrogen bonding interactions.54 This intriguing absence of inter-chain chemical linkages can thus be exploited during the synthetic processing of the SRT proteins. As a result, SRT are soluble under mildly acidic conditions, can be reversibly melted by simple heating just like a thermoplastic polymer, and in turn, can be molded into complex shapes across multiple length scales, with β-sheets spontaneously forming upon solidification or solution evaporation.55 The design lessons obtained from investigating this thermoplastic-like behavior and water-based processability has the potential to provide “green” approaches for the synthetic fabrication of high-performance protein-based polymers for regenerative medical applications where very high load-bearing properties are required.
Towards our goal to duplicate these systems, a central requirement relates to the need to elucidate the genes that encode their constitutive proteins. While historically such efforts have been extremely time-consuming, there have, however, been exciting developments in recent years that have the potential to dramatically reduce the time constraints needed to sequence and clone such intriguing biomacromolecules, the details of which are described in the following section.
5. Molecular biomimetics: structure–properties relationships at the protein sequence level
5.1 Next gen sequencing for biological structural materials
A central issue – and a major bottleneck in the design of biomimetic materials – has been the time required for obtaining full-length protein sequences of interest. For example, the development of mussel-inspired adhesives, arguably one of the best success stories of biomimetic materials, took decades and was completely dependent on the discovery of the “adhesive” dihydroxyphenylalanine (DOPA) side chain modification56 and on the elucidation of the primary amino acid sequences of these natural protein-based glues.57–61 These characteristics later inspired a wide range of chemical strategies for the development of catechol-based synthetic polymers for biomedical applications,62,63 including those based on the clever use of dopamine chemistry.64 Likewise, the development of genetically engineered silk-like fibers fully relied on the sequencing of silk genes. But while key silk secondary structural motifs were first elucidated in the 1950s,65 it wasn't until more than 30 years later that the first partial silk protein sequence was obtained by Xu and Lewis,66 followed a few years later by partial sequencing of other silk genes.67–69 Of course, this understanding has been intimately linked to parallel advances in molecular cloning technologies, since the discovery and characterization of novel proteins from extracellular biological materials was intrinsically limited by sequencing bottlenecks. There have also been other limiting factors that have slowed progress on this front. First, proteins from structural biological materials are often difficult to extract, owing to their intrinsic chemical stability. They are often covalently cross-linked,70–72 precluding their isolation with common extraction buffers and also tend to form insoluble aggregates. Furthermore many contain often unknown post-translational modifications which are critical for their proper function in the extracellular environment. To further complicate this situation, the elucidation of these modifications is not straightforward and cannot be obtained through standard molecular cloning. Second, while an increasing number of genomes have been sequenced in recent years73 there is still very limited genomic information for many interesting model systems that are currently being explored in the biomimetic materials community. In turn, obtaining full-length sequences of novel proteins though either direct N-terminal and internal fragmentation sequencing or through the use of more traditional molecular biological techniques is perceived as a challenging endeavor, and is thus not often undertaken in the bioinspired materials community.With remarkable advances achieved in the past few years with Next Generation Sequencing Technologies,74–76 many of the constraints described above have been lifted. In light of these recent discoveries, we have employed Next Generation RNA-sequencing (RNA-seq) in the context of biological materials characterization.55 This approach involves the following steps, summarized in Fig. 3. The transcriptome of a specific tissue can be rapidly obtained from mRNA extraction and isolation, from which a cDNA library can be generated. Transcripts are then assembled using bioinformatic tools such as the Trinity software suite,77 which assembles transcripts without a reference genome library. In parallel, proteomic analyses of the tissue or glands secreting the protein of interest are conducted with classical and modern proteomic tools, including amino acid analysis (AAA), N-terminal sequencing, or high throughput tandem mass spectroscopy (as a side note, it is interesting to mention that traditional analytical techniques such as AAA or Edman-based sequencing, which have tended to vanish from Life Sciences laboratories in the past decade, have proved extremely useful in order to mine biological material transcriptomic libraries, especially when looking for novel proteins). Genes of interest are then obtained by probing the transcriptome library with the proteomics data, including protein amino acid composition, isoelectric point and molecular weight, as well as N-terminal and internal sequence information. Once candidate transcripts have been identified, standard PCR techniques may still be needed in some cases to confirm the final full-length sequences, especially for the highly repetitive sequences that are common in structural proteins. This step, however, is extremely accelerated through prior knowledge of the candidate transcripts and in our experience, has not led to any ambiguities or major difficulties in obtaining the genes of interest.
 | ||
| Fig. 3 An integrative approach combining Next Gen sequencing (RNA-seq) with proteomics and materials characterization for the discovery of novel molecular designs and architectures for the development of new biomimetic materials. Adapted from ref. 55. | ||
Using this approach, we have been able to successfully obtain full-length sequences from our targeted model systems. Other groups have also recently made use of similar approaches to sequence caddisfly silks52 or the underwater adhesives from barnacles, flatworms, and sea stars,78–80 indicating an increasing awareness towards this integrative research platform. Significantly, most genes of interest from a given model system have been sequenced with only a few months of work. Until recently, such critical sequence information – which is key for subsequent synthetic biomimetic efforts through protein engineering81,82 or peptide synthesis – would have typically taken many years of tedious molecular cloning work, often to obtain only a few genes of interest.83 Overall, this research platform is extremely promising since it permits the genes from a newly identified model system to be rapidly sequenced, significantly reducing or eliminating the many hurdles faced when tackling protein sequencing from natural biological materials.
5.2 Sequence analysis of model tissues
We now return to the model systems described in Section 4 and briefly describe the key molecular features gleaned from the sequencing of their constituent proteins.Silk has generated intense interest in recent years, with many successfully demonstrated biomedical and engineering applications.9,93,94 One challenge with silk processing, however, is the required use of rather harsh solvents in order to solubilize native and high molecular-weight recombinant silks. Furthermore, owing to their extremely high molecular weights (>400 kDa) and the heavy bias towards Gly and Ala, expression of full-length recombinant silk proteins remains challenging, although there has been recent progress on this front.95 In comparison, the recombinant expression of full-length suckerin proteins was not met with major technical difficulties, and just like the native SRT proteins, can be processed from aqueous solutions at various length scales and into various shapes, while spontaneously forming a high percentage of β-sheets. Sequence analysis of the suckerin proteins has also revealed the regular placement of tyrosine, which was exploited to tune the mechanical properties under hydrated conditions over 2-orders of magnitude by forming di-tyrosine cross-links using a straightforward photo-activation method.55 As illustrated in a materials selection chart (Fig. 4), the combination of hardness and modulus of SRT proteins can match or even exceed those of the strongest synthetic polymers. Interestingly, the inspiration for the di-tyrosine cross-linking came from another natural bioelastomer, namely resilin96 from insect wings, which had previously been shown to contain di-tyrosine and whose properties can be artificially mimicked through photo-induced cross-linking.97
 | ||
| Fig. 4 Ashby plot of H vs. E of common engineering materials along with recombinant suckerin (Rec-suckerin 39, wet conditions) after cross-linking. Rec-suckerin exhibits very high elastic modulus and hardness that can compete with the best structural synthetic polymers, with the final properties depending on the di-tyrosine cross-link density. Such materials could find usage in restorative biomedical applications requiring high load-bearing capability. Reproduced from ref. 55. | ||
6. Towards the incorporation of bio-inspired modular peptides for the design of structural biomedical materials
The identification of full-length sequences from load-bearing, elastomeric, self-healing, and other intriguing biological model systems provides completely new libraries of biomimetic proteins and peptides that can readily be combined or fused with previously available proteinaceous materials,81 or grafted into other hybrid systems such as those based on synthetic polymers or polysaccharides. As described in the previous sections, this molecular toolbox thus has the potential to facilitate the development of novel biopolymeric material systems that offer combinations of properties not currently available in existing materials. Using these approaches, we come one step closer to filling the “empty spaces” present in the soft materials landscape depicted in Fig. 2, not only in terms of structural performance but also with the capacity to incorporate novel properties such as stimuli-responsiveness, controlled degradation,98 or anti-oxidative activity.61One recent trend, for example, has been to combine different classes of polymers to generate composite matrices with enhanced mechanical or biological properties. Examples of this approach include the fabrication of interpenetrating networks of covalently cross-linked acrylamide and ionically cross-linked alginate gels that are both tough and highly extensible.99 In a second example, interpenetrating networks of reconstituted basement membrane matrix and alginate have been used to modulate matrix stiffness independent of ligand density.100 These types of multi-phasic biopolymeric constructs could be especially relevant in the field of cartilage tissue engineering due to cartilage's low intrinsic capacity to heal and the continuous, long term, and relatively high dynamic stresses (∼20 MPa)101 that this tissue experiences.102 Compared to natural cartilaginous tissues, however, engineered cartilage equivalents generally exhibit a nearly 10-fold reduction in mechanical performance in terms of their toughness and tensile and compressive moduli.103 While much still remains to be learned regarding the synthesis methods to custom-tailor the mechanical properties of biomedical materials that function as cartilage replacements, recent advances in the use of blended gels of fibrin-alginate have been used to form fibrocartilage tissue equivalents in vitro that exhibit greater extensibility, while still possessing the capacity to promote chondrogenesis and cartilage matrix production by mesenchymal stem cells.104 Recently developed tough hydrogels (displayed in the materials selection chart in Fig. 2a) also exhibit promising mechanical properties as cartilage replacement materials, with Young's moduli under hydrated conditions ranging from 0.3–0.4 MPa![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 105 to greater than 1 MPa.39 On the other hand, however, if we consider the lessons learned from investigating structural biological materials, marine snail egg capsules, for example, exhibit a mechanical response that could potentially be highly suitable for such applications, including a high elastic modulus of 50 to 70 MPa combined with a fully reversible extensibility of 150% (a combination of properties that exceeds that of traditional tough synthetic elastomers106), while simultaneously exhibiting high mechanical energy absorption and a high fatigue resistance.44
105 to greater than 1 MPa.39 On the other hand, however, if we consider the lessons learned from investigating structural biological materials, marine snail egg capsules, for example, exhibit a mechanical response that could potentially be highly suitable for such applications, including a high elastic modulus of 50 to 70 MPa combined with a fully reversible extensibility of 150% (a combination of properties that exceeds that of traditional tough synthetic elastomers106), while simultaneously exhibiting high mechanical energy absorption and a high fatigue resistance.44
Tough hydrogels typically rely on a double cross-linked network as a primary toughening mechanism, where weak “sacrificial bonds” such as hydrogen bonds provide energy dissipation, while a second covalently cross-linked (and stronger) network is responsible for the load bearing capability and increased elasticity. Conceptually, it is interesting to note that the mechanical properties of marine snail egg capsules can also be attributed to a double-network system: during mechanically-induced unfolding of the coiled-coil rods, breaking of intra-chain hydrogen bonds that stabilize the coiled coil parallel to the rod axes occur. As the strain is further increased, transient, inter-chain hydrogen bonds within the extended state of β-sheet-like domains are formed. The intra-chain bonds reform almost instantaneously upon unloading, similar to hydrogen bonds within ureidopyrimidinone (UPy) segments reported by Guo et al.105 and to ionic bonds in ampholyte-based hydrogels.39 The elasticity and improved load-bearing properties, on the other hand, are largely attributed to the presence of lysine-derived covalent cross-links which render the egg capsule not only mechanically, but also highly chemically stable. Exhibiting a nearly 50-fold increase in modulus compared to the stiffest known synthetic gels, the impressive mechanical properties of the egg capsules are likely related to their low water content as well as the intrinsically stiffer nature of the coiled-coil structural motif.107,108 Tough hydrogels, on the other hand, are more extensible (ranging from 500 to 1000%) vs. ca. 150% for the egg capsules. Since the work of fracture scales linearly with E and exhibits a power of two dependence with εf (eqn (1)), the egg capsules and tough hydrogels are found along roughly the same energy to failure guideline in Fig. 2a. Thus, in terms of fracture energy, the lower strain to failure of the egg capsules is compensated by their high moduli. A useful target for future development would be to tailor the properties between these boundaries, perhaps by creating hybrid systems that are based on both synthetic and natural design strategies of the respective materials. We envision that either full-length recombinant proteins or the shorter modular peptides described above could be included in such composite systems.
Another important challenge towards the use of bioinspired materials with tailored bulk mechanical properties for tissue engineering applications will be to design materials that present appropriate biophysical and biochemical cues to direct cell fate or function at the nanoscale. Integrin binding cell adhesion ligands are necessary for the survival of many cell types, and the altered density and type of cell adhesion ligands coupled to biomedical materials can profoundly influence the resulting cell fate and behavior.109 In addition to integrin binding ligands, tethered growth factors can also modulate the functionality of implanted scaffolds. Indeed, recent work has demonstrated that the tethering of high affinity growth factors to fibrin gels can be used to enhance bone regeneration and wound healing.110 In addition to biochemical cues, biophysical cues such as material stiffness can impact various cellular behaviors. For example, mesenchymal stem cells undergo osteogenic differentiation optimally in ionically cross-linked alginate matrices with an elastic modulus of 10–30 kPa, and adipogenesis in gels with a modulus of 1–10 kPa.109 On the other hand, no effect of stiffness is observed when MSCs are encapsulated in covalently cross-linked gels, with adipogenesis occurring over a range of elastic moduli of 4–90 kPa.111 In these gels, osteogenesis only occurs when the gel is engineered to be degradable by cellular enzymes, demonstrating the additional importance of crosslinking type and degradability on stem cell fate. Research efforts such as those described above highlight how nanoscale tuning of the biochemical and biophysical environment of polymeric scaffolds can be utilized to promote desired outcomes in the context of tissue engineering. Our ability to precisely manipulate hybrid materials at the molecular scale by integrating some of the designs discovered in natural systems adds an extra dimension to this tailorability, similar to modular concepts previously used to confer multi-functionality of bioelastomers112 and other soft materials.113 For example, one can imagine incorporating the coiled coil heptad repeats from snail egg capsules into other recombinant protein constructs in order to mimic the high stiffness, elasticity, and energy absorption associated with the reversible α–β phase transition. Similarly, the modular sequence design of the suckerins could be exploited in several ways to achieve novel performance metrics. Specifically, the nanoconfined β-sheet forming modules from the suckerins could be used to control the relative content of the β-sheet reinforcement phase in hybrid biopolymers, and in turn, their mechanical response. In addition, photo-induced di-tyrosine cross-linking could also be used to modulate the mechanical properties of recombinant suckerin-based materials.
7. Conclusions
The diversity of materials employed for biomedical applications (synthetic, natural, and hybrid systems) have been expanding rapidly in recent years, with many developments in areas such as cell recognition, controlled degradation, or mechanical tuning. The molecular design of natural materials provide countless design lessons to chemists and materials scientists, at all length scales of their structural hierarchy, to further improve the mechanical performance and expand the multi-functionality of their synthetic analogs. Until recently, elucidating the sequences of intriguing model proteinaceous materials, which constitutes a prerequisite for many molecular-based biomimicry approaches, was considered a challenging and time-consuming endeavor. Recent advances in Next Gen sequencing technologies, however, have considerably reduced this time constraint, while at the same time, high-throughput sequencing costs are now a fraction of what they were just five years ago. Taken together, they point to the remarkable potential of vast libraries of biological materials waiting to be elucidated, and whose properties could fill up the empty materials properties landscape, particularly if one is able to recapitulate them in artificial systems using polymer synthesis and/or protein engineering approaches. It is our view that curiosity-driven discoveries of intriguing biological systems, combined with a keen eye towards the biological function of model tissues and careful multi-scale characterization all the way down to the molecular level, holds great potential to discover a wide range of novel materials with potential usage in tissue engineering and regenerative medicine applications.Acknowledgements
We thank Dr Paul Guerette (NTU) and Dr Shawn Hoon (A*STAR, Singapore) for insightful comments and discussions during the preparation of this manuscript. A.M. acknowledges financial support from the Singapore National Research Foundation (NRF) through a NRF Fellowship, and the Singapore Ministry of Education (MOE) for a Tier 2 grant (grant # MOE 2011-T2-2-044) supporting the sucker ring teeth work.References
- P. Y. Chen, J. McKittrick and M. A. Meyers, Prog. Mater. Sci., 2012, 57, 1492–1704 CrossRef CAS PubMed.
- S. Amini and A. Miserez, Acta Biomater., 2013, 9, 7895–7907 CrossRef CAS PubMed.
- M. A. Meyers, J. McKittrick and P. Y. Chen, Science, 2013, 339, 773–779 CrossRef CAS PubMed.
- J. Dunlop, R. Weinkamer and P. Fratzl, Mater. Today, 2010, 14, 70–78 CrossRef.
- C. Guo and M. Spector, in Scaffolding in Tissue Engineering, ed. P. X. Ma and J. H. Elisseeff, CRC Press, Taylor&Francis group, Boca Raton, FL, 2006, pp. 385–411 Search PubMed.
- Principles of Tissue Engineering, ed. R. Lanza, R. Langer and J. Vacanti, Elsevier Academic Press, Burlington, MA, 3rd edn, 2007 Search PubMed.
- E. A. Sander and V. H. Barocas, in Collagen: Structure and Mechanics, ed. P. Fratzl, Springer-US, New York, 2008, pp. 475–504 Search PubMed.
- L. E. R. O'leary, J. A. Fallas, E. L. Bakota, M. K. Kang and J. D. Hartgerink, Nat. Chem., 2011, 3, 821–828 CrossRef PubMed.
- F. G. Omenetto and D. Kaplan, Science, 2010, 329, 528–531 CrossRef CAS PubMed.
- M. Ehrbar, S. C. Rizzi, R. Hlushchuk, V. Djonovb, A. H. Zisch, J. A. Hubbell, F. E. Weber and M. P. Lutolf, Biomaterials, 2007, 3856–3866 CrossRef CAS PubMed.
- N. Annabi, S. M. Mithieux, G. Camci-Unal, M. R. Dokmeci, A. S. Weiss and A. Khademhosseini, Biochem. Eng. J., 2013, 77, 110–118 CrossRef CAS PubMed.
- N. A. Peppas, J. Z. Hilt, A. Khademhosseini and R. Langer, Adv. Mater., 2006, 18, 1345–1360 CrossRef CAS.
- M. P. Lutolf and J. A. Hubbell, Nat. Biotechnol., 2005, 23, 47–55 CrossRef CAS PubMed.
- C. J. Kearney and D. J. Mooney, Nat. Mater., 2013, 12, 1004–1017 CrossRef CAS PubMed.
- O. A. Ali, N. Huebsch, L. Cao, G. Dranoff and D. J. Mooney, Nat. Mater., 2009, 8, 151–158 CrossRef CAS PubMed.
- C. Storm, J. J. Pastore, F. C. MacKintosh, T. C. Lubensky and P. A. Janmey, Nature, 2005, 435, 191–194 CrossRef CAS PubMed.
- A. E. X. Brown, R. I. Litvinov, D. E. Discher, P. K. Purohit and J. W. Weisel, Science, 2009, 325, 741–744 CrossRef CAS PubMed.
- J. R. Houser, N. E. Hudson, L. Ping, E. T. O'Brien, R. Superfine, S. T. Lord and M. R. Falvo, Biophys. J., 2010, 99, 3038–3047 CrossRef CAS PubMed.
- W. F. McKay, S. M. Peckham and J. M. Badura, Int. Orthop., 2007, 31, 729–734 CrossRef PubMed.
- C. A. Simmons, E. Alsberg, S. Hsiong, W. J. Kim and D. J. Mooney, Bone, 2004, 35, 562–569 CrossRef CAS PubMed.
- L. S. Nair and C. T. Laurencin, Prog. Polym. Sci., 2007, 32, 762–798 CrossRef CAS PubMed.
- P. Fratzl, K. Misof, I. Zizak, G. Rapp, H. Amenitsch and S. Bernstorff, J. Struct. Biol., 1998, 122, 119–122 CrossRef CAS PubMed.
- A. A. Jalan and J. D. Hartgerink, Curr. Opin. Chem. Biol., 2013, 17, 960–967 CrossRef CAS PubMed.
- H. S. Gupta, in Collagen: Structure and Mechanics, ed. P. Fratzl, Springer-US, New York, 2008, pp. 155–173 Search PubMed.
- T. J. Wess, in Collagen: Structure and Mechanics, ed. P. Fratzl, Springer-US, New York, 2008, pp. 49–80 Search PubMed.
- S. V. Madihally and H. W. Matthew, Biomaterials, 1999, 20, 511–521 CrossRef.
- L. Q. Wu, R. Ghodssi, Y. A. Elabd and G. F. Payne, Adv. Funct. Mater., 2005, 15, 189–195 CrossRef CAS.
- M. Benjamin, H. Toumi, J. R. Ralphs, G. Bydder, T. M. Best and S. Milz, J. Anat., 2006, 208, 471–490 CrossRef CAS PubMed.
- H. L. Lu and S. Thomopoulos, Annu. Rev. Biomed. Eng., 2013, 15, 201–226 CrossRef CAS PubMed.
- A. Miserez, T. Schneberk, C. Sun, F. W. Zok and J. H. Waite, Science, 2008, 319, 1816–1819 CrossRef CAS PubMed.
- K. U. Claussen, T. Scheibel, H.-W. Schmidt and R. Giesa, Macromol. Mater. Eng., 2012, 297, 938–957 CrossRef CAS.
- J. D. Fox, J. R. Capadona, P. D. Marasco and S. J. Rowan, J. Am. Chem. Soc., 2013, 135, 5167–5174 CrossRef CAS PubMed.
- M. F. Ashby, Materials Selection and Process in Mechanical Design, Butterworth Heinemann, Oxford, UK, 3rd edn, 2005 Search PubMed.
- U. Wegst and M. F. Ashby, Philos. Mag., 2004, 84, 2167–2181 CrossRef CAS.
- P. X. Ma and J. H. Elisseeff, Scaffolding in Tissue Engineering, CRC Press, 2006 Search PubMed.
- J. M. Gosline, M. A. Lillie, E. Carrington, P. Guerette, C. Ortlepp and K. Savage, Philos. Trans. R. Soc., B, 2002, 357, 121–132 CrossRef CAS PubMed.
- S. Lv, D. M. Dudek, Y. Cao, M. M. Balamurali, J. Gosline and H. Li, Nature, 2010, 465, 69–73 CrossRef CAS PubMed.
- X. Zhao, Soft Matter, 2014, 10, 672–687 RSC.
- T. L. Sun, T. Kurokawa, S. Kuroda, A. Bin Ihsan, T. Akasaki, K. Sato, A. Haque, T. Nakajima and J. P. Gong, Nat. Mater., 2013, 12, 932–937 CrossRef CAS PubMed.
- A. Miserez, Y. Li, J. H. Waite and F. Zok, Acta Biomater., 2007, 3, 139–149 CrossRef CAS PubMed.
- A. Miserez, S. Wasko, C. Carpenter and J. H. Waite, Nat. Mater., 2009, 8, 910–916 CrossRef CAS PubMed.
- A. Miserez, Y. Li, J. Cagnon, J. C. Weaver and J. H. Waite, Biomacromolecules, 2012, 12, 332–341 CrossRef PubMed.
- H. S. Rapoport and R. E. Shadwick, Biomacromolecules, 2002, 3, 42–50 CrossRef CAS PubMed.
- H. S. Rapoport and R. E. Shadwick, J. Exp. Biol., 2007, 210, 12–26 CrossRef CAS PubMed.
- P. A. Guerette, G. Z. Tay, S. Hoon, J. J. Loke, A. F. Hermawan, C. N. Z. Schmitt, M. J. Harrington, A. Masic, A. Karunaratne, H. S. Gupta, K. S. Tan, A. Schwaighofer, C. Nowak and A. Miserez, Biomater. Sci., 2014, 2, 710–722 RSC.
- M. J. Harrington, S. Wasko, A. Masic, F. D. Fisher, H. S. Gupta and P. Fratzl, J. R. Soc., Interface, 2012, 9, 2911–2922 CrossRef CAS PubMed.
- A. Miserez and P. A. Guerette, Chem. Soc. Rev., 2013, 42, 1973–1995 RSC.
- A. Zhmurov, O. Kononova, R. I. Litvinov, R. I. Dima, V. Barsegov and J. W. Weisel, J. Am. Chem. Soc., 2012, 134, 20396–20402 CrossRef CAS PubMed.
- Z. Qin and M. J. Buehler, Phys. Rev. Lett., 2010, 104, 198304 CrossRef.
- I. Schwaiger, C. Sattler, D. R. Hostetter and M. Rief, Nat. Mater., 2002, 1, 232–235 CrossRef CAS PubMed.
- J. B. Addison, N. N. Ashton, W. S. Weber, R. J. Stewart, G. P. Holland and J. L. Yarger, Biomacromolecules, 2013, 14, 1140–1148 CrossRef CAS PubMed.
- N. N. Ashton, D. R. Roe, R. B. Weiss, T. E. Cheatham and R. J. Stewart, Biomacromolecules, 2013, 14, 3668–3681 CrossRef CAS PubMed.
- A. Miserez, J. C. Weaver, P. B. Pedersen, T. Schneberk, R. T. Hanlon, D. Kisailus and H. Birkedal, Adv. Mater., 2009, 21, 401–406 CrossRef CAS.
- P. A. Guerette, S. Hoon, D. Ding, S. Amini, A. Masic, B. Venkatesh, V. Ravi, J. C. Weaver and A. Miserez, ACS Nano, 2014, 7, 7170–7179 CrossRef PubMed.
- P. A. Guerette, S. Hoon, Y. Seow, F. T. Wong, V. H. B. Ho, M. Raida, A. Masic, M. C. Demirel, F. Abdon, S. Amini, G. Z. Tay, D. Ding and A. Miserez, Nat. Biotechnol., 2013, 31, 908–915 CrossRef CAS PubMed.
- J. H. Waite and M. Tanzer, Science, 1981, 212, 1038–1040 CAS.
- J. H. Waite, T. J. Housley and M. L. Tanzer, Biochemistry, 1985, 24, 5010–5014 CrossRef CAS.
- K. J. Coyne, X. X. Qin and H. Waite, Science, 1997, 277, 1830–1832 CrossRef CAS.
- J. H. Waite and X.-X. Qin, Biochemistry, 2001, 40, 2887–2893 CrossRef CAS PubMed.
- H. Zhao and J. H. Waite, Biochemistry, 2006, 45, 14223–14231 CrossRef CAS PubMed.
- J. Yu, W. Wei, E. Danner, R. K. Ashley, J. N. Israelachvili and J. H. Waite, Nat. Chem. Biol., 2011, 7, 588–590 CrossRef CAS PubMed.
- P. L. Lee, P. B. Messersmith, J. N. Israelachvili and J. H. Waite, Annu. Rev. Mater. Res., 2011, 41, 99–132 CrossRef PubMed.
- J. Sedó, J. Saiz-Poseu, F. Busqué and D. Ruiz-Molina, Adv. Mater., 2012, 25, 653–701 CrossRef PubMed.
- H. Lee, S. M. Dellatore, W. M. Mille and P. B. Messersmith, Science, 2007, 318, 426–430 CrossRef CAS PubMed.
- J. O. Warwicker, Acta Crystallogr., 1954, 7, 565–573 CrossRef CAS.
- M. Xu and R. V. Lewis, Proc. Natl. Acad. Sci. U. S. A., 1990, 87, 7120–7124 CrossRef CAS.
- P. A. Guerette, D. Ginzinger, B. Weber and J. M. Gosline, Science, 1996, 272, 112–115 CAS.
- C. Y. Hayashi and R. Lewis, Science, 2000, 287, 1477–1479 CrossRef CAS.
- J. Gatesy, C. Y. Hayashi, D. Motriuk, J. Woods and R. Lewis, Science, 2001, 291, 2603–2605 CrossRef CAS PubMed.
- C. C. Broomell, R. K. Khan, D. N. Moses, A. Miserez, M. G. Pontin, G. D. Stucky, F. W. Zok and J. H. Waite, J. R. Soc., Interface, 2007, 4, 19–31 CrossRef PubMed.
- S. A. Andersen, Insect Biochem. Mol. Biol., 2010, 40, 166–178 CrossRef CAS PubMed.
- D. Rubin, A. Miserez and J. H. Waite, in Advances in Insect Physiology, ed. J. Casas, Elsevier, 2010, vol. 38, pp. 75–133 Search PubMed.
- H. Ellegren, Trends Ecol. Evol., 2014, 29, 51–63 CrossRef PubMed.
- M. L. Metzker, Nat. Rev. Genet., 2009, 11, 31–46 CrossRef PubMed.
- Z. Wang, M. Gerstein and M. Snyder, Nat. Rev. Genet., 2009, 10, 57–63 CrossRef CAS PubMed.
- F. Ozsolak and P. M. Milos, Nat. Rev. Genet., 2011, 12, 87–98 CrossRef CAS PubMed.
- M. G. Grabherr, B. J. Haas, M. Yassour, J. Z. Levin, D. A. Thompson, I. Amit, X. Adiconis, L. Fan, R. Raychowdhury, Q. Zeng, Z. Chen, E. Mauceli, N. Hacohen, A. Gnirke, N. Rhind, F. Di Palma, B. W. Birren, C. Nusbaum, K. Lindblad-Toh, N. Friedman and A. Regev, Nat. Biotechnol., 2011, 29, 644–652 CrossRef CAS PubMed.
- Z.-F. Chen, K. Matsumura, H. Wang, S. M. Arellano, X. Yan, I. Alam, J. A. C. Archer, V. B. Bajic and P. Y. Qian, PLoS One, 2011, 7, e22913 Search PubMed.
- B. Lengerer, R. Pjeta, J. Wunderer, M. Rodrigues, R. Arbore, L. Schärer, E. Berezikov, M. W. Hess, K. Pfaller, B. Egger, S. Obwegeser, W. Salvenmoser and P. Ladurner, Front. Zool., 2014, 11, 1–15 CrossRef PubMed.
- E. Hennebert, R. Wattiez, M. Demeuldre, P. Ladurner, D. S. Hwang, J. H. Waite and P. Flammang, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 6317–6322 CrossRef CAS PubMed.
- S. Gomes, I. B. Leonor, J. F. Mano, R. L. Reis and D. L. Kaplan, Prog. Polym. Sci., 2011, 37, 1–17 CrossRef PubMed.
- B. D. Olsen, AIChE J., 2013, 59, 3558–3568 CrossRef CAS.
- R. V. Lewis, Chem. Rev., 2006, 106, 3762–3774 CrossRef CAS PubMed.
- M. Delorenzi and T. Speed, Bioinformatics, 2002, 18, 617–625 CrossRef CAS PubMed.
- C. T. Armstrong, T. L. Vincent, P. J. Green and D. N. Woolfson, Bioinformatics, 2011, 27, 1908–1914 CrossRef CAS PubMed.
- S. Wasko, G. Tay, C. Nowak, A. Schwaighofer, J. H. Waite and A. Miserez, Biomacromolecules, 2014, 15, 30–42 CrossRef CAS PubMed.
- H. Herrmann, H. Bär, L. Kreplak, S. V. Strelkov and U. Aebi, Nat. Rev. Mol. Cell Biol., 2007, 6, 562–573 CrossRef PubMed.
- M. Arslan, Z. Qin and M. J. Buehler, Comput. Meth. Biomech. Biomed. Eng., 2011, 14, 483–489 CrossRef PubMed.
- O. J. L. Rackham, M. Madera, C. T. Armstrong, T. L. Vincent, D. N. Woolfson and J. Gough, J. Mol. Biol., 2010, 403, 480–493 CrossRef CAS PubMed.
- B. Apostolovic, D. Maarten and H.-A. Klok, Chem. Soc. Rev., 2010, 39, 3541–3575 RSC.
- J. M. Gosline, P. A. Guerette, C. S. Ortlepp and K. N. Savage, J. Exp. Biol., 1999, 202, 3295–3303 CAS.
- Z. Z. Shao and F. Vollrath, Nature, 2002, 418, 741 CrossRef CAS PubMed.
- H. Tao, D. L. Kaplan and F. G. Omenetto, Adv. Mater., 2012, 24, 2824–2837 CrossRef CAS PubMed.
- J. Zhang, E. Pritchard, X. Hu, T. Valentin, B. Panilaitis, F. G. Omenetto and D. L. Kaplan, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11981–11986 CrossRef CAS PubMed.
- X. X. Xia, Z. G. Qian, C. S. Ki, Y. H. Park, D. L. Kaplan and S. Y. Lee, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14059–14063 CrossRef CAS PubMed.
- T. Weis-Fogh, J. Mol. Biol., 1961, 3, 648–667 CrossRef CAS.
- C. M. Elvin, A. G. Carr, M. G. Huson, J. M. Maxwell, R. D. Pearson, T. Vuocolo, N. E. Liyou, D. C. C. Wong, D. J. Merritt and N. E. Dixon, Nature, 2005, 437, 999–1002 CrossRef CAS PubMed.
- P. M. Kharkar, K. L. Kiick and A. M. Kloxin, Chem. Soc. Rev., 2013, 42, 7335–7372 RSC.
- J.-Y. Sun, X. Zhao, W. R. K. Illeperuma, O. Chaudhuri, K. H. Oh, D. J. Mooney, J. J. Vlassak and Z. Suo, Nature, 2012, 489, 133–136 CrossRef CAS PubMed.
- O. Chaudhuri, S. T. Koshy, C. Branco da Cunha, J.-W. Shin, C. S. Verbeke, K. H. Allison and D. J. Mooney, Nat. Mater., 2014, 13, 970–978 CrossRef CAS PubMed.
- L. Han, A. J. Grodzinsky and C. Ortiz, Annu. Rev. Mater. Res., 2011, 41, 133–168 CrossRef CAS PubMed.
- D. J. Huey, J. C. Hu and K. A. Athanasiou, Science, 2012, 338, 917–921 CrossRef CAS PubMed.
- A. H. Huang, M. J. Farrell and R. L. Mauck, J. Biomech., 2010, 43, 128–136 CrossRef PubMed.
- K. Ma, A. L. Titan, M. Stafford, C. H. Zheng and M. E. Levenston, Acta Biomater., 2012, 8, 3754–3764 CrossRef CAS PubMed.
- M. Guo, L. M. Pitet, H. M. Wyss, M. Vos, P. Y. W. Dankers and E. W. Meijer, J. Am. Chem. Soc., 2014, 136, 6969–6977 CrossRef CAS PubMed.
- E. Ducrot, Y. Chen, M. Bulters, R. P. Sijbesma and C. Creton, Science, 2014, 344, 186–189 CrossRef CAS PubMed.
- D. S. Fudge, K. H. Gardner, V. T. Forsyth, C. Riekel and J. M. Gosline, Biophys. J., 2003, 85, 2015–2027 CrossRef CAS.
- C. W. Wolgemuth and S. X. Sun, Phys. Rev. Lett., 2006, 97, 248101 CrossRef.
- N. Huebsch, P. R. Arany, A. S. Mao, D. Shvartsman, O. A. Ali, S. A. Bencherif, J. Rivera-Feliciano and D. J. Mooney, Nat. Mater., 2010, 9, 518–526 CrossRef CAS PubMed.
- M. M. Martino, P. S. Briquez, E. Güç, F. Tortelli, W. W. Kilarski, S. Metzger, J. J. Rice, G. A. Kuhn, R. Müller, M. A. Swartz and J. A. Hubbell, Science, 2014, 343, 885–888 CrossRef CAS PubMed.
- S. Khetan, M. Guvendiren, W. R. Legant, D. M. Cohen, C. S. Chen and J. A. Burdick, Nat. Mater., 2013, 12, 458–465 CrossRef CAS PubMed.
- L. Li, M. Charati and K. L. Kiick, Polym. Chem., 2010, 1, 1160–1170 RSC.
- A. M. Kushner and Z. Guan, Angew. Chem., Int. Ed. Engl., 2011, 50, 9026–9057 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2015 |