
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence5,20-Bis(α-oligothienyl)-substituted [26]hexaphyrins possessing electronic circuits strongly perturbed by meso-oligothienyl substituents†
Hirotaka
Mori
a,
Masaaki
Suzuki
a,
Woojae
Kim
b,
Jong Min
Lim
b,
Dongho
Kim
*b and
Atsuhiro
Osuka
*a
aDepartment of Chemistry, Graduate School of Science, Kyoto University Sakyo-ku, Kyoto 606-8502, Japan. E-mail: osuka@kuchem.kyoto-u.ac.jp
bSpectroscopy Laboratory for Functional π-Electronic Systems and Department of Chemistry Yonsei University, Seoul 120-749, Korea. E-mail: dongho@yonsei.ac.kr
First published on 2nd December 2014
Abstract
A series of [26]hexaphyrins(1.1.1.1.1.1) bearing two α-oligothienyl substituents at 5,20-positions have been synthesised and are shown to have a dumbbell hexaphyrin conformation, to which the α-oligothienyl groups are linked with small dihedral angles to form an acyclic helix-like conjugated network. While their distinct diatropic ring currents and four reversible reduction waves characteristic of aromatic [26]hexaphyrins indicate that the [26]hexaphyrin aromatic circuits are viable, the absorption spectra and excited state dynamics are significantly perturbed, which becomes increasingly evident with elongation of the oligothienyl substituents. DFT calculations of these hexaphyrins indicated that the LUMO and LUMO + 1 are localised on the hexaphyrin circuit and the HOMO and HOMO − 1 are spread over the acyclic helix-like conjugation network, which can explain the perturbed absorption spectra.
Introduction
In the last two decades, the chemistry of expanded porphyrins has gained increasing popularity, in light of their flexible structures, multi-metal coordination properties, anion sensing abilities, facile interconversions between multiple and neutral redox states, and unprecedented chemical reactivities such as drastic skeletal rearrangements.1 Fairly flexible electronic systems of meso-aryl expanded porphyrins have been demonstrated by the exploration of versatile electronic states such as Hückel aromatic, Hückel antiaromatic,2 Möbius aromatic,3 Möbius antiaromatic,4 stable monoradical,5 and singlet biradicaloid species.6 As an additional example, internally 5,20-aromatic-bridged [26]hexaphyrins have been recently developed as dual electronic systems consisting of [18]porphyrin and/or [26]hexaphyrin, which can be modulated by the internal bridging aromatic units (Fig. 1a).7 In this paper, we report the synthesis of a series of 5,20-bis(α-oligothienyl) [26]hexaphyrins(1.1.1.1.1.1) (T2–T4), which display characteristic optical and electronic properties due to the presence of 5,20-bis(α-oligothienyl) substituents (Fig. 1b).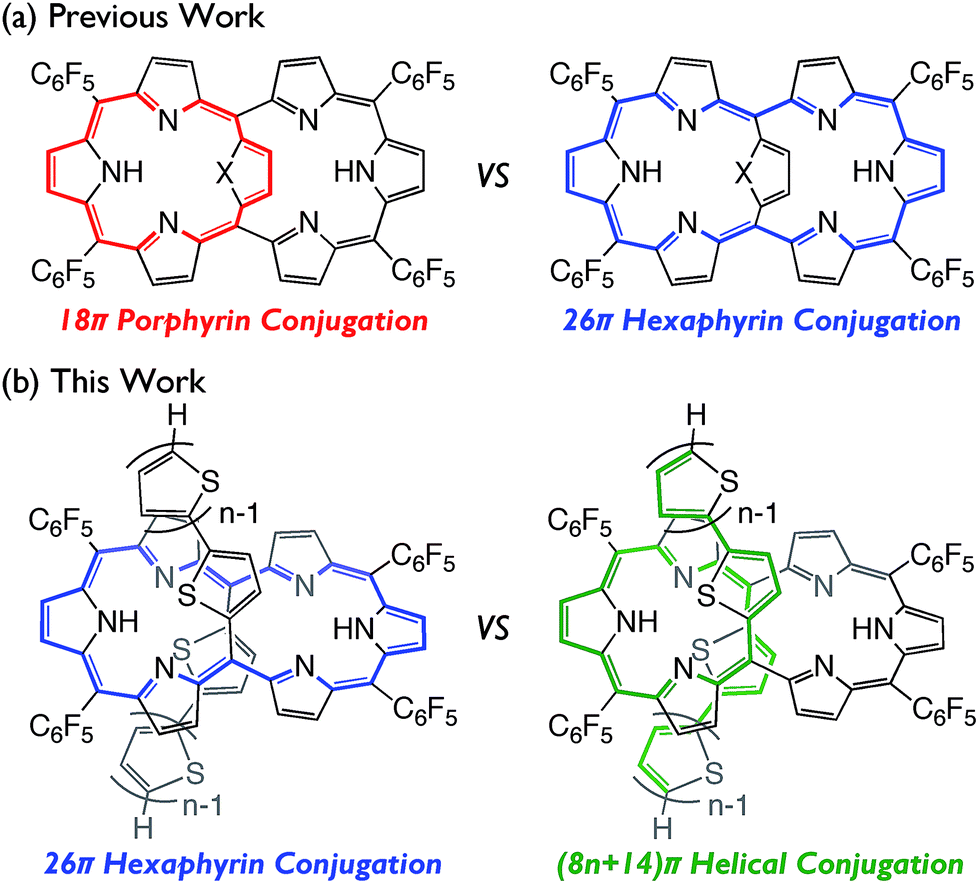 | ||
| Fig. 1 [26]Hexaphyrins possessing dual electronic systems; (a) internally aromatic-bridged [26]hexaphyrins (X = S, NH) and (b) bis(α-oligothienyl) [26]hexaphyrins. Effective conjugated networks are indicated by the coloured lines. | ||
Results and discussion
Synthesis of 5,20-bis(α-oligothienyl) [26]hexaphyrins(1.1.1.1.1.1)
[26]Hexaphyrins T2–T4 have been prepared by a modified method used for the synthesis of 5,20-(2-thienyl) [26]hexaphyrin(1.1.1.1.1.1) T1.8 Acid-catalysed condensation of 5,10-bis(pentafluorophenyl)tripyrrane 1 with corresponding α-oligothiophene-5-carbaldehydes followed by oxidation with 2,3-dichloro-5,6-dicyano-1,4-benzoquinone (DDQ) gave T2–T4 in 22, 10, and 15% yields, respectively (Scheme 1). High-resolution electrospray ionisation time-of-flight (HR ESI-TOF) mass measurements showed the parent ion peaks at m/z 1457.0685 ([M+H]+; calcd for C70H25N6F20S4: 1457.0699) for T2, m/z 1621.0462 ([M+H]+; calcd for C78H29N6F20S6: 1621.0453) for T3, and m/z 1783.0059 ([M−H]−; calcd for C86H31N6F20S8: 1783.0062) for T4 (see ESI†). 5,15-Bis(α-oligothienyl)-substituted porphyrins P1–P4 were also prepared (ESI†) to examine the effects of α-oligothienyl substituents on the electronic system of porphyrins, which highlight the characteristically flexible electronic properties of [26]hexaphyrins.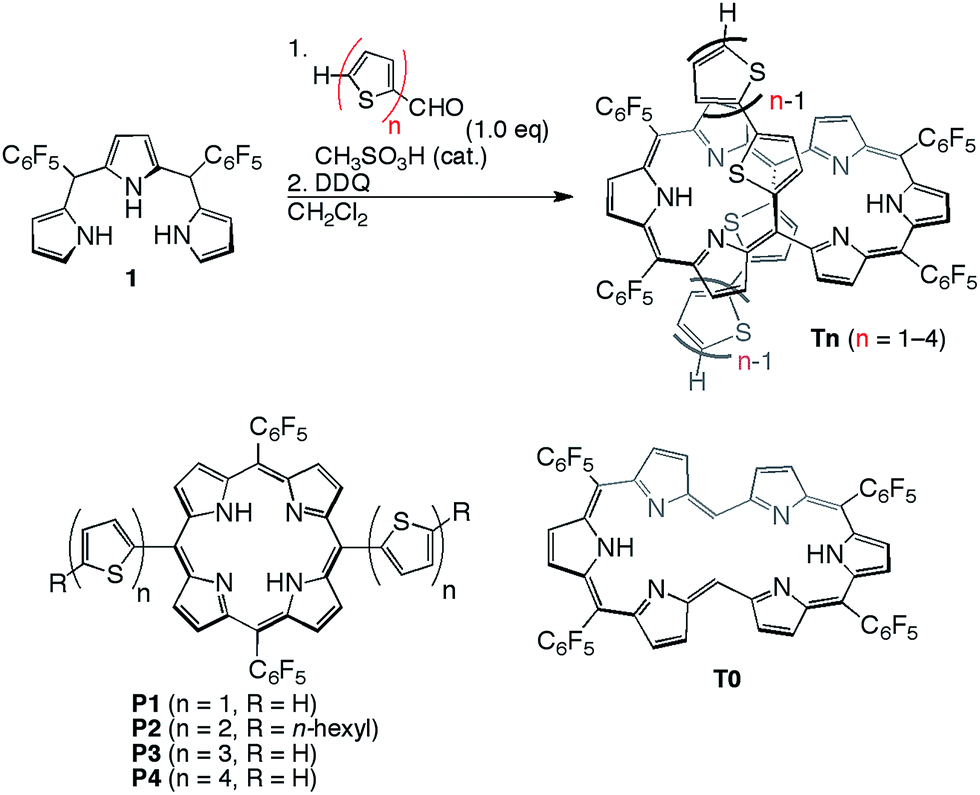 | ||
| Scheme 1 Synthesis of 5,20-(α-oligothienyl)-substituted [26]hexaphyrins T1–T4 and structures of P1–P4. | ||
The solid-state structures of T2–T4 have been unambiguously determined by single crystal X-ray diffraction analysis.9 Hexaphyrins T2–T4 all show a dumbbell hexaphyrin conformation, to which the two oligothienyl groups are appended with small dihedral angles, being favorable for conjugation with the hexaphyrin macrocycle (Fig. 2 and ESI†). The hexaphyrin frames of T2–T4 are essentially the same as those of 5,20-unsubstituted hexaphyrin (T0)5a and T1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 8 with regard to the planar dumbbell structure. The dumbbell conformation is intrinsically the most stable for [26]hexaphyrins, because of four possible energy-stabilising intramolecular hydrogen bonding interactions, but is only allowed for [26]hexaphyrins bearing small substituents at 5,20-positions.5a,8 Thus, T0 is the most stable, having a strain-free fairly planar conformation with the largest diatropic ring current. The observed dumbbell structures of T1–T4 have been similarly ascribed to small 2-thienyl and α-oligothienyl substituents.8 The two oligothienyl substituents are positioned above and below the macrocycle and are oriented toward the same side of the hexaphyrin to form a helix-like conjugated network involving the tripyrrodimethene segments of the hexaphyrin.
8 with regard to the planar dumbbell structure. The dumbbell conformation is intrinsically the most stable for [26]hexaphyrins, because of four possible energy-stabilising intramolecular hydrogen bonding interactions, but is only allowed for [26]hexaphyrins bearing small substituents at 5,20-positions.5a,8 Thus, T0 is the most stable, having a strain-free fairly planar conformation with the largest diatropic ring current. The observed dumbbell structures of T1–T4 have been similarly ascribed to small 2-thienyl and α-oligothienyl substituents.8 The two oligothienyl substituents are positioned above and below the macrocycle and are oriented toward the same side of the hexaphyrin to form a helix-like conjugated network involving the tripyrrodimethene segments of the hexaphyrin.
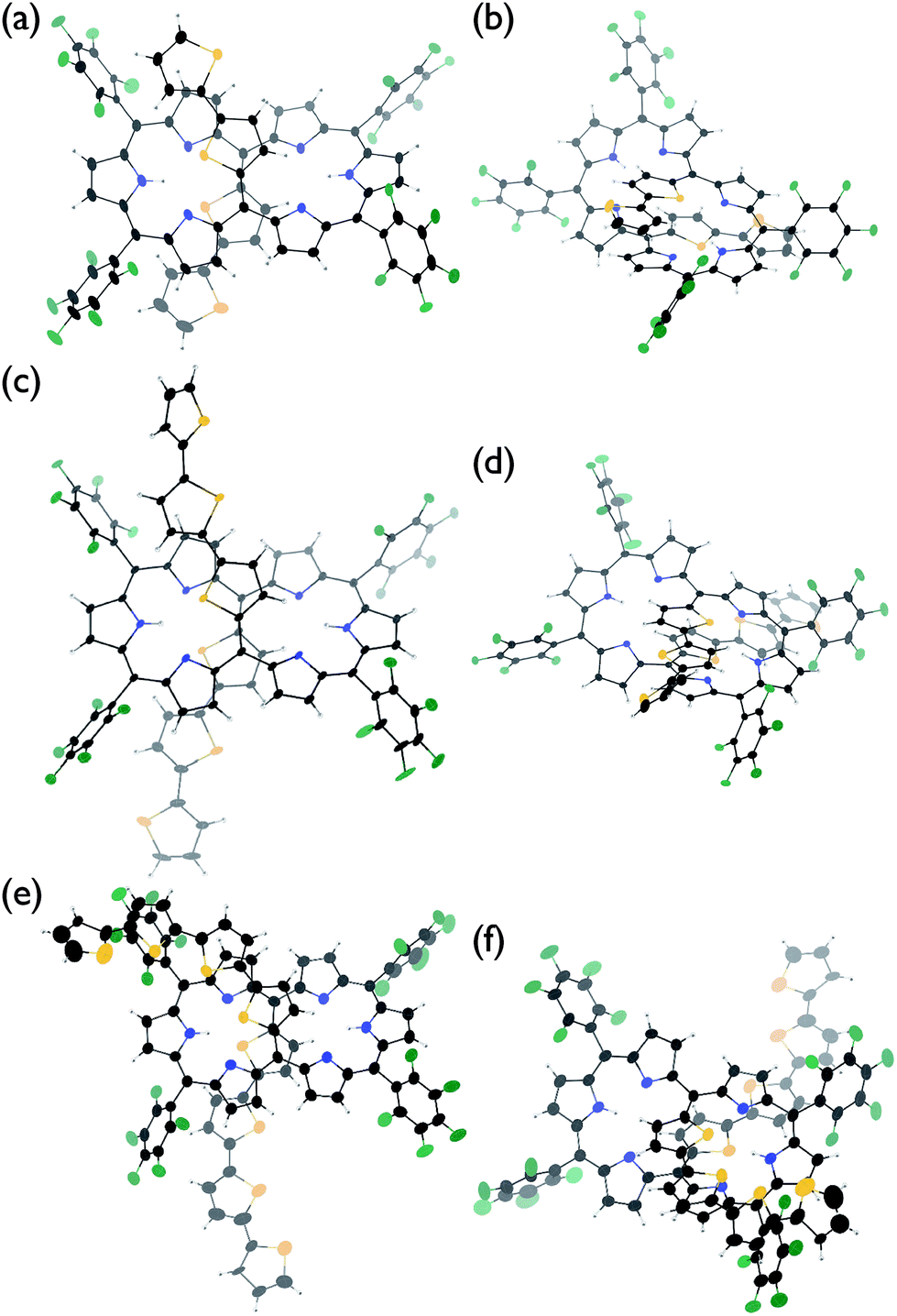 | ||
| Fig. 2 X-Ray crystal structures of (a and b) T2, (c and d) T3, and (e and f) T4. Solvent molecules are omitted for clarity. The thermal ellipsoids are scaled to a 30% probability level. One of the two molecules in the unit cell is shown for T2 and T4. | ||
The 1H NMR spectra of T0 and T1 indicate sharp signals at room temperature, which are consistent with the dumbbell structures.5a,8 The 1H NMR spectrum of T1 exhibits only three signals at 7.38, 7.72, and 8.16 ppm due to the outer β-pyrrolic protons, indicating a very rapid rotation of the meso-thienyl substituents. In contrast, the 1H NMR spectra of T2–T4 taken at room temperature are interesting, in that the protons of the hexaphyrin periphery appear as very broad signals but the protons of the oligothienyl substituents appear as sharp signals. At −60 °C, these 1H NMR spectra became sharp, featuring six signals due to the outer β-pyrrolic protons in the range δ = 8.21–5.76 ppm and signals due to the meso-oligothienyl groups in the range δ = 7.25–4.06 ppm. These data indicate substantial diatropic ring currents for the hexaphyrin in T2–T4 in accordance with the solid-state dumbbell structures. Importantly, two singlets due to the two inner NH protons appeared differently around δ = 8.4–8.2 and 5.4–5.1 ppm, implying that the right and left tripyrrodimethene segments of the hexaphyrin are electronically different, owing to the influence of the meso-oligothienyl substituents. Most probably the rotational dynamics of the meso-oligothienyl substituents are slow at room temperature as compared with the 1H NMR time scale.
Redox potentials and UV/Vis/NIR absorption spectra
The electrochemical properties of T1–T4 were examined by cyclic voltammetry (CV) in CH2Cl2 containing 0.1 M Bu4NPF6 as a supporting electrolyte versus ferrocene/ferrocenium cation (see Fig. S20 and Table S1†). All the cyclic voltammograms of T1–T4 showed four reversible reduction waves almost at the same potential, that correspond to stepwise reductions reaching their 30π states, hence indicating that the 26π hexaphyrin electronic circuits are all viable. On the other hand, the first oxidation potentials appeared at 0.38 V for T1, 0.27 V for T2, 0.22 V for T3, and 0.17 V for T4, indicating the rising of the HOMO levels upon elongation of the meso-oligothienyl chains.The absorption spectrum of T0 displays a sharp and intense Soret-like band at 549 nm and weak but distinct Q-bands in the range 700–1100 nm as characteristic features of typical aromatic porphyrinoids. The absorption spectrum of T1 exhibits an ill-defined Soret-like band at 616 nm and broad Q-bands in the range 700–1150 nm, which is significantly different from that of T0, indicating that the 2-thienyl substituents cause significant perturbation on the electronic state of the [26]hexaphyrin. This trend is increasingly evident upon elongation of the oligothienyl chains. Namely, a Soret-like band, a characteristic attribute of aromatic [26]hexaphyrins, almost disappears in the absorption spectra of T2–T4, and instead a broad high-energy band is observed around 350 nm for T1, which is red-shifted and intensified with the elongation of the meso-oligothienyl chains.10 In addition, the spectra of T2–T4 show broad Q-bands in the NIR region. Collectively, these absorption features of T2–T4 are radically different from those of aromatic porphyrinoids, but are rather similar to those of reported acyclic oligopyrromethenes.11 The absorption spectra of T1–T4 became sharpened upon lowering the temperature (ESI†), indicating the importance of the dynamic motion of the oligothienyl substituents for broadening the absorption spectra. In sharp contrast, no such drastic changes were observed in the absorption spectra of the porphyrin counterparts P1–P4 (Fig. 3b), underlining the unique and flexible electronic properties of the [26]hexaphyrins.
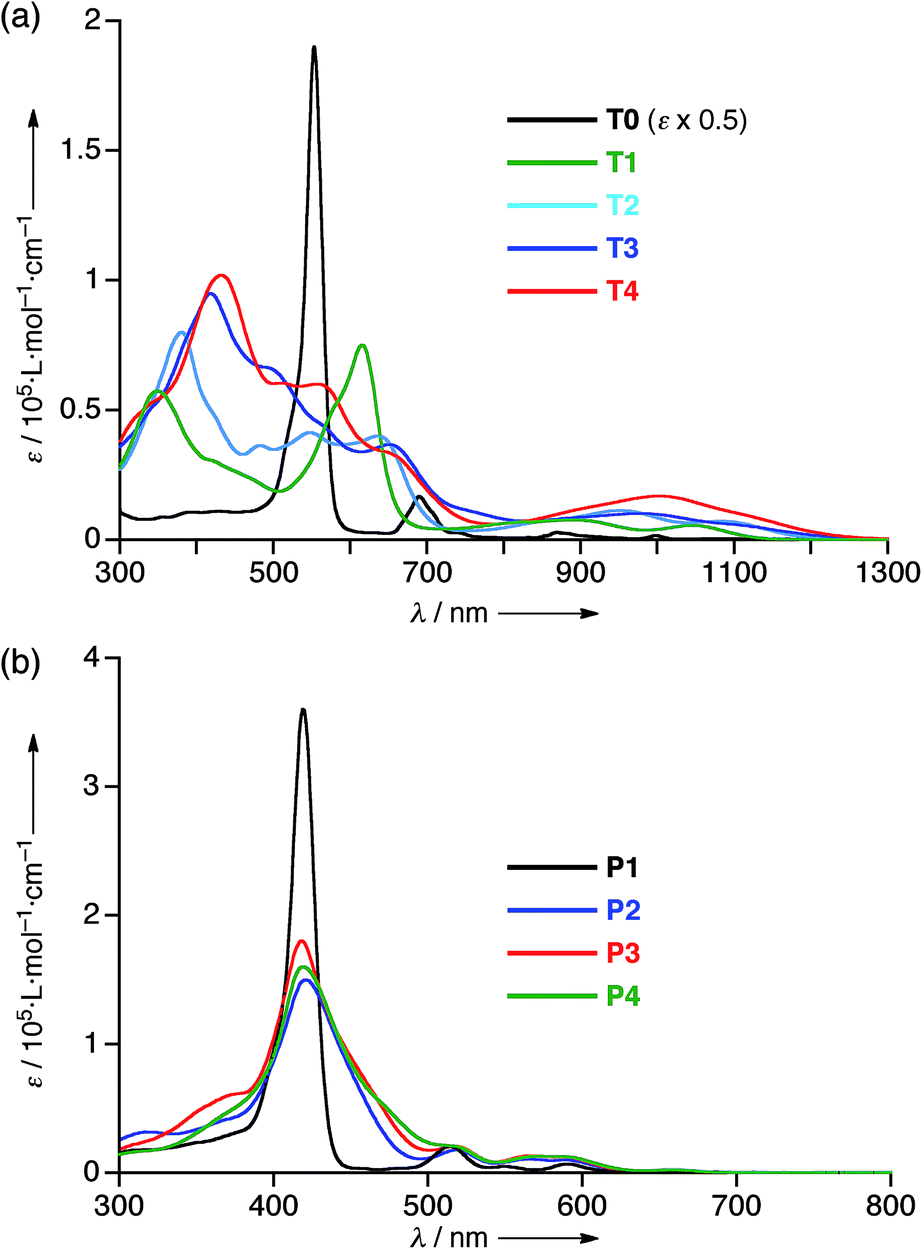 | ||
| Fig. 3 UV/Vis/NIR absorption spectra of (a) T0–T4 and (b) P1–P4 in CH2Cl2. | ||
Theoretical calculations and excited state dynamics
To understand the anomalous absorption spectra, density functional theory (DFT) calculations of T0–T4 were carried out using the Gaussian 09 program at the B3LYP/6-31G(d) level.12 Geometry optimisations produced dumbbell structures that were respectively similar to those obtained via X-ray diffraction analyses. Since the calculated MO diagrams of T2–T4 are quite similar (ESI†), we discuss here the MOs of T3 in comparison with those of T0 (Fig. 4). Similar to the rectangular shaped [26]hexaphyrin,13 the HOMO and LUMO of T0 are, respectively, similar to the a1u and a2u HOMOs and two degenerate e*g LUMOs of porphyrins with a HOMO–LUMO gap of 1.88 eV. The LUMO and LUMO + 1 of T3 are nearly localised on the hexaphyrin circuit, and their energy levels are only slightly destabilised from those of T0. In contrast, the HOMO − 1 is spread over the helix-like network and the HOMO is mainly spread over the same network, and both are largely destabilised, which causes a small HOMO–LUMO gap of 1.340 eV. The MO features of T2 and T4 are nearly the same as those of T3, which are all consistent with the CV results. These MO features compelled us to consider a dual contribution of the 26π-hexaphyrin network and an acyclic conjugation along the helix-like structure (Fig. 1b), which plays an important role in the electronic excitation to the excited states.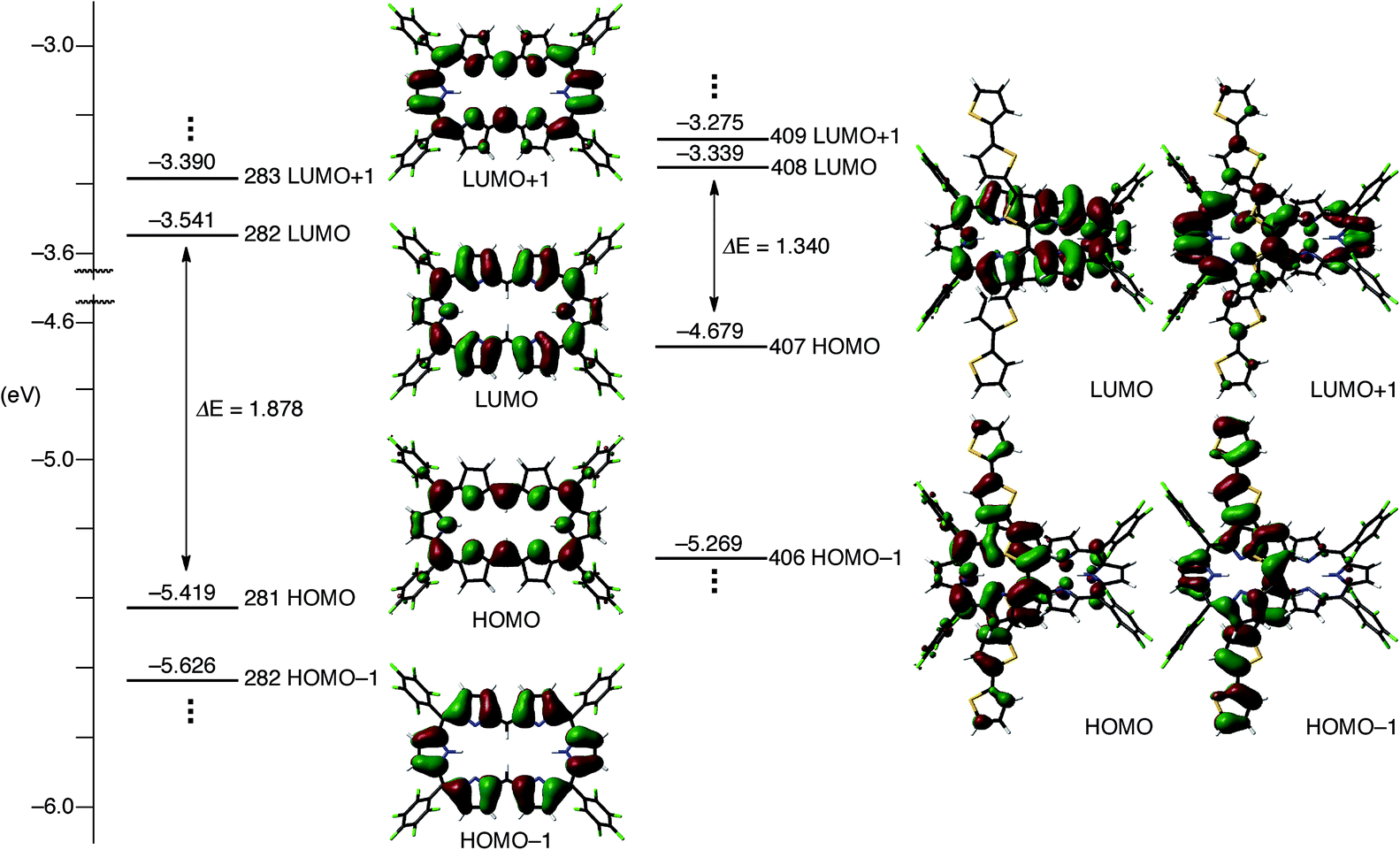 | ||
| Fig. 4 Kohn–Sham MO diagrams of T0 and T3 calculated with the Gaussian 09 package. All energy levels were calculated at the B3LYP/6-31G(d) level. | ||
The nucleus-independent chemical shift (NICS) values14 inside the [26]hexaphyrin macrocycle were calculated to be −15.54, −12.21, −8.50, −8.30, and −8.26 for T0–T4, respectively. The harmonic oscillator model of aromaticity (HOMA) values15 for the [26]hexaphyrin circuit were calculated from the real crystal structures and the optimised structures to be 0.783 and 0.737 for T0, 0.568 and 0.604 for T1, 0.464 (0.425) and 0.518 for T2, and 0.397 and 0.498 for T3. The HOMA value of T4 was only calculated for the optimised structure to be 0.498 due to insufficient crystal data to discuss the bond length. These results indicate the increasing perturbation by the 5,20-oligothienyl substituents of T1–T4.
We have also investigated the excited state dynamics of T1–T4 by using femtosecond transient absorption (fs-TA) spectroscopy (Fig. S46 and 47†) to confirm the electronic perturbation by the 5,20-oligothienyl substituents. S1-state lifetimes have been determined to be 35.4 ps for T1, 15.2 ps for T2, 10.0 ps for T3, and 8.5 ps for T4, respectively, which show a dramatic decrease compared to the value of 138 ps for T0.5a These overall observations also confirmed the increasing electronic perturbation by the oligothienyl chains.
Collectively, these experimental and theoretical calculations indicate that the perturbation provided by the oligothienyl substituents is delicate, being not strong enough to spoil the aromatic [26]hexaphyrin network but large enough to alter the absorption properties and mitigate the aromatic characters of the [26]hexaphyrin segments as a rare case.
Conclusions
5,20-Bis(α-oligothienyl)-substituted [26]hexaphyrins(1.1.1.1.1.1) T1–T4 were prepared and were shown to possess characteristic optical and electronic properties. The observed diatropic ring currents and reversible reduction waves indicate that the aromatic [26]hexaphyrin networks are viable in T1–T4. Nevertheless, the absorption spectra and the excited state dynamics of T1–T4 are significantly perturbed by the appended oligothienyl substituents. DFT calculations indicated that the LUMO and LUMO + 1 are localised at the [26]hexaphyrin network but that the HOMO and HOMO − 1 are significantly spread over the helix-like network and are largely destabilised. Further explorations of expanded porphyrins possessing dual electronic networks are actively under way in our group.Acknowledgements
The work at Kyoto was supported by JSPS KAKENHI of Grant Numbers 25220802 and 25620031. H.M. acknowledges a JSPS Fellowship for Young Scientists. The work at Yonsei was supported by Global Research Laboratory (GRL) Program (2013-8-1472) of the Ministry of Education, Science, and Technology (MEST) of Korea.Notes and references
- (a) A. Jasat and D. Dolphin, Chem. Rev., 1997, 97, 2267 CrossRef CAS PubMed; (b) H. Furuta, H. Maed and A. Osuka, Chem. Commun., 2002, 1795 RSC; (c) J. L. Sessler and D. Seidel, Angew. Chem., Int. Ed., 2003, 42, 5134 CrossRef CAS PubMed; (d) T. K. Chandrashekar and S. Venkatraman, Acc. Chem. Res., 2003, 36, 676 CrossRef CAS PubMed; (e) M. Stępień, N. Sprutta and L. Latos-Grażyński, Angew. Chem., Int. Ed., 2011, 50, 4288 CrossRef PubMed; (f) S. Saito and A. Osuka, Angew. Chem., Int. Ed., 2011, 50, 4342 CrossRef CAS PubMed; (g) A. Osuka and S. Saito, Chem. Commun., 2011, 47, 4330 RSC.
- (a) S. Mori and A. Osuka, J. Am. Chem. Soc., 2005, 127, 8030 CrossRef CAS PubMed; (b) S. Mori, K. S. Kim, Z. S. Yoon, S. B. Noh, D. Kim and A. Osuka, J. Am. Chem. Soc., 2007, 129, 11344 CrossRef CAS PubMed; (c) K. Naoda, H. Mori and A. Osuka, Chem.–Asian J., 2013, 8, 1395 CrossRef CAS PubMed; (d) H. Mori, Y. M. Sung, B. S. Lee, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2012, 51, 12459 CrossRef CAS PubMed.
- (a) M. Stępień, L. Latos-Grażyński, N. Sprutta, P. Chwalisz and L. Szterenberg, Angew. Chem., Int. Ed., 2007, 46, 7869 CrossRef PubMed; (b) Y. Tanaka, S. Saito, S. Mori, N. Aratani, H. Shinokubo, N. Shibata, Y. Higuchi, Z. S. Yoon, K. S. Kim, S. B. Noh, J. K. Park, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 008, 47, 681 CrossRef PubMed; (c) J. Sankar, S. Mori, S. Saito, H. Rath, M. Suzuki, Y. Inokuma, H. Shinokubo, K. S. Kim, Z. S. Yoon, J.-Y. Shin, J. M. Lim, Y. Matsuzaki, O. Matsushita, A. Muranaka, N. Kobayashi, D. Kim and A. Osuka, J. Am. Chem. Soc., 2008, 130, 13568 CrossRef CAS PubMed; (d) Z. S. Yoon, A. Osuka and D. Kim, Nat. Chem., 2009, 1, 113 CrossRef CAS PubMed.
- (a) E. Pacholska-Dudziak, J. Skonieczny, M. Pawlicki, L. Szterenberg, Z. Ciunik and L. Latos-Grażyński, J. Am. Chem. Soc., 2008, 130, 6182 CrossRef CAS PubMed; (b) T. Higashino, J. M. Lim, T. Miura, S. Saito, J.-Y. Shin, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 4950 CrossRef CAS PubMed; (c) T. Higashino, B. S. Lee, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2012, 51, 13105 CrossRef CAS PubMed.
- (a) T. Koide, G. Kashiwazaki, M. Suzuki, K. Furukawa, M.-C. Yoon, S. Cho, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2008, 47, 9661 CrossRef CAS PubMed; (b) H. Rath, S. Tokuji, N. Aratani, K. Furukawa, J. M. Lim, D. Kim, H. Shinokubo and A. Osuka, Angew. Chem., Int. Ed., 2010, 49, 1489 CrossRef CAS PubMed.
- T. Koide, K. Furukawa, H. Shinokubo, J.-Y. Shin, K. S. Kim, D. Kim and A. Osuka, J. Am. Chem. Soc., 2010, 132, 7246 CrossRef CAS PubMed.
- (a) H. Mori, J. M. Lim, D. Kim and A. Osuka, Angew. Chem., Int. Ed., 2013, 52, 12997 CrossRef CAS PubMed; (b) G. Karthik, M. Sneha, V. P. Raja, J. M. Lim, D. Kim, A. Srinivasan and T. K. Chandrashekar, Chem.–Eur. J., 2013, 19, 1886 CrossRef CAS PubMed.
- M. Suzuki and A. Osuka, Chem.–Eur. J., 2007, 13, 196 CrossRef CAS PubMed.
-
(a) Crystallographic data for T2: 2(C70H24F20N6S4)·1.82(C6H14)·1.10(CH2Cl2), Mr = 3165.36, triclinic, space group P
![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) (no. 2), a = 18.9073(16), b = 20.7452(6), c = 21.091(2) Å, α = 67.38(2), β = 77.21(2), γ = 65.257(13)°, V = 6917.2(9) Å3, T = 93 K, ρcalcd = 1.520 g cm−3, Z = 2, 87882 reflections measured, 23494 unique (Rint = 0.0351), R1 = 0.0639 (I > 2σ(I)), Rw = 0.1805 (all data), GOF = 1.042;
(b) Crystallographic data for T3: C78H28F20N6S6·0.51(C7H16)·C7·1.89(CH2Cl2), Mr = 1882.68, monoclinic, space group C2/c (no. 15), a = 49.998(16), b = 12.402(3), c = 35.242(11) Å, β = 131.613(5)°, V = 16338(8) Å3, T = 93 K, ρcalcd = 1.513 g cm−3, Z = 8, 51126 reflections measured, 14608 unique (Rint = 0.0547), R1 = 0.0850 (I > 2σ(I)), Rw = 0.2299 (all data), GOF = 1.034;
(c) Crystallographic data for T4: 2(C86H32F20N6S8)·2(C7H16)·0.44(C7)·1.53(CHCl3)·O, Mr = 4006.31, triclinic, space group P (no. 2), a = 17.2734(8), b = 18.9028(5), c = 30.1289(17) Å, α = 105.000(14), β = 92.92(2), γ = 107.23(3)°, V = 8988.7(7) Å3, T = 93 K, ρcalcd = 1.481 g cm−3, Z = 2, 99137 reflections measured, 27782 unique (Rint = 0.0733), R1 = 0.1104 (I > 2σ(I)), Rw = 0.3750 (all data), GOF = 1.073.
(no. 2), a = 18.9073(16), b = 20.7452(6), c = 21.091(2) Å, α = 67.38(2), β = 77.21(2), γ = 65.257(13)°, V = 6917.2(9) Å3, T = 93 K, ρcalcd = 1.520 g cm−3, Z = 2, 87882 reflections measured, 23494 unique (Rint = 0.0351), R1 = 0.0639 (I > 2σ(I)), Rw = 0.1805 (all data), GOF = 1.042;
(b) Crystallographic data for T3: C78H28F20N6S6·0.51(C7H16)·C7·1.89(CH2Cl2), Mr = 1882.68, monoclinic, space group C2/c (no. 15), a = 49.998(16), b = 12.402(3), c = 35.242(11) Å, β = 131.613(5)°, V = 16338(8) Å3, T = 93 K, ρcalcd = 1.513 g cm−3, Z = 8, 51126 reflections measured, 14608 unique (Rint = 0.0547), R1 = 0.0850 (I > 2σ(I)), Rw = 0.2299 (all data), GOF = 1.034;
(c) Crystallographic data for T4: 2(C86H32F20N6S8)·2(C7H16)·0.44(C7)·1.53(CHCl3)·O, Mr = 4006.31, triclinic, space group P (no. 2), a = 17.2734(8), b = 18.9028(5), c = 30.1289(17) Å, α = 105.000(14), β = 92.92(2), γ = 107.23(3)°, V = 8988.7(7) Å3, T = 93 K, ρcalcd = 1.481 g cm−3, Z = 2, 99137 reflections measured, 27782 unique (Rint = 0.0733), R1 = 0.1104 (I > 2σ(I)), Rw = 0.3750 (all data), GOF = 1.073. - The intense bands of T1–T4 in the UV region are red-shifted in a manner similar to those of the α-oligothiophenes as below; 353/354 nm for T1 and terthiophene, 385/392 nm for T2 and quaterthiophene, 414/417 nm for T3 and quinquethiophene, and 432/436 nm for T4 and sexithiophene. R. S. Becker, J. Seixas de Melo, L. Macanita and F. Elisei, J. Phys. Chem., 1996, 100, 18683 CrossRef CAS . One of the reviewers suggested charge transfer interactions between the [26]hexaphyrin networks and the oligothienyl substituents, but the solvent polarity effects were only marginal for T1–T4 (ESI†).
- (a) P. Morosini, M. Scherer, S. Meyer, V. Lynch and J. L. Sessler, J. Org. Chem., 1997, 62, 8848 CrossRef CAS; (b) J.-Y. Shin, S. S. Hepperle, B. O. Patrick and D. Dolphin, Chem. Commun., 2009, 2323 RSC; (c) S. Saito, K. Furukawa and A. Osuka, J. Am. Chem. Soc., 2010, 132, 2128 CrossRef CAS PubMed.
- For the full Gaussian citation, see ESI.†.
- T. K. Ahn, J. H. Kwon, D. Y. Kim, D. W. Cho, D. H. Jeong, S. K. Kim, M. Suzuki, S. Shimizu, A. Osuka and D. Kim, J. Am. Chem. Soc., 2005, 127, 12856 CrossRef CAS PubMed.
- Z. Chen, C. S. Wannere, C. Corminboeuf, R. Puchta and P. v. R. Schleyer, Chem. Rev., 2005, 105, 3842 CrossRef CAS PubMed.
- T. M. Krygowski and K. M. Cryański, Chem. Rev., 2001, 101, 1385 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: General experimental methods, HR-ESI-TOF mass spectra, UV/Vis absorption spectra, NMR spectra, cyclic voltammograms, X-ray crystal structures and results of DFT calculations. CCDC 1025770, 1025771 and 1025772. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c4sc03394a |
| This journal is © The Royal Society of Chemistry 2015 |