
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePhotostick: a method for selective isolation of target cells from culture†
Miao-Ping
Chien
,
Christopher A.
Werley
,
Samouil L.
Farhi
and
Adam E.
Cohen
*
Departments of Chemistry and Chemical Biology and Physics, Harvard Stem Cell Institute, Harvard University, Howard Hughes Medical Institute, Cambridge, MA 02138, USA. E-mail: cohen@chemistry.harvard.edu
First published on 21st January 2015
Abstract
Sorting of target cells from a heterogeneous pool is technically difficult when the selection criterion is complex, e.g. a dynamic response, a morphological feature, or a combination of multiple parameters. At present, mammalian cell selections are typically performed either via static fluorescence (e.g. fluorescence activated cell sorter), via survival (e.g. antibiotic resistance), or via serial operations (flow cytometry, laser capture microdissection). Here we present a simple protocol for selecting cells based on any static or dynamic property that can be identified by video microscopy and image processing. The “photostick” technique uses a cell-impermeant photochemical crosslinker and digital micromirror array-based patterned illumination to immobilize selected cells on the culture dish. Other cells are washed away with mild protease treatment. The crosslinker also labels the selected cells with a fluorescent dye and a biotin for later identification. The photostick protocol preserves cell viability, permits genetic profiling of selected cells, and can be performed with complex functional selection criteria such as neuronal firing patterns.
The ability to select a small number of cells from a heterogeneous population is fundamental to many aspects of biological research. Selections form the basis of genetic screens, of protein engineering and directed evolution, and of protocols to produce stably transformed or genome-edited cell lines. In many instances, one would like to select cells on the basis of complex dynamic or morphological features. For example, in a culture of olfactory neurons, one might screen for calcium flux in response to a specific odorant; and then wish to select responsive cells for subsequent transcriptional profiling. Or in a culture with single genes knocked down by an siRNA library,1,2 one might find cells with unusual shapes, organelle sizes, or metabolic responses; and then wish to select these cells to determine which gene had been knocked down. These types of selections are difficult to perform with existing tools.
The most common selection technique uses fluorescence-activated cell sorting (FACS),3 which requires a robust static fluorescence signal. Laser-capture microdissection4,5 selects cells or tissue regions one at a time, and so can have limited throughput, and is usually performed on samples that have been chemically fixed. Imaging cytometry6,7 typically functions in a flow-through geometry, and so is not compatible with selections of surface-bound cells such as neurons; nor with selections that probe dynamic cellular responses.
Spatially patterned photochemistry is becoming widely applied in cell biology for its ability to induce specific reactions in complex patterns of space and time.8 Photochemical pre-patterning of cell adhesion molecules enables cell growth in complex morphologies,9–11 and photopatterned hydrogels are now used to direct cell culture in three dimensions.12–14 In these applications the pattern is defined prior to plating the cells. For screening purposes one would like to define the adhesion pattern after plating the cells, only retaining cells with a user-specified phenotype. Two recent demonstrations showed photochemical release of cells from a photodegradable surface,15,16 but in these protocols the surface had to be specially prepared prior to cell culture.
Photochemical radical initiators have been used in macroscopic tissue bonding applications17 and found to produce minimal toxicity.18 Here we describe a photochemical tissue bonding scheme to capture single cells grown on a standard tissue culture substrate. We synthesized a cell-impermeable photochemical crosslinker that also labels selected cells with a fluorescent marker and a biotin group. By using a custom ultra-wide field epi-fluorescence microscope equipped with a digital micromirror device (DMD) to pattern the violet illumination, multiple single cells were selected in parallel from fields of view containing up to ∼4000 cells.
Fig. 1A illustrates the photostick protocol. Cells are cultured on glass-bottom dishes coated with fibronectin or other cell adhesion protein. Cells of interest are selected by video microscopy and computational image processing. A cell-impermeant photochemical crosslinker (Fig. 1B) is added to the dish. A digital micromirror device (DMD) projects patterned violet illumination targeting the cells of interest (Fig. 1C), typically with 3.25 μm spatial resolution over a 6 mm × 3 mm field of view. The crosslinker immobilizes these cells on the dish. The dish is then rinsed with buffer to remove unreacted crosslinker. To develop the pattern, the dish is incubated with accutase, a mild protease. Cells outside the illuminated region are washed away, while the illuminated cells remain adherent.
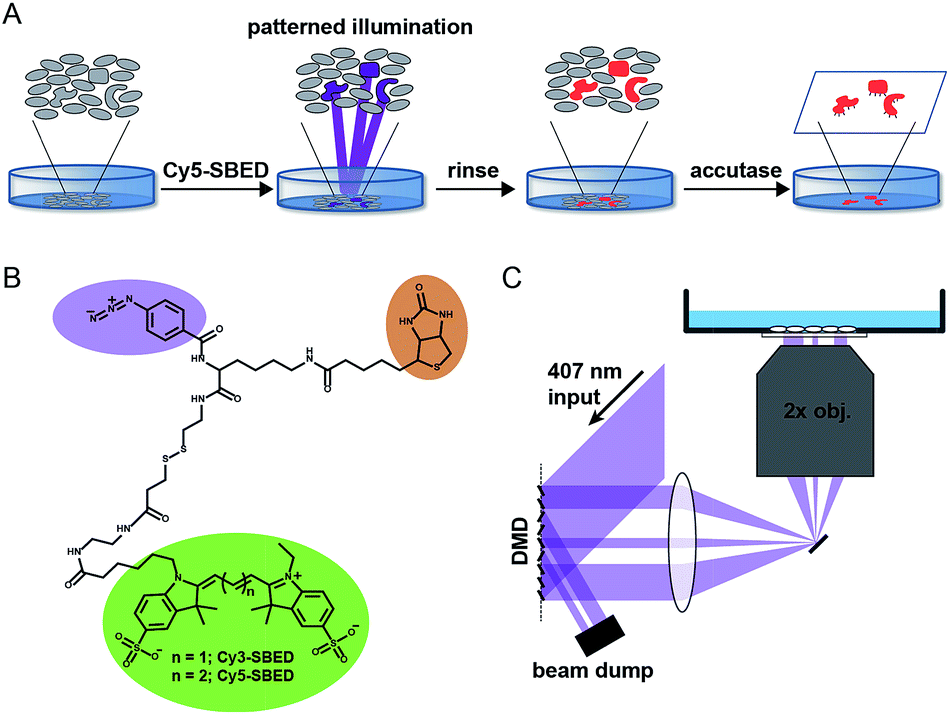 | ||
| Fig. 1 Components of the photostick protocol. (A) Sequence of steps in photostick method. Photochemical immobilization retains target cells while others are washed away under mild protease treatment. (B) Trifunctional crosslinkers Cy3- and Cy5-SBED for photochemical immobilization with simultaneous fluorescent labeling and biotynilation. (C) Digital micromirror device (DMD) for patterned violet illumination to activate photo-crosslinker. | ||
Initially we tested the photostick protocol with a water soluble phenyl azide radical initiator, 4-fluoro-3-nitrophenyl azide (FNPA, Fig. S1†). Upon exposure to violet light (407 nm) this compound releases N2 and produces a nitrene radical19–21 that reacts with protein functional groups via a sequential abstraction–recombination mechanism.22,23 Radical formation on both fibronectin and cellular surface proteins led to covalent cross-linking of cells to the dish surface. We added FNPA at a concentration of 4 μM to cultures of epithelial MDCK cells and exposed to patterned 407 nm light (825 J cm−2). The pattern was developed via incubation with accutase (3 min, 37 °C) followed by rinsing with buffer. The remaining cells clearly followed the illumination pattern (Fig. S1†).
FNPA has a calculated octanol/water partition coefficient of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P = 3.0, implying high membrane permeability.24 We were thus concerned that the initiator could enter the cells, crosslinking internal components and perturbing cell physiology. Furthermore, other than location on the dish, there was no clear indication of which cells had been targeted for selection. Therefore we synthesized two trifunctional photochemical crosslinkers, Cy3- and Cy5-SBED, by reacting an aminated fluorescent dye (Cy3 or Cy5) with sulfo-SBED (sulfo-N-hydroxysuccinimidyl-2-(6-[biotinamido]-2-(p-azido-benzamido)-hexanoamido)-ethyl-1,3′-dithioproprionate) (Fig. 1B). The product contained a fluorescent group, a biotin group, and an aryl azide photochemical radical initiator. The two sulfate groups and the large size of the construct suggested that it would show poor membrane permeability, while the dye allowed easy tracking. The biotin gave the option for downstream labeling with streptavidin, but was not used in this study.
P = 3.0, implying high membrane permeability.24 We were thus concerned that the initiator could enter the cells, crosslinking internal components and perturbing cell physiology. Furthermore, other than location on the dish, there was no clear indication of which cells had been targeted for selection. Therefore we synthesized two trifunctional photochemical crosslinkers, Cy3- and Cy5-SBED, by reacting an aminated fluorescent dye (Cy3 or Cy5) with sulfo-SBED (sulfo-N-hydroxysuccinimidyl-2-(6-[biotinamido]-2-(p-azido-benzamido)-hexanoamido)-ethyl-1,3′-dithioproprionate) (Fig. 1B). The product contained a fluorescent group, a biotin group, and an aryl azide photochemical radical initiator. The two sulfate groups and the large size of the construct suggested that it would show poor membrane permeability, while the dye allowed easy tracking. The biotin gave the option for downstream labeling with streptavidin, but was not used in this study.
We quantified the selectivity of the photostick protocol as a function of Cy3-SBED concentration and illumination dose (Fig. S2 and S3†). The optimal conditions depended on cell type, e.g. 4 μM Cy3/5-SBED at light dose 825 J cm−2 was optimal for MDCK cells (Fig. 2 and S2†), while 15 μM Cy3/5-SBED at light dose 2200 J cm−2 was optimal for neurons (Fig. 3C and D). To test the viability of cells after a photostick procedure, we returned a dish of patterned MDCK cells to the incubator. A live–dead stain showed 98% live cells subsequent to a photostick protocol (Fig. S4†). The cells continued to migrate and divide (Fig. S5†) with a doubling time of 34 h.
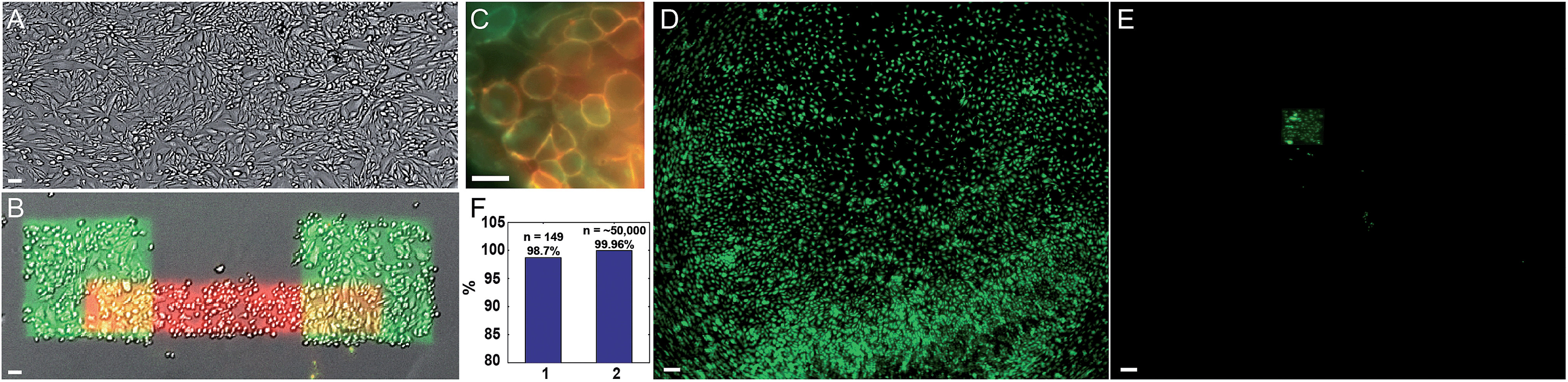 | ||
| Fig. 2 Photostick of MDCK cells with sequential addition of Cy3-SBED and Cy5-SBED. (A) Transmitted light image of MDCK cells. (B) Cells were exposed to Cy3-SBED (4 μM) and illuminated with two squares of violet light. The cells were then exposed to Cy5-SBED and exposed to a bar of violet light. After development with accutase, cells that had been illuminated were preferentially retained. Image shows a composite of transmitted light (grey), fluorescence of Cy3 (green) and fluorescences of Cy5 (red) after this protocol. (C) Close-up of the overlap of the Cy3- and Cy5-labeled regions, showing absence of intracellular labeling by either dye. Image acquired on cells fixed immediately after the photostick procedure. (D) A partial low-magnification field of view from a dish of MDCK cells labeled with 2 μM calcein AM. (E) The same field of view as (D) after photostick with 4 μM Cy3-SBED and accutase development. Cells in the illuminated region remained, while most others were washed away. (F) Quantification of photostick selectivity and specificity. Cells within the illuminated region were retained with high efficiency (147/149, 98.7%, n = 9 experiments; bar 1), and cells in the dark region were removed with high efficiency (99.96%, 21 false positives of ∼50000 cells; bar 2). Scale bar: (A and B), 50 μm; (C), 30 μm; (D and E), 200 μm. | ||
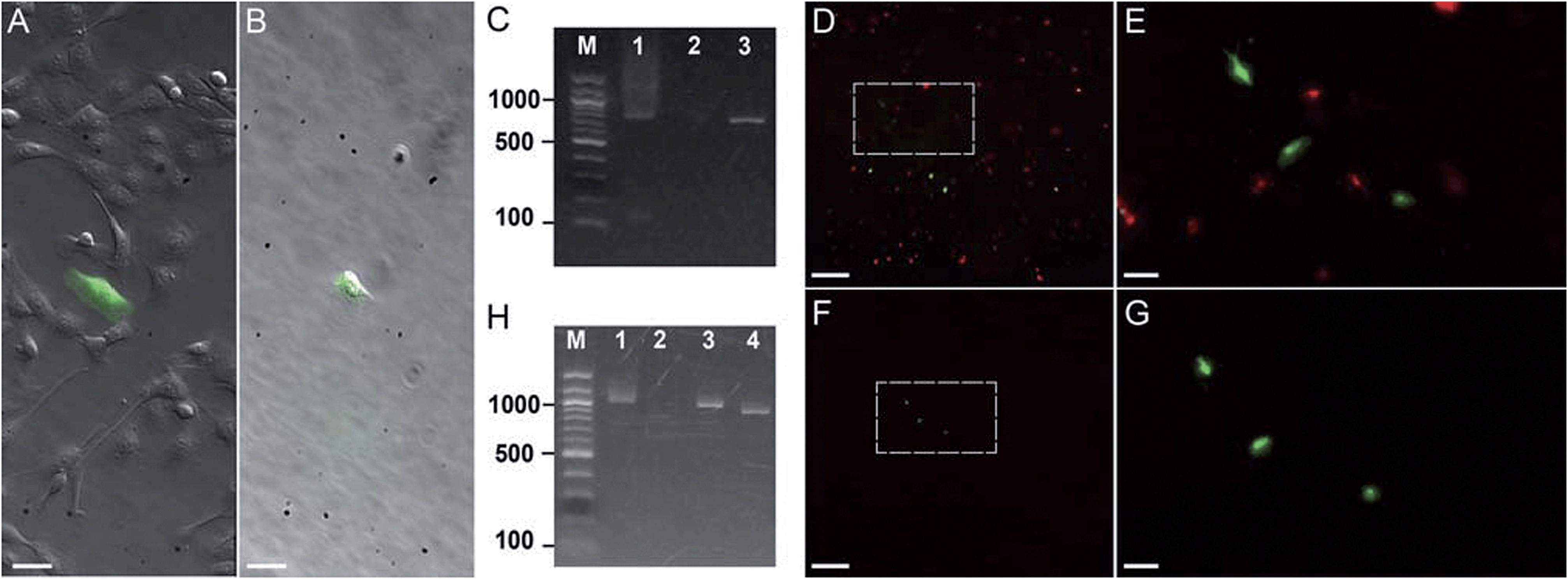 | ||
| Fig. 3 Photostick of target cells. (A and B) Photostick of a single YFP-expressing MDCK cell, using 4 μM Cy5-SBED. (A) Merged bright field and YFP image prior to photostick. (B) Merged image after photostick. (C) PCR detection of YFP gene from the cell in panel B. Lane 1: YFP gene (713 bp) amplified from the cell selected by photostick. Lane 2: PCR amplification of a non-fluorescent cell did not produce a band. Lane 3: PCR of purified YFP gene. (D–G) Photostick of three YFP-expressing MDCK cells, using 4 μM Cy5-SBED, from a mixed culture of cells expressing either YFP or mOrange. (D) Merged image before photostick. (E) Zoom-in image from panel D. (F) Merged image after photostick. (G) Zoom-in image from panel F. (H) PCR detection of YFP in cells selected by photostick from panel G. Lane 1: YFP (1017 bp) amplified with consensus primers for YFP and mOrange (Con-primers, see ESI†). Lane 2: PCR amplification with mOrange specific primers (mO2 primers, see ESI†). No mOrange gene was detected. Lane 3: PCR of purified YFP gene with Con-primers (See ESI†). Lane 4: PCR of purified mOrange gene with Con-primers (see ESI†). Scale bar: (A and B): 30 μm; (D and F): 500 μm; (E and G): 100 μm. | ||
We performed successive photostick protocols on a sample of MDCK cells (Fig. 2A and B), first with a green dye, Cy3-SBED (4 μM, 825 J cm−2), and then with a red dye, Cy5-SBED. Upon wash-out of the dyes, the illuminated cells showed strong fluorescence corresponding to the color dye with which they were exposed. We did not detect a difference in Cy5 labeling efficiency between the cells that had already been labeled with Cy3 and the cells that had not, indicating that a small fraction of reactive sites on the cell surface were occupied by each dye. Incubation with accutase (3 min, 37 °C) detached the un-exposed cells while leaving the exposed cells (Fig. 2B). High magnification images (Fig. 2C) showed that the fluorescence was localized to the cell membrane. Absence of intracellular fluorescence confirmed that the dye-SBED compounds did not enter the cells. In nine repeated trials 98.7% (147 of 149) photostuck cells remained (Fig. 2F, bar 1), while 0.04% (21 of ∼50000) of non-photostuck cells remained (Fig. 2F, bar 2; see also Fig. S6†). Thus the photostick method has high selectivity, specificity and accuracy for the targeted cells (Fig. 2F). Fig. 2D and E show a low-magnification field of view of MDCK cells before (Fig. 2D) and after (Fig. 2E) the photostick protocol.
A natural application of the photostick technique is to select single clones from a genetically heterogeneous culture. These clones could be produced e.g. by library lentiviral knockdown of endogenous genes,25 or by overexpression of a library of functional endogenous or heterologous genes. Thus we sought to test the suitability of the photostick protocol for genetic profiling of single cells selected from a heterogeneous culture.
First we tested whether genetic information could be retrieved from a single cell selected by photostick. MDCK cells expressing YFP were plated sparsely in a background of non-expressing cells (Fig. 3A). A single YFP-positive cell was selected by photostick (4 μM Cy5-SBED). After accutase treatment (3 min, 37 °C), only the single targeted cell was visible (Fig. 3B). The selected cell was then released via trypsinization, and its genetic content was analyzed by single-cell PCR (ESI methods†). The YFP gene product was detected (Fig. 3C lane 1). The experiment was repeated with selection of a cell lacking YFP expression. No YFP gene product was detected (Fig. 3C lane 2).
Next we tested whether genetic information from surrounding cells could contaminate the genetic material amplified from the cell selected by photostick. Such contamination could arise, for instance, by lysis of surrounding cells; or by surrounding cells remaining adhered during the accutase treatment but then being released by trypsin. In Fig. 3, three YFP-positive cells were selected by photostick (4 μM Cy5-SBED) out of a background population dominated by cells expressing mOrange. After accutase treatment only these three cells remained (Fig. 3D–G). These cells were released with trypsin. Amplification with consensus primers for YFP and mOrange led to a single band (Fig. 3H, lane 1). Amplification with primers selective for mOrange did not produce a product (Fig. 3H, lane 2). These results established that mOrange DNA from the surrounding cells did not contaminate the photostick-selected YFP-expressing cells, despite the large number of mOrange-expressing cells initially in the population.
Finally, we tested the ability to select cells on the basis of a complex functional parameter. Our lab recently developed a platform for all-optical electrophysiology (“Optopatch”) in cultured neurons.26 We expressed the Optopatch construct in cultured rat hippocampal neurons, and used a wide-field imaging system for simultaneous optical stimulation and optical recording from a field of view containing ∼40 neurons expressing the Optopatch construct. We selected a neuron that showed a rapidly adapting firing pattern (Fig. 4C), added Cy3-SBED (15 μM) to the imaging medium, and selected the cell via photostick (Fig. 4A). After incubation with accutase (6 min, 37 °C), the selected neuron remained, while the other neurons had been washed away (Fig. 4B). The photostick protocol kept the cell body, but not the distal neurites. PCR recovered the Optopatch genes from the selected cell (Fig. 4D) and subsequent sequencing recovered the complete gene sequence.
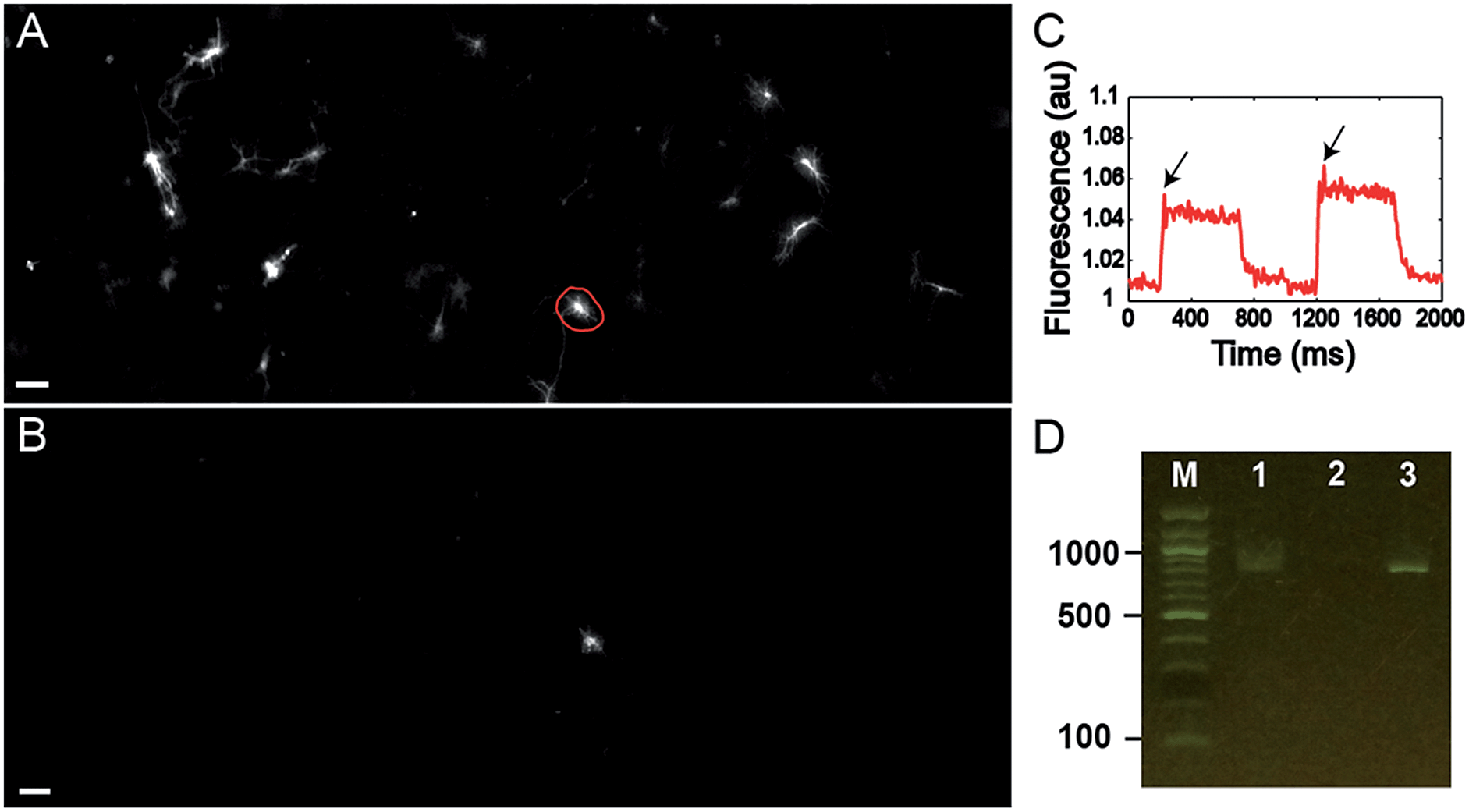 | ||
| Fig. 4 (A and B) Photostick of a single neuron with rapidly adapting firing pattern (C, arrows) as determined by Optopatch measurement. (A) Image of GFP fluorescence in neurons expressing the Optopatch construct prior to cell selection via photostick. (B) GFP image after photostick. (D) Single-cell PCR detection of partial Optopatch gene (745 bp) in a single neuron selected by photostick. Lane 1: Optopatch gene (745 bp) amplified from a single cell selected by photostick. Lane 2: PCR negative control without cell lysate. Lane 3: PCR of purified Optopatch gene. | ||
Conclusions
The 407 nm light used for photostick is not directly absorbed by proteins or nucleic acids, but could excite cofactors such as FAD. We demonstrated that the photostick protocol preserved viability of MDCK cells, but one may worry about more subtle cellular perturbations or stress associated either with the violet light exposure or with the covalently bound dyes. The significance of these perturbations depends on the application. When the selected cells are immediately fixed or lysed for biochemical analysis (e.g. DNA or RNA sequencing, or proteomics) optical perturbation effects will likely be minor, due to the short interval between violet illumination and cell harvest. When the selected cells are to be grown into a stable cell line, optical perturbation effects will also likely be minor, due to the many generations of growth required before use, providing time for cells to recover. However, when the selected cells will be used for functional assays shortly after selection, we advise appropriate control experiments to test for illumination artefacts.Each cell type and culture protocol will likely require optimization of the parameters. We recommend a two-step procedure: first, without using the photostick protocol, one should determine the minimum accutase incubation time to lift the cells. This determination can be performed in a single dish by gently pipetting the accutase solution and periodically checking for cell detachment. Second, one should determine the concentration of Cy3- or Cy5-SBED and illumination dose to achieve accutase-resistant adhesion, starting from the parameters presented here. Cells adhered by the photostick protocol will not be detached by the shear associated with gentle rinsing. The photostick protocol worked with substrates coated with either fibronectin or poly-D-lysine, and we anticipate similar results with any surface presenting primary amines (e.g. lysine) or hydroxyl groups (e.g. serine).
For screening applications, one is particularly concerned about the proportion of false positives among the selected cells. Suppose there are N cells initially on the dish, the false-positive rate is f (cells that should be washed away but remain), and the true positive rate is p (cells that should remain and do remain). To achieve a ratio, R, of true-positive to false-positive cells, one should select n = RNf/p cells. In our experiments, the false positive rate ranged from 0 to 2%, with the undesired cells often adhering around a defect in the dish. Through careful attention to preparation of the dish one can minimize f. By increasing the accutase incubation time one can further decrease f at the expense of a modest decrease in p. One can increase the illumination dose or the concentration of crosslinker to maximize p. Under strong illumination, scattered light can crosslink cells adjacent the desired cell. We found that selections worked best with MDCK cells, whose large size facilitated single-cell selection. In highly confluent cultures of HEK cells, application of photostick to a single cell often retained one or more of its adjacent neighbors as well.
Modern high-resolution cameras and advanced image processing can characterize biochemically significant numbers of cells in experimentally reasonable timescales. In principle, computational methods could select based on a vastly larger set of parameters than can be selected by biochemical or pharmacological means, but an unresolved challenge has been how to physically isolate cells of interest from a complex culture. The photostick approach could be used to identify genes whose over- or under-expression affects complex aspects of cell morphology, dynamics, or response to perturbations. This method could also be useful to select antibodies or other functional proteins expressed from a library at one copy per cell. Finally, photostick could be used in the generation of stable cell lines, where gene expression is detected by a morphological or functional parameter rather than fluorescence or antibiotic resistance.
Acknowledgements
This work was supported by the Howard Hughes Medical Institute, the Harvard Center for Brain Science, PECASE award N00014-11-1-0549, US National Institutes of Health grants 1-R01-EB012498-01 and New Innovator grant 1-DP2-OD007428, and the Gordon and Betty Moore Foundation.References
- D. E. Root, N. Hacohen, W. C. Hahn, E. S. Lander and D. M. Sabatini, Nat. Methods, 2006, 3, 715–719 CrossRef CAS PubMed.
- J. Moffat, D. A. Grueneberg, X. Yang, S. Y. Kim, A. M. Kloepfer, G. Hinkle, B. Piqani, T. M. Eisenhaure, B. Luo and J. K. Grenier, Cell, 2006, 124, 1283–1298 CrossRef CAS PubMed.
- M. T. Anderson, I. M. Tjioe, M. C. Lorincz, D. R. Parks, L. A. Herzenberg, G. P. Nolan and L. A. Herzenberg, Proc. Natl. Acad. Sci. U. S. A., 1996, 93, 8508–8511 CrossRef CAS.
- M. R. Emmert-Buck, R. F. Bonner, P. D. Smith, R. F. Chuaqui, Z. Zhuang, S. R. Goldstein, R. A. Weiss and L. A. Liotta, Science, 1996, 274, 998–1001 CrossRef CAS.
- V. Espina, J. D. Wulfkuhle, V. S. Calvert, A. VanMeter, W. Zhou, G. Coukos, D. H. Geho, E. F. Petricoin and L. A. Liotta, Nat. Protoc., 2006, 1, 586–603 CrossRef CAS PubMed.
- M. Henriksen, Nat. Methods, 2010, 7, 331–332 CrossRef PubMed.
- Y. Ozaki, S. Uda, T. H. Saito, J. Chung, H. Kubota and S. Kuroda, PLoS One, 2010, 5, e9955 Search PubMed.
- B. P. Fors, J. E. Poelma, M. S. Menyo, M. J. Robb, D. M. Spokoyny, J. W. Kramer, J. H. Waite and C. J. Hawker, J. Am. Chem. Soc., 2013, 135, 14106–14109 CrossRef CAS PubMed.
- T. Matsuda and T. Sugawara, J. Biomed. Mater. Res., 1995, 29, 749–756 CrossRef CAS PubMed.
- K. M. El Muslemany, A. A. Twite, A. M. ElSohly, A. C. Obermeyer, R. A. Mathies and M. B. Francis, J. Am. Chem. Soc., 2014, 136, 12600–12606 CrossRef CAS PubMed.
- H. Onoe, S. C. Hsiao, E. S. Douglas, Z. J. Gartner, C. R. Bertozzi, M. B. Francis and R. A. Mathies, Langmuir, 2012, 28, 8120–8126 CrossRef CAS PubMed.
- A. M. Kloxin, M. W. Tibbitt and K. S. Anseth, Nat. Protoc., 2010, 5, 1867–1887 CrossRef CAS PubMed.
- P. Soman, P. H. Chung, A. P. Zhang and S. Chen, Biotechnol. Bioeng., 2013, 110, 3038–3047 CrossRef CAS PubMed.
- K. C. Hribar, P. Soman, J. Warner, P. Chung and S. Chen, Lab Chip, 2014, 14, 268–275 RSC.
- D. Shin, J. You, A. Rahimian, T. Vu, C. Siltanen, A. Ehsanipour, G. Stybayeva, J. Sutcliffe and A. Revzin, Angew. Chem., Int. Ed., 2014, 53, 8221–8224 CrossRef CAS PubMed.
- M. Tamura, F. Yanagawa, S. Sugiura, T. Takagi, K. Sumaru, H. Matsui and T. Kanamori, Sci. Rep., 2014, 4, 4793–4798 Search PubMed.
- Y. Kamegaya, W. A. Farinelli, A. V. Vila Echague, H. Akita, J. Gallagher, T. J. Flotte, R. Anderson, R. W. Redmond and I. E. Kochevar, Lasers Surg. Med., 2005, 37, 264–270 CrossRef PubMed.
- M. Yao, A. Yaroslavsky, F. P. Henry, R. W. Redmond and I. E. Kochevar, Lasers Surg. Med., 2010, 42, 123–131 CrossRef PubMed.
- E. Leyva, M. S. Platz, G. Persy and J. Wirz, J. Am. Chem. Soc., 1986, 108, 3783–3790 CrossRef CAS.
- M. J. Travers, D. C. Cowles, E. P. Clifford and G. B. Ellison, J. Am. Chem. Soc., 1992, 114, 8699–8701 CrossRef CAS.
- V. Voskresenska, R. M. Wilson, M. Panov, A. N. Tarnovsky, J. A. Krause, S. Vyas, A. H. Winter and C. M. Hadad, J. Am. Chem. Soc., 2009, 131, 11535–11547 CrossRef CAS PubMed.
- M. S. Platz, R. A. Moss and J. Maitland, in Reviews of reactive intermediate chemistry, ed. R. S. Sheridan, John Wiley & Sons, 2007, p. 415 Search PubMed.
- A. Reiser, F. Willets, G. Terry, V. Williams and R. Marley, Trans. Faraday Soc., 1968, 64, 3265–3275 RSC.
- MolInspiration, 2013, http://www.molinspiration.com/cgi-bin/properties.
- D. Sims, A. M. Mendes-Pereira, J. Frankum, D. Burgess, M. Cerone, C. Lombardelli, C. Mitsopoulos, J. Hakas, N. Murugaesu and C. M. Isacke, Genome Biol., 2011, 12, R104 CrossRef CAS PubMed.
- D. R. Hochbaum, Y. Zhao, S. Farhi, N. Klapoetke, C. A. Werley, V. Kapoor, P. Zou, J. M. Kralj, D. Maclaurin, N. Smedemark-Margulies, J. Saulnier, G. Boulting, Y. Cho, M. Melkonian, G. K. Wong, D. J. Harrison, V. N. Murthy, B. Sabatini, E. S. Boyden, R. E. Campbell and A. E. Cohen, Nat. Methods, 2014, 11, 825–833 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c4sc03676j |
| This journal is © The Royal Society of Chemistry 2015 |