
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
H2-driven biotransformation of n-octane to 1-octanol by a recombinant Pseudomonas putida strain co-synthesizing an O2-tolerant hydrogenase and a P450 monooxygenase†
Thomas H.
Lonsdale‡
ab,
Lars
Lauterbach‡
*a,
Sumire
Honda Malca
c,
Bettina M.
Nestl
c,
Bernhard
Hauer
c and
Oliver
Lenz
*a
aDepartment of Chemistry, Technische Universität Berlin, Straße des 17. Juni 135, 10623 Berlin, Germany. E-mail: lars.lauterbach@tu-berlin.de; oliver.lenz@tu-berlin.de
bDepartment of Chemistry, University of Oxford, Inorganic Chemistry Laboratory, South Parks Rd, Oxford, OX1 3QR, UK
cInstitute of Technical Biochemistry, Universität Stuttgart, Allmandring 31, 70569, Stuttgart, Germany
First published on 11th September 2015
Abstract
An in vivo biotransformation system is presented that affords the hydroxylation of n-octane to 1-octanol on the basis of NADH-dependent CYP153A monooxygenase and NAD+-reducing hydrogenase heterologously synthesized in a bacterial host. The hydrogenase sustains H2-driven NADH cofactor regeneration even in the presence of O2, the co-substrate of monooxygenase.
Cytochrome P450 monooxygenases (CYPs) have the extraordinary capability to introduce oxygen into non-activated C–H bonds in a regio- and stereoselective manner, which is still a challenging task for synthetic catalysts. The continuous discovery of novel CYPs and their modification by protein engineering paves new avenues for their utilization as biocatalysts in the production of pharmaceuticals and fine chemicals.1–3
In particular the regioselective terminal hydroxylation of alkanes by CYPs is of significant interest because the corresponding alkanols can be used as surfactants, plasticizers, solvents or polymer precursors.4 In this study we used the tripartite alkane-oxidizing CYP153A system from Polaromonas sp. JS666 for selective oxidation of n-octane to 1-octanol.5 The system is composed of the catalytically active cytochrome (CYP153A), the electron carrier ferredoxin (Fd) and the FAD-containing ferredoxin reductase (FRN). The latter two proteins are necessary for electron transfer from NADH to the P450 active site (Fig. 1, see ESI,† for details). CYP153 enzymes are promising biocatalysts as they can be heterologously produced in active and soluble form.4 For efficient use of CYPs both in vitro and in vivo, a sufficient supply of the reduced cofactor NADH is required. In whole-cell systems, NAD(P)H is generated either from the basic metabolic routes or via a recombinant regeneration system, generally based on glucose dehydrogenase relying on the supplementation of the growth medium with glucose.6 Whole-cell catalysis for chemical synthesis is attractive because enzymes are generally more stable in their natural environment. Recently, a hydrogenase that oxidizes dihydrogen (H2) and transfers the released electrons to NAD+ has complemented the so-far established systems for NADH regeneration.7–10 In contrast to glucose, H2 readily diffuses through cell membranes, and hydrogenase-based NADH recycling has the advantage of proceeding without a carbon-based reducing agent. Moreover, when coupled with CYP-mediated catalysis water is the only by-product (Fig. 1).11
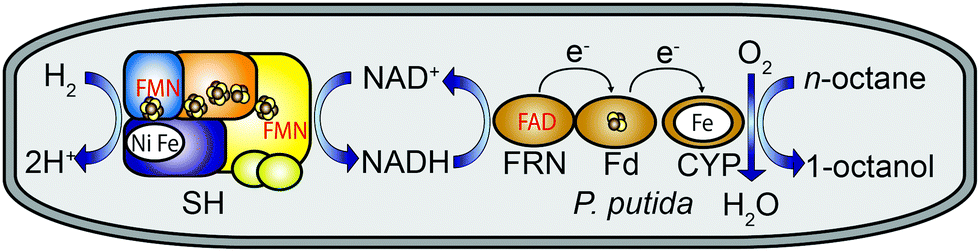 | ||
| Fig. 1 H2-driven conversion of n-octane to 1-octanol in recombinant Pseudomonas putida KT2440 cells producing the soluble NAD+-reducing hydrogenase (SH) from Ralstonia eutropha in addition to CYP153A (CYP), NADH-ferredoxin reductase (FRN), and ferredoxin (Fd) from Polaromonas sp. JS666. The SH contains two flavin mononucleotide (FMN) molecules, and FRN contains one flavin adenine dinucleotide (FAD). Electron-conducting iron–sulfur clusters are shown as cluster-forming spheres coloured in brown and yellow. | ||
Exposure of most hydrogenases to O2 leads to their irreversible inactivation.12 However, a few O2-tolerant [NiFe]-hydrogenases sustain catalytic activity in the presence of O2. One of the most prominent examples is the NAD+-reducing hydrogenase (SH) from the “Knallgas” bacterium Ralstonia eutropha H16 consisting of two functionally distinct heterodimeric moieties.13 The H2-dependent NAD+ reduction activity of the SH is unaffected even at ambient O2 concentrations.14 These features make the SH particularly promising for NADH regeneration in cascade reactions requiring O2, e.g. those involving CYPs.
In this study, we have designed an electron transfer pathway for the H2-driven ω-hydroxylation of n-octane to 1-octanol in recombinant Pseudomonas putida.§ Pseudomonads provide an attractive platform for new metabolic pathways due to their solvent resistance, metabolic versatility and simple genetic accessibility.15 Biosynthesis of the [NiFe] active site of the SH is highly complex and requires a set of auxiliary proteins involved in nickel uptake, metal centre assembly and insertion, site-specific endoproteolysis and transcriptional regulation.16 The corresponding set of 13 genes is arranged as an operon and was set under control of the alkB promoter that controls alkane degradation in Pseudomonas putida GPo1.17 The three genes required for biosynthesis of the CYP153A system from Polaromonas sp. JS666 were likewise equipped with the alkB promoter. Both recombinant operons were inserted into a broad-host-plasmid replicating in P. putida KT2440 (see footnote § and ESI,† for details). In order to obtain the optimal growth conditions for the biotransformations, both the SH activity and the CYP concentration were determined dependent on the length of the induction period. Recombinant P. putida cells were first grown for 12 hours in a glucose–glycerol (GGN) minimal medium.
Gene expression was induced through addition of dicyclopropyl ketone (DCPK), which cannot be metabolized by P. putida. In the course of the induction period (24 h), the CYP concentration showed an almost continuous increase reaching a final value of 0.58 nmol mg−1 (of total protein) after 24 h (Table S1, ESI†). The SH activity, by contrast, showed a considerable increase during the first 4 h of induction and then stayed relatively stable at 0.15 U mg−1 for a further 4 h. However, less than 50% of the maximal activity was left after 24 h (Table S1, ESI†). Henceforth, all subsequent biotransformations were carried out with resting cells derived from a recombinant P. putida culture grown for 12 hours in GGN minimal medium followed by an induction period of 6 h.
After induction the cells were centrifuged, and the cell pellet was re-suspended in carbon-free H16 minimal medium containing n-octane. Biotransformations with 15% (v/v) n-octane were performed in gas-tight reaction vessels containing a gas mixture of H2 and air in the headspace. The same setup without H2 was used to evaluate the biotransformation capacity of the system without support of the SH.
The addition of H2 led to a significantly higher yield of 1-octanol (Fig. 2), demonstrating the supportive role of SH-generated NADH during the biotransformation. After 24 h, 101 mg L−1 and 36 mg L−1 of 1-octanol were formed in the presence and absence of H2, respectively. The maximum concentration of 1-octanol in the biotransformation mixture was approximately five times higher than that achieved previously in in vitro experiments with purified Polaromonas sp. JS666 CYP153A.5
 | ||
| Fig. 2 H2-supported production of 1-octanol by recombinant P. putida KT2440 cells synthesizing CYP153A and SH. Bioconversions were monitored either in the absence (open bars) or presence (dashed bars) of H2. The bar height represents the mean value of three replicates, and the corresponding standard deviation is indicated. | ||
Further oxidation products such as octanoic acid and 2-octanol/octanal were found to constitute 39% of the total octane oxidation products (Table 1). Overoxidation is frequently observed in biotransformations involving P450 enzymes, and in a previous in vitro study, up to 10% of the octane was shown to be overoxidized by CYP153A.5 In contrast to the latter study, however, no α,ω-diols were detected in our in vivo approach. From these observations, it seems likely that the accumulation of octanoic acid and 2-octanol/octanal resulted from a combination of direct overoxidation by CYP153A and native catabolic pathways for 1-octanol degradation present in P. putida. Indeed, in contrast to wild-type P. putida KT2440, the recombinant strain synthesizing the CYP153A system was able to grow with n-octane as the sole carbon and energy source (Fig. S2, ESI†). This clearly indicates catabolic degradation of 1-octanol in vivo. Future mutagenesis studies18 are envisaged to identify potential alcohol dehydrogenases involved in 1-octanol conversion. A subsequent knock-out of the corresponding gene(s) is expected to reduce the amount of overoxidation products.
| Time (h) | 1-Octanol (mg L−1) | 2-Octanol/octanal (mg L−1) | Octanoic acid (mg L−1) |
|---|---|---|---|
| 2 | 28 ± 3 | 4 ± 0.1 | 8 ± 3 |
| 4 | 29 ± 10 | 8 ± 0.6 | 9 ± 3 |
| 6 | 37 ± 8 | 15 ± 1 | 16 ± 5 |
| 24 | 101 ± 1 | 32 ± 9 | 32 ± 14 |
Recombinant P. putida KT2440 cells synthesizing both the CYP153A system and SH grew even slightly faster when H2 was added (Fig. S3, ESI†). This result in combination with the increased 1-octanol production in the presence of H2 point out that NADH-regeneration in carbon-free minimal medium is significantly facilitated by the SH. This is in marked contrast to the results obtained for cells grown in glucose-supplemented mineral medium, where P. putida is able to adjust its energy and reductant demand according to the metabolic burden.15,19
In our proof-of-concept study, we investigated the applicability of the R. eutropha SH to increase the NADH supply in whole cell systems. H2-supported NADH recycling led to an approximately 3-fold increase in the 1-octanol yield in a whole-cell biotransformation system, which uses a dedicated P450 monooxygenase as octane-converting biocatalyst. In order to prevent formation of hazardous gas mixtures during in vivo biotransformation, the application of sub-critical gas concentrations, such as <8 vol% O2 and <5 vol% H2, is recommended.20,21 Our study clearly shows that the SH is capable of overcoming potential bottlenecks of cofactor supply in whole-cell systems. Moreover, H2 represents a viable alternative to carbon-based reductants currently used for in vivo cofactor recycling strategies. Thus, our H2-driven in vivo cofactor regeneration system holds considerable potential for application in other cascades reactions that rely on sustainable supply of NAD(P)H as the reducing agent.
This work was supported by an European Research Council (ERC) proof of concept grant 297503 (to L.L.) and the Deutsche Forschungsgemeinschaft (DFG) through the cluster of excellence “Unifying Concepts in Catalysis”, Berlin (to L.L. and O.L.). T.H.L. would like to thank the Deutscher Akademischer Austauschdienst (DAAD) for a one year scholarship. We are indebted to Andreas Schmid and Bruno Bühler for providing us plasmid pSPZ10. We thank Kylie A. Vincent, Holly A. Reeve and Leland B. Gee for helpful discussions.
Notes and references
- J. B. Y. H. Behrendorff, W. L. Huang and E. M. J. Gillam, Biochem. J., 2015, 467, 1–15 CrossRef CAS PubMed.
- J. M. Caswell, M. O'Neill, S. J. C. Taylor and T. S. Moody, Curr. Opin. Chem. Biol., 2013, 17, 271–275 CrossRef CAS PubMed.
- V. B. Urlacher and M. Girhard, Trends Biotechnol., 2012, 30, 26–36 CrossRef CAS PubMed.
- J. B. van Beilen and E. G. Funhoff, Curr. Opin. Biotechnol., 2005, 16, 308–314 CrossRef CAS PubMed.
- D. Scheps, S. Honda Malca, H. Hoffmann, B. M. Nestl and B. Hauer, Org. Biomol. Chem., 2011, 9, 6727–6733 CAS.
- H. Schewe, B. A. Kaup and J. Schrader, Appl. Microbiol. Biotechnol., 2008, 78, 55–65 CrossRef CAS PubMed.
- A. K. Holzer, K. Hiebler, F. G. Mutti, R. C. Simon, L. Lauterbach, O. Lenz and W. Kroutil, Org. Lett., 2015, 17, 2431–2433 CrossRef CAS PubMed.
- T. Oda, K. Oda, H. Yamamoto, A. Matsuyama, M. Ishii, Y. Igarashi and H. Nishihara, Microb. Cell Fact., 2013, 12, 2 CrossRef CAS PubMed.
- F. Rundbäck, M. Fidanoska and P. Adlercreutz, J. Biotechnol., 2012, 157, 154–158 CrossRef PubMed.
- H. A. Reeve, L. Lauterbach, P. A. Ash, O. Lenz and K. A. Vincent, Chem. Commun., 2012, 48, 1589–1591 RSC.
- L. Lauterbach, O. Lenz and K. A. Vincent, FEBS J., 2013, 280, 3058–3068 CrossRef CAS PubMed.
- W. Lubitz, H. Ogata, O. Rüdiger and E. Reijerse, Chem. Rev., 2014, 114, 4081–4148 CrossRef CAS PubMed.
- L. Lauterbach, Z. Idris, K. A. Vincent and O. Lenz, PLoS One, 2011, 6, 1–9 Search PubMed.
- L. Lauterbach and O. Lenz, J. Am. Chem. Soc., 2013, 135, 17897–17905 CrossRef CAS PubMed.
- L. M. Blank, G. Ionidis, B. E. Ebert, B. Bühler and A. Schmid, FEBS J., 2008, 275, 5173–5190 CrossRef CAS PubMed.
- J. Schiffels and T. Selmer, Biotechnol. Bioeng., 2015 DOI:10.1002/bit.25658.
- J. B. van Beilen, S. Panke, S. Lucchini, A. G. Franchini, M. Rothlisberger and B. Witholt, Microbiology, 2001, 147, 1621–1630 CrossRef CAS PubMed.
- H. A. Vrionis, A. J. Daugulis and A. M. Kropinski, Appl. Microbiol. Biotechnol., 2002, 58, 469–475 CrossRef CAS PubMed.
- T. Vallon, M. Glemser, S. H. Malca, D. Scheps, J. Schmid, M. Siemann-Herzberg, B. Hauer and R. Takors, Chem. Eng. Technol., 2013, 85, 841–848 CAS.
- A. Schmid, A. Kollmer, B. Sonnleitner and B. Witholt, Bioprocess Eng., 1999, 20, 91–100 CrossRef CAS.
- Z. M. Shapiro and T. R. Moffette, US. Department of Energy, 1957, OSTI ID:4327402 Search PubMed.
- M. M. Bradford, Anal. Biochem., 1976, 72, 248–254 CrossRef CAS.
- W. A. Johnston, W. Huang, J. J. De Voss, M. A. Hayes and E. M. Gillam, J. Biomol. Screening, 2008, 13, 135–141 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Construction of SH and CYP153A production systems, GC analysis, CYP153A and SH production in recombinant strains and catabolism assays. See DOI: 10.1039/c5cc06078h |
| ‡ These authors contributed equally to this work. |
| § The heterologous overproduction system for the SH from R. eutropha and the CYP153A system from Polaromonas sp. JS666 was assembled as described in the ESI.† Main cultures were grown at 30 °C in baffled 2 L-Erlenmeyer flasks including 300 mL of mineral salts medium14 containing 0.2% (w/v) glucose and 0.2% (v/v) glycerol as carbon and energy sources (GGN medium). Inoculation was done with 3 mL of a starter culture grown previously for 48 h in mineral salts medium with 0.4% (w/v) glucose. Induction of the alkB promoter was carried out by addition of 0.05% (v/v) dicyclopropyl ketone, and CYP production was enhanced through addition of 0.5 mM 5-aminolevulinic acid and 1 mM FeCl2.5 H2 oxidation activity in soluble extracts was measured spectrophotometrically at 30 °C by following the conversion of NAD+ into NADH at 365 nm.14 Protein concentration was determined using the Bradford assay.22 The CYP153A concentration was measured in whole cells using the method of Johnston et al.23 Biotransformations were performed in a final volume of 2.2 mL in 110 mL gas-tight, thick-walled glass flasks. Cells obtained from the main cultures were harvested and re-suspended to a final OD436 of 20 in carbon-free, five times concentrated, mineral salts medium.14 After addition of 15% (v/v) n-octane and 2% (v/v) DMSO the flasks were sealed with a rubber bung. The headspace of the flasks was filled with a gas mixture of 20% H2 in air. Control experiments were carried out under air. Aliquots of the biotransformation suspensions were analyzed using GC-FID, see ESI,† for details. |
| This journal is © The Royal Society of Chemistry 2015 |