Mechanism of electrophilic fluorination with Pd(IV): fluoride capture and subsequent oxidative fluoride transfer†‡
Jochen R.
Brandt
,
Eunsung
Lee
,
Gregory B.
Boursalian
and
Tobias
Ritter
*
Department of Chemistry and Chemical Biology, Harvard University, 12 Oxford Street, Cambridge, Massachusetts 02138, USA. E-mail: ritter@chemistry.harvard.edu
First published on 31st October 2013
Abstract
Electrophilic fluorinating reagents derived from fluoride are desirable for the synthesis of 18F-labeled molecules for positron emission tomography (PET). Here, we study the mechanism by which a Pd(IV)–complex captures fluoride and subsequently transfers it to nucleophiles. The intermediate Pd(IV)–F is formed with high rates even at the nano- to micromolar fluoride concentrations typical for radiosyntheses with 18F due to fast formation of an outer-sphere complex between fluoride and Pd(IV). The subsequent fluorine transfer from the Pd(IV)–F complex is proposed to proceed through an unusual SET/fluoride transfer/SET mechanism. The findings detailed in this manuscript provide a theoretical foundation suitable for addressing a more general approach for electrophilic fluorination with high specific activity 18F PET imaging.
Introduction
Nucleophilic fluorination reactions using [18F]fluoride are currently the method of choice for the synthesis of 18F-labeled molecules for positron emission tomography (PET).1 Electrophilic fluorinating reagents (“F+”) would complement nucleophilic fluorinations,2 but their use in radiofluorinations is currently limited due to technical constraints. Almost all F+ reagents are derived from fluorine gas (Fig. 1), and [18F]F2 is more difficult to make and handle than [18F]fluoride.1e Moreover, [18F]fluoride is generally available in higher isotopic enrichment, referred to as specific activity (SA), than [18F]F2. High specific activity is typically a requirement for PET imaging of molecular targets that are of low endogenous abundance. Tracers of low specific activity contain a larger fraction of the non-PET-active 19F isotopologue, which can saturate the molecular target.1d | ||
| Fig. 1 Synthesis of electrophilic fluorinating reagent 1 from fluoride.3a | ||
In 2011, our group reported a conceptually distinct approach for electrophilic fluorination with applications in PET.3 Pd(IV) complex 1 is the first reagent that can be synthesized from fluoride and, subsequently, transfers its entire fluorine content in an electrophilic fluorination reaction suitable for late-stage 18F fluorination.4 Here, we describe an experimentally substantiated proposal regarding the unusual mechanism by which electrophilic fluorination proceeds and outline the scope and limitations of complex 1 as electrophilic fluorinating reagent for fluorination of several nucleophiles. Mechanistic investigation suggests that efficient fluoride capture, even for the low concentration typical for 18F chemistry (nM), is made possible by pre-organization of fluoride and bis-cationic Pd(IV) 2 in an outer-sphere complex. Subsequent electrophilic fluorination is proposed to proceed by a single electron transfer (SET), fluoride ion transfer from a Pd(III)–F intermediate, followed by a second SET. We anticipate that knowledge about how 1 operates can serve as guiding principle to expand the development of high-specific activity PET tracer synthesis by electrophilic fluorination.
Conventional radiofluorination is generally accomplished by well-established nucleophilic substitution reactions with [18F]fluoride as nucleophile.1 Modern fluorination reactions, largely developed over the past few years, hold significant promise for PET tracer synthesis. Some transition metal-catalysed or -mediated nucleophilic fluorination reactions have employed [18F]fluoride directly,5 and the adoption of other modern [19F] fluorination reactions to radiochemistry could dramatically expand the scope of accessible PET tracers.6 Most prominently, the palladium-catalyzed fluorination of aryl triflates7 developed by the Buchwald group falls into this category, and other nucleophilic aromatic fluorinations mediated by copper8 hold promise as well. However, translation from 19F to 18F is challenging and significantly more complicated and unpredictable than would be anticipated from the synthesis of a mere isotopologue. Concentration and reaction conditions, most notably stoichiometry, dramatically change as a consequence of the small molar amount of fluoride (nmol) used in radiosyntheses. An excellent 19F fluorination reaction may or may not be suitable for 18F chemistry.
Advances in electrophilic 18F fluorination beyond those with reagents such as fluorine gas9 and acetyl hypofluorite10 have been accomplished by Gouverneur through the synthesis of useful and versatile modern reagents such as [18F]NFBS11 and [18F]Selectfluor.12 The group of Solin has developed a process that can afford [18F]F2 in significantly higher specific activity than was achievable before,13 which allows [18F]Selectfluor-mediated fluorinations to proceed in up to 0.6 Ci μmol−1.12a Nevertheless, the use of [18F]fluoride is generally preferred over [18F]F2, for reasons of simplicity and practicality.1a,1e,6a
Pd(IV)–fluoride reagent 1 can be synthesized in four steps and 84% overall yield from palladium acetate and has been routinely prepared in batches larger than 5 g. The multi-dentate ligands of complex 1 reduce the rate of reductive elimination from 1 and potential nucleophilic attack at Pd(IV):3a Pd(IV)fluoride 1 can exist for hours at 100 °C or in the presence of water, which is beneficial for [18F] chemistry because 18F is made from [18O]water.1 No decomposition of complex 1 is observed over 3 h in 10% aqueous acetonitrile, and the synthesis of alkyl fluorides with Pd(IV)–F 1 can be performed in the presence of water (0.2 M in DMF). For imaging studies, radiofluorinations mediated by [18F]–1 have been translated to automated syntheses which were carried out in less than 100 min to afford >10 mCi of purified and formulated 18F–radiolabeled aryl fluorides at up to 13 Ci μmol−1 specific activity.3b
Late-stage [18F]fluorination via fluoride capture to afford 1, followed by electrophilic fluorination by 1 provides a new opportunity to address PET tracer synthesis. To better understand the principles of fluorinations with 1 and aid the development of new, similarly functioning reagents, we aimed to answer two questions in this manuscript: first, how can the formation of Pd(IV)–[18F] proceed in less than ten minutes despite the nano- to micromolar concentration of fluoride encountered in radiochemistry? Second, how does the fluorine transfer from the Pd(IV)–F 1 to the nucleophile occur? Our results suggest that the fluoride capture is facilitated by the formation of an outer-sphere complex in a pre-equilibrium, and that fluorination is initiated by an SET and involves transfer of fluoride ion. Finally, we demonstrate an extended reaction scope for 1.
Results
The fluorination from fluoride mediated by 2 is a two-step sequence: (1) substitution of the picoline ligand of Pd(IV)–complex 2 with fluoride to form Pd(IV)–F 1 (fluoride capture) and (2) electrophilic fluorination of a nucleophile with Pd(IV)–F 1. The mechanism of fluoride capture by Pd(IV)–picoline complex 2 was investigated by measuring the rates of ligand exchanges and the activation parameters of fluoride capture. Moreover, an intermediate observed in the formation of Pd(IV)–F 1 was characterized by 1H NMR spectroscopy. For the subsequent electrophilic fluorine transfer, the viability of nucleophilic attack on 1 was compared to SET reduction of 1. Additionally, it was investigated whether the fluoride substituent is transferred to the nucleophile as anionic fluoride or as a radical fluorine atom by using a model substrate exhibiting charge–spin separation upon oxidation.Pyridine dissociation from Pd(IV)
The competence of dissociative pathways in the formation of Pd(IV)–F 1 was investigated first. To assess the rate of pyridine dissociation from Pd(IV)–pyridine complex 3, a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixture of 3 and pyridine-d5 in acetonitrile-d3 was monitored by 1H NMR spectroscopy (Fig. 2B). The formation of free pyridine proceeded at a rate of 3.3 × 10−5 M s−1. The rate determined for the degenerate pyridine exchange represents an upper limit for the rate of picoline dissociation from 2. Due to its methyl substituent, 4-picoline is a better Lewis base than pyridine and should dissociate more slowly from Pd(IV).
:1 mixture of 3 and pyridine-d5 in acetonitrile-d3 was monitored by 1H NMR spectroscopy (Fig. 2B). The formation of free pyridine proceeded at a rate of 3.3 × 10−5 M s−1. The rate determined for the degenerate pyridine exchange represents an upper limit for the rate of picoline dissociation from 2. Due to its methyl substituent, 4-picoline is a better Lewis base than pyridine and should dissociate more slowly from Pd(IV).
 | ||
| Fig. 2 Comparison of rates for (A) Pd(IV)–F 1 formation (fluoride capture), (B) degenerate pyridine exchange and (C) intramolecular pyrazole exchange. The rates are relative to the calculated rate of fluoride capture (A) at 26 (B) and 68 °C (C), respectively. | ||
Intramolecular pyrazole exchange
The rate of pyrazole ligand dissociation was investigated. Only sharp signals were observed in the 1H NMR spectrum of Pd(IV)–complex 3 up to 68 °C. A 1H NMR magnetization transfer experiment14 showed an exchange between the three bound pyrazole ligands, but not with the unbound pyrazole. The rate constant for the pyrazole exchange at 68 °C was determined as 2.0 × 10−1 s−1.Fluoride capture by Pd(IV)–picoline 2
The formation of Pd(IV)–F 1 from Pd(IV)–picoline complex 2 and anhydrous TMAF was studied by 1H NMR spectroscopy in a 5:3 mixture of CD2Cl2 and acetone-d6 below −50 °C. Reactions performed in pure CD2Cl2 led to the formation of several different Pd(IV)–complexes. Additionally, pure CD3CN or acetone-d6 solutions could not be employed because anhydrous fluoride is highly basic and deprotonates the solvent at ambient temperature. Therefore, an acetone-d6 solution of 2 and a CD2Cl2 solution of TMAF were mixed at −80 °C (see ESI‡ for details). At this temperature, solvent deprotonation was negligible and no Pd(IV)–F 1 was formed over the course of several hours. However, an immediate chemical shift change (Δδ) of approximately 0.10 ppm was observed for two peaks in the 1H NMR (Fig. 3). The hydrogen atoms corresponding to the shifted peaks were identified by 1H-COSY and nOe measurements as those of the 2-position of benzo[h]quinoline and the 3-position of the pyrazole ring cis to the heterocyclic moiety of benzo[h]quinoline (Fig. 3A). A 19F NMR signal for fluoride was not observed.
 | ||
| Fig. 3 (A) 1H NMR spectra of a 2/TMAF mixture with constant total concentration [2] + [TMAF] at −80 °C and decreasing χ(Pd(IV)–picoline 2). (B) Job plot for a 2 + TBACl mixture at −80 °C in acetone-d6: product of palladium mole fraction and normalized chemical shift difference Δδnorm = Δδobs/Δδmax is plotted over the mole fraction of chloride. | ||
Similar chemical shift changes of up to 0.20 ppm were observed for the benzo[h]quinoline and pyrazole ligands when TBACl instead of TMAF was added to acetone-d6 solutions of 1. The higher solubility of TBACl compared to TMAF facilitated further 1H NMR studies and a Job plot of Δδ(H) in the presence of TBACl afforded a maximum at χ(TBACl) = 0.5, which is consistent with a 1:1 and 2:2 binding stoichiometry of the chloride anions and complex 1. The addition of silver triflate to a mixture of Pd(IV)–picoline 2 and TBACl at −80 °C restored the chemical shifts to those observed in the absence of TBACl.
The measurement of the activation parameters of the Pd(IV)–F formation (Fig. 4) by 1H NMR spectroscopy was complicated by the low solubility of anhydrous fluoride at low temperatures. Due to low fluoride concentration, up to 14 scans were required for reliable peak integration and the initial rates of the fast fluoride anation could not be measured accurately. This problem was overcome by using an oxygen atmosphere because the presence of the paramagnetic gas lowered15 the T1 of the diagnostic peaks from around three seconds to less than one second and allowed the collection of initial rates for reactions performed between −55 °C and −35 °C. The activation parameters for the formation of 1 are ΔH‡ = 22.8 ± 0.9 kcal mol−1, ΔS‡ = 36 ± 4 e.u. and ΔG‡298 = 12.0 ± 0.2 kcal mol−1.
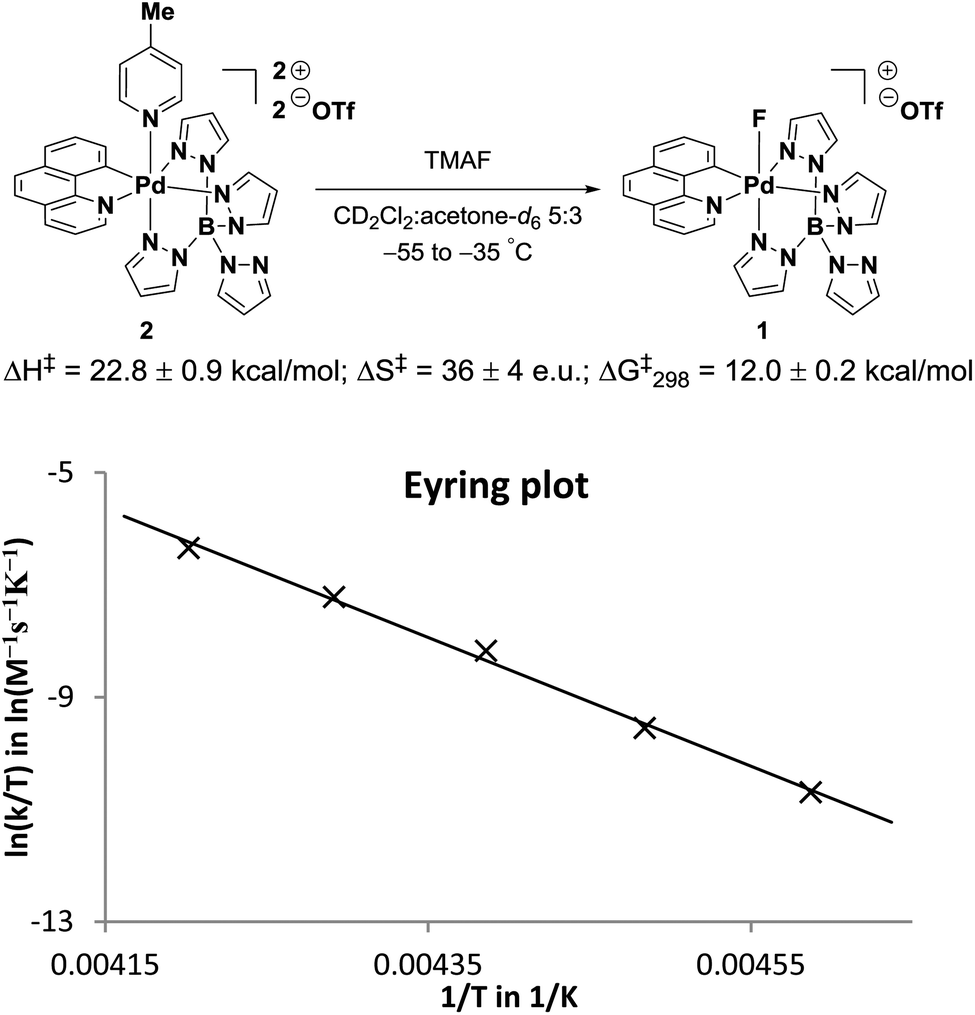 | ||
| Fig. 4 Activation parameters for the formation of Pd(IV)–F 1. | ||
Reduction of Pd(IV)–F 1
To investigate the mechanism of fluorine transfer, 1 was treated with one-electron reductants with reduction potentials between 0.45 and 1.37 V vs. SCE. More strongly reducing reductants, i.e. those that possess a lower E0′, correlated with higher reduction rates of 1 (Fig. 5). A sharp drop in the reduction rate was observed with reductants of E0′ around 1.1 V vs. SCE and no reduction was observed when E0′ ≥ 1.26 V. This strong decrease in reactivity correlates well with the measured peak potential of Pd(IV)–F 1 (Ep = 1.12 V, vide infra). | ||
| Fig. 5 Correlation of Pd(IV)–F 1 reduction rate with reagents' E0′ and nucleophilicity. avs. SCE.16 The dotted line at 1.12 V corresponds to the E0′ of Pd(IV)–F vs. SCE. b Monitored by 1H NMR spectroscopy using THF as internal standard. c Mayr nucleophilicity parameter N.17 | ||
We did not observe a positive correlation of the rate of reduction of 1 with the nucleophilicity of the nucleophile. The influence of nucleophilicity (Mayr nucleophilicity parameter N)18 was investigated using the structurally related bicyclic, tertiary amines DABCO and quinuclidine (Fig. 5). DABCO (weaker nucleophile) reduced 1 quantitatively in less than ten minutes at 23 °C, while quinuclidine (better nucleophile) led to only 58% conversion after three hours. This reactivity trend does not positively correlate with the amines' nucleophilicity: based on the logarithmic Mayr nucleophilicity parameter N, quinuclidine (N = 20.5) is a more reactive nucleophile than DABCO (18.8).17 In contrast, the reduction rate correlates well with the stronger reducing ability of DABCO (E0′ = 0.6 V) compared to quinuclidine (1.1 V).16a
A differential pulse voltammetry (DPV) experiment of Pd(IV)–F 1 showed a peak potential (Ep) of 1.12 V vs. SCE, which is well in line with the ability to oxidize quinuclidine16a (E0′ = 1.1 V) and N(4-BrC6H4)3 (E0′ = 1.16 V) but not thianthrene (E0′ = 1.26 V).16b The oxidation of Pd(II)–complex 8a (Scheme 2, Ar = 3-BnO-C6H4) displayed Ep = 1.07 V in the DPV, 50 mV lower than the Ep value of Pd(IV)–F 1. For both palladium complexes, irreversible redox behavior prevented the measurement of meaningful cyclic voltammetry (CV) graphs.
Fluorine transfer after SET
After initial single electron reduction of cationic Pd(IV)–F 1, the generated neutral Pd(III)–F intermediate could transfer the fluoride substituent to the nucleophile through either ionic or radical pathways. To differentiate between these two mechanisms, we chose E-stilbene 4 (Table 1) as a mechanistic probe. SET from 4 to 1 would generate a charge–spin separated radical cation19 that should afford different constitutional isomers for ionic or radical fluorine transfer (vide infra). To allow the isolation and characterization of the constitutional isomers, the reaction was performed in the presence of 4-methoxypyridine, which intercepted the carbocation intermediate formed after fluorine transfer.20 Subsequent demethylation of the pyridinium moiety with lithium bromide provided the two constitutional isomers 5a and 5b as a 7:1 mixture. When Selectfluor was used as electrophilic fluorinating reagent under otherwise identical conditions, the selectivity was inverted and a 1:>20 mixture of 5a and 5b was obtained.
|
|
||
|---|---|---|
| Reagent | Yield [%] 5a | Yield [%] 5b |
| 1 | 46 | 6 |
| Selectfluor | <2 | 55 |

Scope and limitations of electrophilic fluorinations with 1
The original reports on Pd(IV)–F 1 only describe the electrophilic fluorination of Pd(II)–aryl complexes.3 To evaluate the scope of the fluorination with 1, we investigated substrate classes that had previously been successfully fluorinated with fluorine gas-derived reagents such as F-TEDA. We first turned our attention to the reaction of arylsilver compounds21 with 1 because of its potentially high functional group tolerance.21c Purified mesitylsilver reacted with Pd(IV)–F 1 in the presence of bathophenanthroline,22 to afford mesitylfluoride (7a) in 91% yield (Table 2). The ortho-substituted substrates 2-fluorobiphenyl (7b) and naphthalene derivative 7c were obtained in >99% and 60% yield, respectively. The yield for arylsilver substrates that lack ortho substitution is considerably lower; for example, para-substituted 4-biphenylfluoride (7d) was obtained in 41%. In the absence of bathophen, only 22% yield of 7a were obtained. The reaction of Pd(IV)–F 1 with more basic aryl metal reagents20b,23 was not successful: neither mesitylmagnesium bromide (6e) nor phenylzinc bromide (6f) led to detectible arylfluoride formation. Fluorination with 1 is not limited to aryl fluorides; alkyl fluorides can be obtained from activated double bonds. When treated with Pd(IV)–F 1, the enamine derived from pyrrolidine and 1-tetralone (6g) was fluorinated in 85% yield. TMS-enol ether 6h afforded the desired α-fluoro carbonyl compound 7g in 82%. In addition to the enamine and silyl enol ethers, activated alkenes also reacted with Pd(IV)–F 1. 2-Phenylindene (6i) afforded the allyl fluoride 7i in 60% yield, presumably via deprotonation of the benzyl cation intermediate formed after electrophilic fluorination.
Discussion
Fluoride capture
For the displacement of picoline from Pd(IV)–complex 2 by fluoride (anation), we evaluated five potential reaction mechanisms (Fig. 6). Due to the high rate of ligand exchange observed even at low temperatures, high-energy pathways such as dissociation of the carbon ligand of benzo[h]quinoline or the dissociation of two or more ligands were not included in the list.24 In a dissociative (D-type) mechanism, the dissociation of a ligand occurs before an interaction between the resulting complex and the new ligand exists; thus, the dissociation rate is independent of the nature of the entering group. Therefore, the degenerate exchange of pyridine at palladium complex 3 should occur at a rate that is no smaller than fluoride or chloride anation. In contrast, the rate observed for the formation of Pd(IV)–F 1 is more than five orders of magnitude larger than pyridine exchange. The large rate differences cannot reasonably be explained by possible dissociation rate differences between the pyridine and picoline ligands of complexes 3 and 2. Although the rates of bimolecular (fluoride capture) and monomolecular reactions (pyridine dissociation) cannot rigorously be compared quantitatively, the differences of more than five orders of magnitude suggest that mechanism 1 (Fig. 6) does not operate. | ||
| Fig. 6 Potential mechanism pathways for the formation of Pd(IV)–F 1 from Pd(IV)–picoline 2. aLangford–Gray nomenclature; D: dissociative process with identifiable intermediate of lower coordination number. A: associative process with identifiable intermediate of higher coordination number. Id: dissociatively activated interchange mechanism from a pre-formed aggregate; bond breaking of the leaving group is more advanced than bond formation to the entering group.26 pic: 4-picoline. | ||
Dissociation of one of the three bound pyrazole ligands is not kinetically competitive as a pathway in fluoride capture either. No peak broadening due to fast intramolecular pyrazole exchange is observed in the 1H NMR spectrum of 3. The transfer magnetization by the inversion recovery 1H NMR experiment showed a pyrazole exchange that was more than five orders of magnitude slower than the rate calculated for fluoride capture at the same temperature (Fig. 2C). Additionally, dissociation of pyrazole ligands is likely even slower than the measured pyrazole exchange. Dissociation of a pyrazole ligand would lead to exchange of all four pyrazoles through a “tumbling” process.25 However, exchange was only observed for bound pyrazole ligands, which suggests a twist of the pyrazole ligands around the Pd–B axis. This ligand twist has been reported for other transition metal complexes ligated by κ3-coordinated tetra(pyrazole)borates.25 Similarly, a dissociation of the benzo[h]quinolyl ligand (pathway 4) can be disregarded due to the chelating nature of the benzo[h]quinolyl ligand and the data obtained for the rate of dissociation of the picoline ligand.
The formation of Pd(IV)–F 1 through an associative mechanism (Fig. 6, pathway 2) is unlikely considering the large, positive value determined for ΔS‡ (36 ± 4 e.u.).26c,27 Moreover, heptacoordinate complexes of late transition metals of the second and third row are rare.28 Based on the activation parameters and the rarity of heptacoordinate palladium complexes, A-type mechanism 2 can be disregarded.
Of the five considered pathways, the Eigen–Wilkins-type interchange mechanism26b,c,27c,29 5 is consistent with all experimental data: equilibrium formation of an outer-sphere complex [2·F], followed by dissociative interchange substitution of picoline. Based on the large positive value of the entropy of activation ΔS‡ = 36 ± 4 e.u., associative interchange pathways (Ia), in which new bond formation is more advanced than bond breaking in the transition state, were disregarded. In the dissociative interchange mechanism (Id, path 5) bond breaking of the leaving group is more advanced than bond formation to fluoride entering from the outer-sphere complex [2·F]. Evidence for the formation of an outer-sphere complex between the Pd(IV)–picoline complex and the halide ions was obtained by 1H NMR spectroscopy at −80 °C. The addition of halide ions to Pd(IV)–picoline complex 2 caused a change in chemical shift of 0.1 (fluoride) or 0.2 ppm (chloride) of hydrogen atoms of the benzo[h]quinoline and pyrazole ligands (Fig. 3). In contrast, a chemical shift change of only <0.01–0.03 ppm (fluoride) or <0.01–0.05 ppm (chloride) was observed for the remaining peaks, implying that the shift change was not caused by a general ionic strength effect or by an exchange or structural reorganization of ligands. Similar outer-sphere complexes exhibiting halide binding to heterocyclic ligands of octahedral Pd(IV) complexes have been reported previously.30
The 1H NMR studies also established that the formation of the outer-sphere complexes [2·Hal] is fast and reversible. Firstly, the equilibrium was shifted to the starting complex 2 by the addition of silver triflate to the 2/TBACl mixture. Secondly, in the presence of substoichiometric amounts of halide, each hydrogen substituent of the palladium complex displayed only one sharp signal in the 1H NMR spectrum. Therefore, even at −80 °C the forward and backward rates of the equilibrium in Fig. 3A are too fast on the NMR time scale to allow the bound and unbound form to be resolved into two separate peaks. It is not clear, why the fluoride ion is not observed by 19F NMR spectroscopy. However, the chemical shift of TMAF is strongly dependent on the chemical environment and can change by over 60 ppm, depending on the solvent.31 It is possible that uncoordinated fluoride and [2·F] possess such large chemical shift differences that coalescing peaks could be too broad to be detectible by 19F NMR. In our experience, the absence of fluoride signals in 19F NMR spectra is not uncommon.
The participation of an outer-sphere complex accelerates the formation of 1 in two ways. Firstly, the intermediacy of complex [2·F] means that the rate of ligand exchange will be higher the larger the concentration of [2·F] is.26c,27c Thus, the strong electrostatic interaction32 between the doubly cationic palladium complex 2 and the anionic fluoride ion drives the formation of outer-sphere complex [2·F] and therefore leads to high rates of formation of 1. Secondly, the pre-organization of the two reagents into the outer-sphere complex [2·F] and the subsequent dissociation of picoline contribute to the large, positive entropy of activation of ΔS‡ = 36 ± 4 e.u. The high reaction rates for the formation of Pd(IV)–F 1 at the nano- to micromolar fluoride concentrations typical for radiosyntheses can therefore be largely attributed to the preorganization of the oppositely charged fluoride ion and Pd(IV)–complex 2. A rate law for the formation of Pd(IV)–F 1 was not established due to low solubility of fluoride at temperatures at which meaningful rate data could be measured. The obtained data suggest that the reaction is first order in fluoride (see ESI‡ for details), but a fractional reaction order or saturation kinetics could not be ruled out. The reaction order for Pd(IV)–picoline complex 2 was not determined because it is not relevant for radiochemical fluorinations. The approximately 104-fold excess of 2 to fluoride in radiofluorinations3 leads to a negligible change in [2] and therefore to pseudo-first-order reactions.
Fluorine transfer
The transfer of fluorine from Pd(IV)–F 1 to nucleophiles could proceed by at least three distinct mechanisms: (1) SN2 at fluoride33 (2) SET followed by fluorine atom transfer34 or (3) SET, fluoride transfer and a second SET (Fig. 7). A mechanism involving two consecutive SET processes was disregarded because it would generate a 1,2-dication as an intermediate in the fluorination of activated double bonds. Nucleophilic pathway (1) is unlikely to occur, as the reduction rates of Pd(IV)–F 1 did not correlate with the nucleophilicity parameter N of quinuclidine and DABCO (Fig. 5). Additional data on the influence of nucleophilicity was difficult to obtain because we were unable to identify other pairs of structurally related compounds with appropriate reduction potentials and opposite trends in nucleophilicity and E0′. In contrast to the nucleophilicity, the reduction rate of 1 strongly correlated with the formal potential of the reductants and a sharp drop in reactivity was observed close to the measured Ep of 1. Moreover, Pd(IV)–F 1 is a competent oxidant of Pd(II)–aryl complex 8a, with ΔEp(1–8a) = 50 mV established through DPV measurements. This kinetic and thermodynamic data suggests that the electrophilic fluorination from 1 most likely proceeds via an SET pathway like (path 2) or (path 3).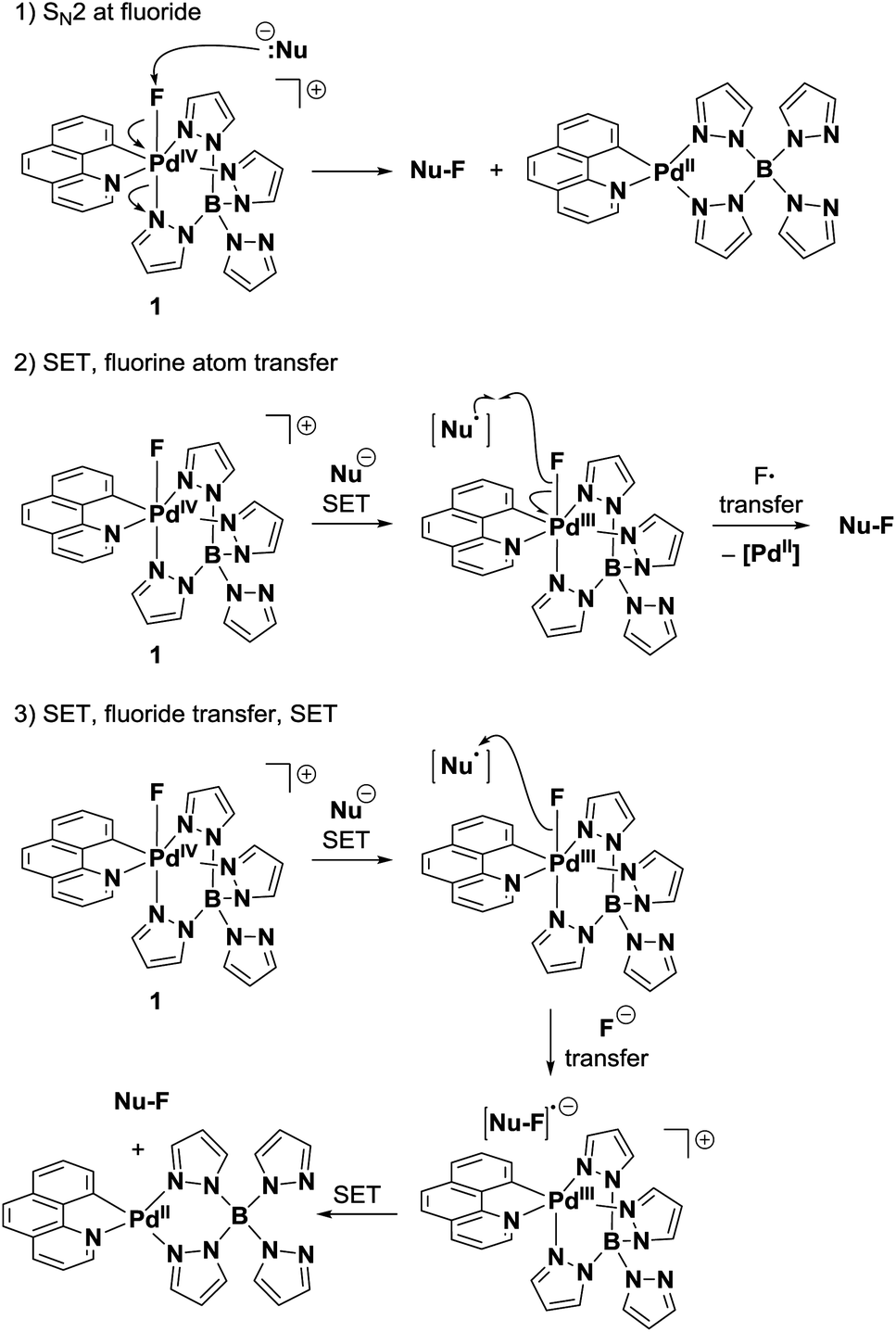 | ||
| Fig. 7 Potential pathways of fluorine transfer. | ||
A distinction between a radical (path 2) or nucleophilic (path 3) transfer of the fluoride substituent is challenging. Electrophilic fluorination of palladium complexes 8 or activated double bonds 6g–ivia pathways (2) or (3) will afford the same products.35 Therefore, we aimed to employ a substrate that would afford different constitutional isomers for the ionic or radical fluorine transfer. E-stilbene 4 (Scheme 1) was chosen as mechanistic probe; it can be assumed to undergo electrophilic fluorination through an SET mechanism, as had been demonstrated for other aryl-substituted olefins.36 Importantly, single electron oxidation of 4 by 1 generates charge–spin separated radical cation I.19 Due to the localized positive charge, ionic fluoride species should transfer preferentially to afford α-fluoro radical IIa. A second SET, pyridine quench and demethylation would then yield constitutional isomer 5a. In contrast, fluorine radical transfer to radical cation I should lead to the formation of 5bvia cation IIb. In the reaction of Pd(IV)–F 1, the observed 7:1 ratio of 5a:5b suggests transfer of a fluoride ion from a Pd(III)–F intermediate to I, implying nucleophilic path (3).
 | ||
| Scheme 1 Distinction between ionic and radical pathways with charge–spin separated radical cation I. | ||
The opposite selectivity (5a:5b 1:>20) was found when Selectfluor was used as electrophilic fluorinating reagent, representing direct experimental support for an SET/fluorine atom transfer sequence from Selectfluor.34 Previous work had shown that Selectfluor can be reduced by one electron, but no data concerning a subsequent fluorine radical transfer was reported.36,37 A charge transfer/fluorine transfer mechanism had been found in computational studies investigating the formation of different constitutional isomers in transannular additions of Selectfluor and NBS.38 Interestingly, the treatment of a radical clock with Selectfluor in the absence of transition metals did not provide evidence for radical intermediates,37a presumably because the fluorine transfer after SET is faster than radical cyclization.34,39 Although it cannot be ruled out, the formation of a cyclic, three-membered fluoronium intermediate is unlikely. Compound 5a should not be formed via a fluoronium intermediate because the nucleophilic attack of 4-methoxypyridine is favored to occur on the position of the more stabilized cation (IIb) to afford 5b. Additionally, bridged fluoronium species are extremely rare in solution phase40 and their formation from an alkyl α-fluoro carbocation has been excluded based on experimental and computational data.41 Moreover, the benzylic stabilization of the cationic intermediates (IIb and the cation derived from IIa) would further decrease the importance of halonium bridging compared to the previously studied alkyl substituents.42
Based on the selectivity observed in the fluorination of stilbene 4, we conclude that the fluorination of Pd(IV)–aryl complexes by an SET/fluoride transfer/SET mechanism is the most likely pathway (Scheme 2), although we cannot exclude the possibility of slight variations of this mechanism depending on the nucleophile. An initial electron transfer from Pd(IV)–aryl complex 8a to Pd(IV)–F 1 generates the two Pd(III) complexes III and IV. Subsequent transfer of fluoride and a second SET affords the reduced benzo[h]quinolinyl (tetrapyrazolylborate)palladium(II) complex and the Pd(IV)–F complex 9, from which reductive elimination to the desired arylfluoride occurs.3a,24,43 To the best of our knowledge, such a SET/fluoride transfer/SET sequence has not been previously proposed for an electrophilic fluorination.
 | ||
| Scheme 2 Proposed mechanism for the fluorine transfer from Pd(IV)–F 1 to Pd(IV)–aryl 8. | ||
Scope of the electrophilic fluorination with Pd(IV)–F 1
The electrophilic fluorination with 1 is not limited to the initially reported Pd(II)–aryl complexes.3 Sterically hindered arylsilver compounds could be fluorinated in 60% to 99% yield (Table 2, 7a–c), but the yield dropped to 41% in the absence of ortho-substitution (7d). This reactivity trend is complementary to that observed with Pd(IV)–aryl complexes, for which the fluorination of ortho-substituted substrates had been challenging. It is unclear, why the fluorination of sterically crowded arylsilver compounds is higher yielding; possible explanations include the higher solubility and thermal stability of sterically hindered arylsilver compounds.44The fluorination of more basic organometallic reagents was unsuccessful. The lack of carbon–fluorine bond formation in the reaction of aryl-magnesium (6e), or -zinc (6f) reagents with Pd(IV)–F 1 can be rationalized by the proposed SET/fluoride transfer mechanism. The radical cations formed from 6e and 6f after the initial SET should be more strongly reducing and less electrophilic than the radical cations obtained from arylsilver compounds or Pd(IV)–aryl complexes. Thus, a one electron reduction of putative intermediate IV may occur faster than fluoride transfer from IV to the radical cation. One might envision that related but less oxidizing high-valent transition metal fluorides would disfavor the second SET and allow fluoride transfer to strongly reducing organometallic reagents.
Activated double bonds (6g–i) were fluorinated with Pd(IV)–F 1 in 60% to 85% yield, affording products consistent with the proposed mechanism.35a,45 Together with the data on the synthesis of aryl fluorides, these results show that the proposed, unusual SET/fluoride transfer/SET mechanism is compatible with different classes of nucleophiles that are weak reductants. Thus, Pd(IV)–F 1 or related reagents could provide access to a wide range of radiolabeled molecules that are currently difficult to obtain through nucleophilic fluorinations.
Summary
The halide capture/electrophilic fluorination sequence from Pd(IV)–picoline complex 2 is facilitated by the complex's positive formal charge and high oxidation state. We propose a mechanism in which an outer-sphere complex is formed between the oppositely charged fluoride ion and palladium complex. Thus, very high anation rates are achieved that allow the formation of Pd(IV)–[18F] complex [18F]–1 under the nano- to micromolar concentrations encountered in radiochemistry. The subsequent fluorine transfer is proposed to proceed by single electron reduction of the Pd(IV)–F 1 followed by fluoride ion transfer and a second SET. The reported fluorinations of activated double bonds and arylsilver compounds with Pd(IV)–F 1 are in line with the proposed mechanism and provide access to alkyl fluorides and sterically hindered aryl fluorides.Conclusions
In the electrophilic fluorination mediated by Pd(IV)–complexes, nucleophilic fluoride is captured and transferred to a range of nucleophiles by an unusual electrophilic fluoride transfer. Pd(IV)–F 1 functions as a combined fluoride shuttle and two electron oxidant. A better mechanistic understanding for electrophilic 18F fluorination with fluoride provides a framework and a new opportunity for the rational development of other electrophilic fluorination reagents to impact 18F PET tracer synthesis in the future.Acknowledgements
Funding was provided by NIH-NIGMS (GM088237) and NIH-NIBIB (EB013042-02). G. B. B. thanks NSF GRFP (DGE0644491) for a graduate fellowship. We thank M. G. Campbell and E. McNeill for helpful discussions and the Kishi group for the use of their preparatory HPLC.Notes and references
- (a) N. S. Mason and C. A. Mathis, in Positron Emission Tomography, ed. D. L. Bailey, D. N. Townsend, P. E. Valk and M. N. Maisey, Springer, London, 2005 Search PubMed; (b) S. M. Ametamey, M. Honer and P. A. Schubiger, Chem. Rev., 2008, 108, 1501–1516 CrossRef CAS PubMed; (c) P. W. Miller, N. J. Long, R. Vilar and A. D. Gee, Angew. Chem., Int. Ed., 2008, 47, 8998–9033 CrossRef CAS PubMed; (d) L. Cai, S. Lu and V. W. Pike, Eur. J. Org. Chem., 2008, 2853–2873 CrossRef CAS; (e) M. Tredwell and V. Gouverneur, Angew. Chem., Int. Ed., 2012, 51, 11426–11437 CrossRef CAS PubMed; (f) S. L. Pimlott and A. Sutherland, Chem. Soc. Rev., 2011, 40, 149–162 RSC.
- (a) T. Liang, C. N. Neumann and T. Ritter, Angew. Chem., Int. Ed., 2013, 52, 8214–8264 CrossRef CAS PubMed; (b) T. Furuya, A. S. Kamlet and T. Ritter, Nature, 2011, 473, 470–477 CrossRef CAS PubMed.
- (a) E. Lee, A. S. Kamlet, D. C. Powers, C. N. Neumann, G. B. Boursalian, T. Furuya, D. C. Choi, J. M. Hooker and T. Ritter, Science, 2011, 334, 639–642 CrossRef CAS PubMed; (b) A. S. Kamlet, C. N. Neumann, E. Lee, S. M. Carlin, C. K. Moseley, N. Stephenson, J. M. Hooker and T. Ritter, PLoS One, 2013, 8, e59187 CAS.
- Other electrophilic fluorinating reagents have been synthesised from fluoride; however, these reagents transfer ≤50% of their fluorine substituents in electrophilic reactions: (a) T. C. Simmons, F. W. Hoffmann, R. B. Beck, H. V. Holler, T. Katz, R. J. Koshar, E. R. Larsen, J. E. Mulvaney, K. E. Paulson, F. E. Rogers, B. Singleton and R. E. Sparks, J. Am. Chem. Soc., 1957, 79, 3429–3432 CrossRef CAS; (b) M. Sawaguchi, S. Ayuba and S. Hara, Synthesis, 2002, 1802–1803 CAS; (c) H. Sun, B. Wang and S. G. DiMagno, Org. Lett., 2008, 10, 4413–4416 CrossRef CAS PubMed.
- (a) J. J. Topczewski, T. J. Tewson and H. M. Nguyen, J. Am. Chem. Soc., 2011, 133, 19318–19321 CrossRef CAS PubMed; (b) C. Hollingworth, A. Hazari, M. N. Hopkinson, M. Tredwell, E. Benedetto, M. Huiban, A. D. Gee, J. M. Brown and V. Gouverneur, Angew. Chem., Int. Ed., 2011, 50, 2613–2617 CrossRef CAS PubMed; (c) E. Lee, J. M. Hooker and T. Ritter, J. Am. Chem. Soc., 2012, 134, 17456–17458 CrossRef CAS PubMed; (d) E. Benedetto, M. Tredwell, C. Hollingworth, T. Khotavivattana, J. M. Brown and V. Gouverneur, Chem. Sci., 2013, 4, 89–96 RSC.
- (a) C. Hollingworth and V. Gouverneur, Chem. Commun., 2012, 48, 2929–2942 RSC; (b) W. Liu, X. Huang, M.-J. Cheng, R. J. Nielsen, W. A. Goddard and J. T. Groves, Science, 2012, 337, 1322–1325 CrossRef CAS PubMed; (c) J. Zhu, G. C. Tsui and M. Lautens, Angew. Chem., Int. Ed., 2012, 51, 12353–12356 CrossRef CAS PubMed; (d) K. B. McMurtrey, J. M. Racowski and M. S. Sanford, Org. Lett., 2012, 14, 4094–4097 CrossRef CAS PubMed; (e) A. M. Lauer and J. Wu, Org. Lett., 2012, 14, 5138–5141 CrossRef CAS PubMed; (f) M.-G. Braun, M. H. Katcher and A. G. Doyle, Chem. Sci., 2013, 4, 1216–1220 RSC; (g) W. Liu and J. T. Groves, Angew. Chem., Int. Ed., 2013, 52, 6024–6027 CrossRef CAS PubMed; (h) Z. Zhang, F. Wang, X. Mu, P. Chen and G. Liu, Angew. Chem., Int. Ed., 2013, 52, 7549–7553 CrossRef CAS PubMed.
- (a) D. A. Watson, M. Su, G. Teverovskiy, Y. Zhang, J. García-Fortanet, T. Kinzel and S. L. Buchwald, Science, 2009, 325, 1661–1664 CrossRef CAS PubMed; (b) T. J. Maimone, P. J. Milner, T. Kinzel, Y. Zhang, M. K. Takase and S. L. Buchwald, J. Am. Chem. Soc., 2011, 133, 18106–18109 CrossRef CAS PubMed; (c) T. Noël, T. J. Maimone and S. L. Buchwald, Angew. Chem., Int. Ed., 2011, 50, 8900–8903 CrossRef PubMed; (d) J. Cardinale, J. Ermert, F. Kügler, A. Helfer, M. R. Brandt and H. H. Coenen, J. Label Compd. Radiopharm, 2012, 55, 450–453 CrossRef CAS.
- (a) M. A. Subramanian and L. E. Manzer, Science, 2002, 297, 1665 CrossRef CAS PubMed; (b) A. Casitas, M. Canta, M. Solà, M. Costas and X. Ribas, J. Am. Chem. Soc., 2011, 133, 19386–19392 CrossRef CAS PubMed; (c) P. S. Fier and J. F. Hartwig, J. Am. Chem. Soc., 2012, 134, 10795–10798 CrossRef CAS PubMed; (d) T. Truong, K. Klimovica and O. Daugulis, J. Am. Chem. Soc., 2013, 135, 9342–9345 CrossRef CAS PubMed.
- (a) V. Casella, T. Ido, A. P. Wolf, J. S. Fowler, R. R. MacGregor and T. J. Ruth, J. Nucl. Med., 1980, 21, 750–757 CAS; (b) R. J. Nickles, M. E. Daube and T. J. Ruth, Int. J. Appl. Radiat. Isot., 1984, 35, 117–122 CrossRef CAS; (c) R. Chirakal, R. M. Adams, G. Firnau, G. J. Schrobilgen, G. Coates and E. S. Garnett, Nucl. Med. Biol., 1995, 22, 111–116 CrossRef CAS.
- (a) S. Rozen, O. Lerman and M. Kol, J. Chem. Soc., Chem. Commun., 1981, 443–444 RSC; (b) C.-Y. Shiue, P. A. Salvadori, A. P. Wolf, J. S. Fowler and R. R. MacGregor, J. Nucl. Med., 1982, 23, 899–903 CAS.
- H. Teare, E. G. Robins, E. Årstad, S. K. Luthra and V. Gouverneur, Chem. Commun., 2007, 2330–2332 RSC.
- (a) H. Teare, E. G. Robins, A. Kirjavainen, S. Forsback, G. Sandford, O. Solin, S. K. Luthra and V. Gouverneur, Angew. Chem., Int. Ed., 2010, 49, 6821–6824 CrossRef CAS PubMed; (b) S. Mizuta, I. S. R. Stenhagen, M. O'Duill, J. Wolstenhulme, A. K. Kirjavainen, S. J. Forsback, M. Tredwell, G. Sandford, P. R. Moore, M. Huiban, S. K. Luthra, J. Passchier, O. Solin and V. Gouverneur, Org. Lett., 2013, 15, 2648–2651 CrossRef CAS PubMed; (c) I. S. R. Stenhagen, A. K. Kirjavainen, S. J. Forsback, C. G. Jørgensen, E. G. Robins, S. K. Luthra, O. Solin and V. Gouverneur, Chem. Commun., 2013, 49, 1386–1388 RSC.
- J. Bergman and O. Solin, Nucl. Med. Biol., 1997, 24, 677–683 CrossRef CAS.
- (a) H. M. McConnell, J. Chem. Phys., 1958, 28, 430–431 CrossRef CAS; (b) S. Forsén and R. A. Hoffman, J. Chem. Phys., 1963, 39, 2892–2901 CrossRef; (c) A. D. Bain and J. A. Cramer, J. Magn. Reson., Ser. A, 1996, 118, 21–27 CrossRef CAS; (d) T. J. Williams, A. D. Kershaw, V. Li and X. Wu, J. Chem. Educ., 2011, 88, 665–669 CrossRef CAS PubMed.
- N. Bloembergen, E. M. Purcell and R. V. Pound, Phys. Rev., 1948, 73, 679–712 CrossRef CAS.
- (a) S. F. Nelsen and P. J. Hintz, J. Am. Chem. Soc., 1972, 94, 7114–7117 CrossRef CAS; (b) N. G. Connelly and W. E. Geiger, Chem. Rev., 1996, 96, 877–910 CrossRef CAS PubMed.
- M. Baidya, S. Kobayashi, F. Brotzel, U. Schmidhammer, E. Riedle and H. Mayr, Angew. Chem., Int. Ed., 2007, 46, 6176–6179 CrossRef CAS PubMed.
- (a) H. Mayr and M. Patz, Angew. Chem., Int. Ed. Engl., 1994, 33, 938–957 CrossRef; (b) H. Mayr, B. Kempf and A. R. Ofial, Acc. Chem. Res., 2003, 36, 66–77 CrossRef CAS PubMed.
- (a) S. Tojo, K. Morishima, A. Ishida, T. Majima and S. Takamuku, J. Org. Chem., 1995, 60, 4684–4685 CrossRef CAS; (b) T. Majima, S. Tojo, A. Ishida and S. Takamuku, J. Org. Chem., 1996, 61, 7793–7800 CrossRef CAS PubMed; (c) T. Majima, S. Tojo, A. Ishida and S. Takamuku, J. Phys. Chem., 1996, 100, 13615–13623 CrossRef CAS.
- (a) M. Zupan and A. Pollak, Tetrahedron Lett., 1974, 12, 1015–1018 CrossRef; (b) G. S. Lal, J. Org. Chem., 1993, 58, 2791–2796 CrossRef CAS.
- (a) T. Furuya, A. E. Strom and T. Ritter, J. Am. Chem. Soc., 2009, 131, 1662–1663 CrossRef CAS PubMed; (b) T. Furuya and T. Ritter, Org. Lett., 2009, 11, 2860–2863 CrossRef CAS PubMed; (c) P. Tang, T. Furuya and T. Ritter, J. Am. Chem. Soc., 2010, 132, 12150–12154 CrossRef CAS PubMed; (d) P. Tang and T. Ritter, Tetrahedron, 2011, 67, 4449–4454 CrossRef CAS PubMed.
- (a) Y. Cui and C. He, J. Am. Chem. Soc., 2003, 125, 16202–16203 CrossRef CAS PubMed; (b) Y. Cui and C. He, Angew. Chem., Int. Ed., 2004, 43, 4210–4212 CrossRef CAS PubMed; (c) Z. Li, D. A. Capretto, R. Rahaman and C. He, Angew. Chem., Int. Ed., 2007, 46, 5184–5186 CrossRef CAS PubMed.
- (a) W. E. Barnette, J. Am. Chem. Soc., 1984, 106, 452–454 CrossRef CAS; (b) T. Furuya, H. M. Kaiser and T. Ritter, Angew. Chem., Int. Ed., 2008, 47, 5993–5996 CrossRef CAS PubMed.
- T. Furuya, D. Benitez, E. Tkatchouk, A. E. Strom, P. Tang, W. A. Goddard and T. Ritter, J. Am. Chem. Soc., 2010, 132, 3793–3807 CrossRef CAS PubMed.
- S. Trofimenko, J. Am. Chem. Soc., 1969, 91, 3183–3189 CrossRef CAS.
- (a) C. H. Langford and H. B. Gray, Ligand Substitution Processes, W. A. Benjamin, Inc, New York, 1966 Search PubMed; (b) M. L. Tobe and J. Burgess, Inorganic Reaction Mechanisms, Longman, 1999 Search PubMed; (c) R. B. Jordan, Reaction Mechanisms of Inorganic and Organometallic Systems, Oxford University Press New York, 2007 Search PubMed.
- (a) I. R. Lantzke and D. W. Watts, J. Am. Chem. Soc., 1967, 89, 815–821 CrossRef CAS; (b) M. V. Twigg, Inorg. Chim. Acta, 1977, 24, L84–L86 CrossRef CAS; (c) R. G. Wilkins, Kinetics and Mechanism of Reactions of Transition Metal Complexes, VCH, Weinheim, 1991 CrossRef; (d) L. Dadci, H. Elias, U. Frey, A. Hörnig, U. Koelle, A. E. Merbach, H. Paulus and J. S. Schneider, Inorg. Chem., 1995, 34, 306–315 CrossRef CAS.
- (a) D. Casanova, P. Alemany, J. M. Bofill and S. Alvarez, Chem.–Eur. J., 2003, 9, 1281–1295 CrossRef CAS PubMed; (b) T. Bollermann, K. Freitag, C. Gemel, M. Molon, R. W. Seidel, M. von Hopffgarten, P. Jerabek, G. Frenking and R. A. Fischer, Inorg. Chem., 2011, 50, 10486–10492 CrossRef CAS PubMed.
- M. Eigen and R. G. Wilkins, Adv. Chem. Ser., 1965, 49, 55–80 CrossRef CAS PubMed.
- (a) P. K. Byers, A. J. Canty, B. W. Skelton and A. H. White, Organometallics, 1990, 9, 826–832 CrossRef CAS; (b) D. G. Brown, P. K. Byers and A. J. Canty, Organometallics, 1990, 9, 1231–1235 CrossRef CAS; (c) A. S. McCall, H. Wang, J. M. Desper and S. Kraft, J. Am. Chem. Soc., 2011, 133, 1832–1848 CrossRef CAS PubMed; (d) A. Maleckis, J. W. Kampf and M. S. Sanford, J. Am. Chem. Soc., 2013, 135, 6618–6625 CrossRef CAS PubMed.
- K. O. Christe and W. W. Wilson, J. Fluorine Chem., 1990, 46, 339–342 CrossRef CAS.
- (a) M. Eigen, Z. Phys. Chem., 1954, 1, 176–200 CrossRef CAS; (b) R. M. Fuoss, J. Am. Chem. Soc., 1958, 80, 5059–5061 CrossRef CAS.
- (a) The fluoride substituent could be attacked by an organic nucleophile: E. Differding and M. Wehrli, Tetrahedron Lett., 1991, 32, 3819–3822 CrossRef CAS; (b) or by a metal in an inner-sphere two electron transfer: H. Taube, H. Myers and R. L. Rich, J. Am. Chem. Soc., 1953, 75, 4118–4119 CrossRef CAS; (c) H. Taube and H. Myers, J. Am. Chem. Soc., 1954, 76, 2103–2111 CrossRef CAS; (d) H. Taube, Science, 1984, 226, 1028–1036 CAS; (e) F. Basolo, M. L. Morris and R. G. Pearson, Discuss. Faraday Soc., 1960, 80–91 RSC.
- P. T. Nyffeler, S. G. Durón, M. D. Burkart, S. P. Vincent and C.-H. Wong, Angew. Chem., Int. Ed., 2005, 44, 192–212 CrossRef CAS PubMed.
- (a) The radical cations derived from heteroatom-substituted double bonds can react in the alpha position with nucleophiles: A. Sutterer and K. D. Moeller, J. Am. Chem. Soc., 2000, 122, 5636–5637 CrossRef CAS; (b) and radicals: T. D. Beeson, A. Mastracchio, J.-B. Hong, K. Ashton and D. W. C. MacMillan, Science, 2007, 316, 582–585 CrossRef CAS PubMed.
- (a) X. Zhang, Y. Liao, R. Qian, H. Wang and Y. Guo, Org. Lett., 2005, 7, 3877–3880 CrossRef CAS PubMed; (b) X. Zhang, H. Wang and Y. Guo, Rapid Commun. Mass Spectrom., 2006, 20, 1877–1882 CrossRef CAS PubMed.
- (a) S. P. Vincent, M. D. Burkart, C.-Y. Tsai, Z. Zhang and C.-H. Wong, J. Org. Chem., 1999, 64, 5264–5279 CrossRef CAS; (b) for an example of a putative fluorine radical transfer from Selectfluor without previous single electron reduction see: M. Rueda-Becerril, C. Chatalova Sazepin, J. C. T. Leung, T. Okbinoglu, P. Kennepohl, J.-F. Paquin and G. M. Sammis, J. Am. Chem. Soc., 2012, 134, 4026–4029 CrossRef CAS PubMed.
- (a) A. M. Nesterenko, M. V. Ponomarenko, L. F. Lur'e and Y. A. Serguchev, Theor. Exp. Chem., 2002, 38, 156–161 CrossRef CAS; (b) Y. A. Serguchev, M. V. Ponomarenko, L. F. Lourie and A. A. Fokin, J. Phys. Org. Chem., 2011, 24, 407–413 CrossRef CAS.
- G. Bucher and J. C. Scaiano, J. Am. Chem. Soc., 1994, 116, 10076–10079 CrossRef CAS.
- M. D. Struble, M. T. Scerba, M. Siegler and T. Lectka, Science, 2013, 340, 57–60 CrossRef CAS PubMed.
- (a) G. A. Olah and J. M. Bollinger, J. Am. Chem. Soc., 1967, 89, 4744–4752 CrossRef CAS; (b) G. A. Olah, G. K. S. Prakash and V. V. Krishnamurthy, J. Org. Chem., 1983, 48, 5116–5117 CrossRef CAS; (c) G. A. Olah, G. K. S. Prakash and G. Rasul, Proc. Natl. Acad. Sci. U. S. A., 2013, 110, 8427–8430 CrossRef CAS PubMed.
- H. Haubenstock and R. R. Sauers, Tetrahedron, 2004, 60, 1191–1196 CrossRef CAS PubMed.
- T. Furuya and T. Ritter, J. Am. Chem. Soc., 2008, 130, 10060–10061 CrossRef CAS PubMed.
- S. Gambarotta, C. Floriani, A. Chiesi-Villa and C. Guastini, J. Chem. Soc., Chem. Commun., 1983, 1087–1089 RSC.
- M. Schmittel and A. Burghart, Angew. Chem., Int. Ed. Engl., 1997, 36, 2550–2589 CrossRef.
Footnotes |
| † Patent applications have been filed through Harvard on reagents 1–3 and the methods presented in ref. 3. SciFluor Life Sciences has licensed several of those patent applications. T.R. has a financial interest in SciFluor Life Sciences. |
| ‡ Electronic supplementary information (ESI) available: detailed experimental procedures and spectroscopic characterization for all new compounds. See DOI: 10.1039/c3sc52367e |
| This journal is © The Royal Society of Chemistry 2014 |