 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Leaf metallome preserved over 50 million years†
N. P.
Edwards
a,
P. L.
Manning
*a,
U.
Bergmann
b,
P. L.
Larson
c,
B. E.
van Dongen
a,
W. I.
Sellers
d,
S. M.
Webb
e,
D.
Sokaras
e,
R.
Alonso-Mori
e,
K.
Ignatyev
f,
H. E.
Barden
a,
A.
van Veelen
a,
J.
Anné
a,
V. M.
Egerton
a and
R. A.
Wogelius
*a
aSchool of Earth, Atmospheric, and Environmental Sciences, Williamson Research Centre for Molecular Environmental Science, University of Manchester, M13 9PL, UK. E-mail: phil.manning@manchester.ac.uk; roy.wogelius@manchester.ac.uk
bSLAC National Accelerator Laboratory, Linac Coherent Light Source, Menlo Park, CA 94025, USA
cBlack Hills Institute of Geological Research, Inc., Hill City, SD 57745, USA
dFaculty of Life Sciences, University of Manchester, Manchester M13 9PL, UK
eSLAC National Accelerator Laboratory, Stanford Synchrotron Radiation Lightsource, Menlo Park, CA 94025, USA
fDiamond Light Source, Didcot, OX11 0DE, UK
First published on 14th January 2014
Abstract
Large-scale Synchrotron Rapid Scanning X-ray Fluorescence (SRS-XRF) elemental mapping and X-ray absorption spectroscopy are applied here to fossil leaf material from the ∼50 Mya Green River Formation (USA) in order to improve our understanding of the chemistry of fossilized plant remains. SRS-XRF of fossilized animals has previously shown that bioaccumulated trace metals and sulfur compounds may be preserved in their original distributions and these elements can also act as biomarkers for specific biosynthetic pathways. Similar spatially resolved chemical data for fossilized plants is sparsely represented in the literature despite the multitude of other chemical studies performed. Here, synchrotron data from multiple specimens consistently show that fossil leaves possess chemical inventories consisting of organometallic and organosulfur compounds that: (1) map discretely within the fossils, (2) resolve fine scale biological structures, and (3) are distinct from embedding sedimentary matrices. Additionally, the chemical distributions in fossil leaves are directly comparable to those of extant leaves. This evidence strongly suggests that a significant fraction of the chemical inventory of the examined fossil leaf material is derived from the living organisms and that original bioaccumulated elements have been preserved in situ for 50 million years. Chemical information of this kind has so far been unknown for fossilized plants and could for the first time allow the metallome of extinct flora to be studied.
Introduction
The ability to resolve the distributions of trace metals (such as copper, zinc etc.) within distinct fossil biological structures can indicate the presence of metal affiliated biological pathways (the metallome) within an extinct organism, as has been shown for melanin pigmentation in fossil animals.1 Synchrotron Rapid Scanning X-ray Fluorescence (SRS-XRF) provides the unique ability to non-destructively map chemical zonation at parts per million (ppm) concentrations within extant and paleontological samples on a large (mm–dm) scale.2 Imaging at this scale allows the visualization of elemental distributions across entire fossil specimens in situ within their geological matrices, allowing the chemical discontinuities between matrix and fossil organism to be rapidly resolved and mapped. Whole specimen imaging reveals the correlation of specific elements with discrete biological structures but also aids in constraining chemical mass transfer and taphonomic alteration within a sample by resolving the distinct patterns that may be inherited either from the original biology or added by post-depositional geochemical processes. Previous studies using SRS-XRF in conjunction with a suite of other analytical methods have shown that the original distribution of the endogenous biochemistry of an organism can be preserved for over 100 million years and that certain elements (trace metals in particular) in discrete distributions can act as biomarkers for specific biosynthetic pathways.1–5 Analyses of fossil animals have already revealed the following: (1) remnant tooth and skin chemistry in ∼50 million year old fossil reptiles from the Green River Formation (USA);2,3 (2) original chemistry preserved within Green River Formation feathers;1,2 (3) pigment residue in feathers of a ∼120 Mya old bird (Confuciusornis sanctus);1 and (4) residual protein and pigment chemistry in an even earlier 150 Mya winged specimen (Archaeopteryx lithographica).4,5 Copper in feathers was identified as a biomarker for eumelanin pigmentation based on its correlation to darkly pigmented regions in extant feathers and its coordination chemistry being similar to that of Cu in modern eumelanin.1 Chemical analysis in such detail allowed the first reconstruction of the patterning of this pigment over an entire extinct organism based on whole specimen mapping with no destructive analyses required.1As we have observed in fossil animals, the chemistry of fossil plant specimens is potentially derived from the living organism and this chemistry may relate to the presence of certain biological processes. This is because plants (like all living organisms) require both macro- and micronutrients that are managed via a multitude of biological processes that regulate, mobilize, utilize and/or sequester elements in ways that allow them to function and sustain good health.6,7 Some elements have specific roles in specific biological structures while others may participate in a range of functions.
Therefore, we applied synchrotron X-ray analyses to fossil leaf material to test whether chemical mapping can improve our understanding of the chemistry of fossilized plant material, and whether this chemistry may relate to that of the living organisms. Previous work has consistently demonstrated the presence of endogenous plant derived biomarkers in plant matter from the Palaeozoic8–10 and demonstrated the survival of plant derived matter in specimens as old as 1 billion years11 so there is already strong evidence that elemental information may also be retained over geological time. However, this current body of work is mostly limited to data obtained via the routinely employed organic geochemistry techniques such as Pyrolysis-Gas Chromatography/Mass Spectrometry (Py-GC/MS), which has yielded little in the way of spatially resolved chemical data. Also, due to the destructive sampling required, extremely precious fossil specimens are often precluded from this type of analysis.
Here, we present data obtained via SRS-XRF, X-ray absorption spectroscopy (XAS) and Fourier Transform Infrared (FTIR) spectroscopy from exceptionally preserved fossil leaf material from the ∼50 Mya (Eocene) Green River Formation (USA) oil shales.12 Fossils from the Green River Formation are excellent candidates for the type of analyses performed in this study because the Green River is well known for abundant exceptionally preserved fossils and also for containing one of the world's largest oil shale reserves. Details of synchrotron, FTIR, and image analysis methods are given below. Additional supporting analytical methods included X-ray diffraction and Py-GC/MS. Information about these supporting methods as well as specimen details and geological setting are presented in the ESI.†
Methods
Synchrotron X-ray fluorescence and absorption spectroscopy
A comprehensive description of SRS-XRF imaging and experimental parameters are provided in Bergmann et al.,5 and subsequent publications.1–4 XRF maps here were obtained at the Stanford Synchrotron Radiation Lightsource (SSRL) wiggler beamline 6-2 and Diamond Light Source (DLS) beamline I-18 with specimens subjected to no sample preparation.Copper and sulfur X-ray Absorption Near Edge Structure (XANES) were recorded at SSRL beam line 6-2. Copper XANES and Extended X-ray Absorption Fine Structure (EXAFS) were also measured at SSRL beamline 10-2. In all cases XAS spectra were monitored during collection to avoid photoreduction of copper by the incident beam. If edge position shifted in successive scans, collection times were decreased and the incident beam was moved by several microns between successive scans in order to minimize photoreduction. A copper metal foil and a K2SO4 standard were used to calibrate the energy of the monochromator position. The energies of S species have been previously determined13,14 and for comparison to our data the peak positions were adjusted to match the sulfate peak energy reported here. S speciation mapping was achieved by setting the incident beam energy to that of a specific XANES resonance and performing the mapping process.
Fourier transform infrared spectroscopy
Infrared maps and point analyses were collected using a Perkin-Elmer Spotlight 400 instrument with a contact Attenuated Total Reflectance (ATR) crystal attachment with a 100 × 100 micron aperture and 4 cm−1 resolution. All spectra and maps were background subtracted and specimens required no preparation. Full details of FTIR mapping are described in Edwards et al.3Image processing and analysis
SRS-XRF maps from SSRL were processed from the raw detector count raster files using a custom MATLAB script which converted the data array into viewable 8 bit tiff images clipped at various contrast percentiles. Photoshop CS5 was used to compose all of the main text figures. The false colour image in Fig. 1I was produced in Photoshop CS5 by overlaying the greyscale Cu, Zn and Ni maps and assigning RGB channels to those individual elements. Curves were then adjusted to produce the best visual representation of the relationship of the three elements and do not show true relative differences in abundance. | ||
| Fig. 1 Optical images plus Cu, Zn and Ni distributions in extant L. styraciflua (A–D) and Green River Formation fossil P. wyomingensis specimen BHI-7032 (E–H). A second P. wyomingensis specimen, BHI-3113, is also shown optically (I) and as a false color composite image (J; Cu = red, Zn = green, and Ni = blue). Elemental capping at serration tips is observed in both the extant and fossil leaves (B and F inset arrows). Inset images in I and J (Cu) are magnified views of the boxed area in I showing the extremely fine detailed physical and chemical preservation of the larvae frass tubes and leaf venation. Note that (J) is not representative of true relative concentrations but is designed to best illustrate the distributions of these elements (see Fig. S2, ESI† and methods for further details). Yellow box in J represents area shown in Fig. 2B and C. Scale bars A, E = 5 mm, I = 1 cm. All maps obtained at SSRL SRS-XRF imaging beamline 6-2 using a 50 micron pinhole except H (100 micron). Black and white images: black = low relative concentrations, white = high relative concentrations. False color image: colour intensity indicates relative concentration. | ||
DLS XRF maps were produced using the PyMCA ROI imaging tool by defining the X-ray emission energy of an element in the recorded spectra and displaying the intensity of X-ray counts for that element. ImageJ was used to produce the sulfate only map of BHI-7032 via the built-in image subtraction function. FTIR absorption map intensities received no further pre-processing.
Results
SRS-XRF
SRS-XRF was performed on multiple fossil leaf specimens from the Green River Formation and extant leaf material (Fig. 1–4 and Fig. S1 and S2, ESI†). Maps of the elements that showed the greatest biological structural control, Cu, Zn and Ni, from a fossilized specimen of extinct Platanus wyomingensis are compared to the morphologically similar extant species Liquidambar styraciflua (Fig. 1). These maps show that the distribution of trace metals in extant plant material is constrained to specific anatomical structures and that comparable distributions occur in the fossil. In the extant leaf (Fig. 1A–D) Zn is restricted to the vascular system. Cu and Ni are more uniformly distributed, but slightly higher intensities in the veins make the vascular system discernible. All maps of extant leaves show high intensity spots of these elements at the tips of the leaf serrations (Fig. 1B arrows, Fig. S1, ESI†), a phenomenon observed in extant leaves elsewhere.16 In this fossil leaf (BHI-7032, Fig. 1E–H) the distributions of Cu, Zn and Ni are also clearly delineated by discrete anatomical structures, including concentrations of Cu at the serration tips (Fig. 1F inset, arrows). In contrast, Mn is not constrained by fossil tissue but shows a distribution as surface deposits (Fig. S2, ESI†), indicating some post-depositional inorganic precipitation has occurred. This shows how large scale mapping allows biological and post-depositional inorganic processes to be distinguished. Without mapping, the presence of elevated Mn concentrations may have been mistaken as being derived from the leaf as Mn is known to accumulate in extant leaves as shown in Fig. S1 (ESI†). Lack of biological control and knowledge of the diagnostic dendritic pattern of Mn oxide precipitates allows us to correctly assign the Mn deposits to a post-depositional geochemical process. Cu, Zn, and Ni, however, reveal biological control.A second much larger P. wyomingensis specimen (BHI-3113, Fig. 1I and J) exhibits large skeletonized areas with clearly visible small veins and curved, tube-like structures. Skeletonization and tube structures are features comparable to those produced by extant Catastega aceriella (Lepidoptera: Olethreutidae) that feed upon and skeletonize the underside of modern maple tree leaves generating distinct ‘trumpet’ shaped cocoons of feces and silk (frass tubes) as they develop.17,18 Maps of Cu, Zn and Ni for the whole of this large specimen (scan height ∼18 cm) are presented as a false color composite image in Fig. 1J. Cu (red), Zn (green) and Ni (blue) are all highly concentrated within the stem and primary venation. Cu is also present in relatively high concentrations both in the uneaten epidermis/parenchyma relative to the primary venation and the non-predated small scale venation, whereas Zn and Ni are present in relatively lower quantities in the epidermis/parenchyma (Fig. S2, ESI†). The frass tubes show high concentrations of all of these trace metals. When magnified, the trace metal distributions resolve fine scale structures such as venation and the individual fecal pellets within the frass tubes (insets of Fig. 1I and J).
Comparison of chemical zonation at higher magnification (Fig. 2) reveals that Cu and Ni in the small veins of an extant Acer pseudoplatanus leaf exhibit nearly identical spacing, diameter and pattern to that of the skeletonized remains of fossil P. wyomingensis. Copper not only shows strong correlation to biological structure but also exhibits the most elevated concentrations relative to the matrix in all fossil leaf specimens analysed (Fig. 3; Fig. S2 and Table S1, ESI†). Having confirmed the affinity between the distributions of several transition metals and original biological structure, we then sought to explore the detailed coordination chemistry of elements concentrated within the fossil to see if further details of fossil chemistry could be resolved.
 | ||
| Fig. 2 Fine scale Cu (A and B) and Ni (C and D) distributions in extant A. pseudoplatanus (A and C) and BHI-3113 (B and D, magnified and rotated area of yellow box in Fig. 1J). Scale bars 1 mm. Images A and C obtained at Diamond Light Source microfocus spectroscopy beamline I18 using a beam spot size of 5 (vertical) × 6.5 (horizontal) microns. (A–C) Blue = low relative concentrations, red = high relative concentrations, D is presented as greyscale (black = low relative concentrations, white = high relative concentrations) instead of colour due to loss of clarity of the fine scale venation using a false colour scheme. Zn distribution in the fine scale venation of BHI-3113 is not resolved due to relatively high Zn concentrations in the matrix (Fig. S2, ESI†). | ||
 | ||
| Fig. 3 Optical images and SRS-XRF maps of Cu distributions in additional Green River Formation leaf material. (A and B) MGSF313 (Zelkova nervosa); (C and D) BHI-045A (P. wyomingensis); (E and F) BHI-3100 (Populas wilmattae); (G and H) BHI-7032 (second species on same specimen as Fig. 1E, Cedrelospermum nervosum tent.). Scale bars = 1 mm. All maps obtained at SSRL SRS-XRF imaging beamline 6-2 using a 100 micron pinhole except H (50 micron), black = low relative concentrations, white = high relative concentrations. These scans confirm that the relationship between preserved plant structure and chemistry is a recurring phenomenon in Green River specimens. | ||
Sulfur XANES data indicate that fossil leaves possess an organosulfur inventory distinct from their embedding matrices (Fig. 4A). In extant leaves inorganic sulfate is the dominant S species, but reduced S species are clearly visible (disulfide, thiol, and sulfoxide). The sedimentary matrices of BHI-7032 and BHI-3113 show only sulfate and sulfonate, whereas thiol and sulfoxide are not detected. The fossil leaf spectra however are directly comparable to extant leaf material with sulfate present and the common reduced organic species of thiol and sulfoxide clearly resolved.13,14
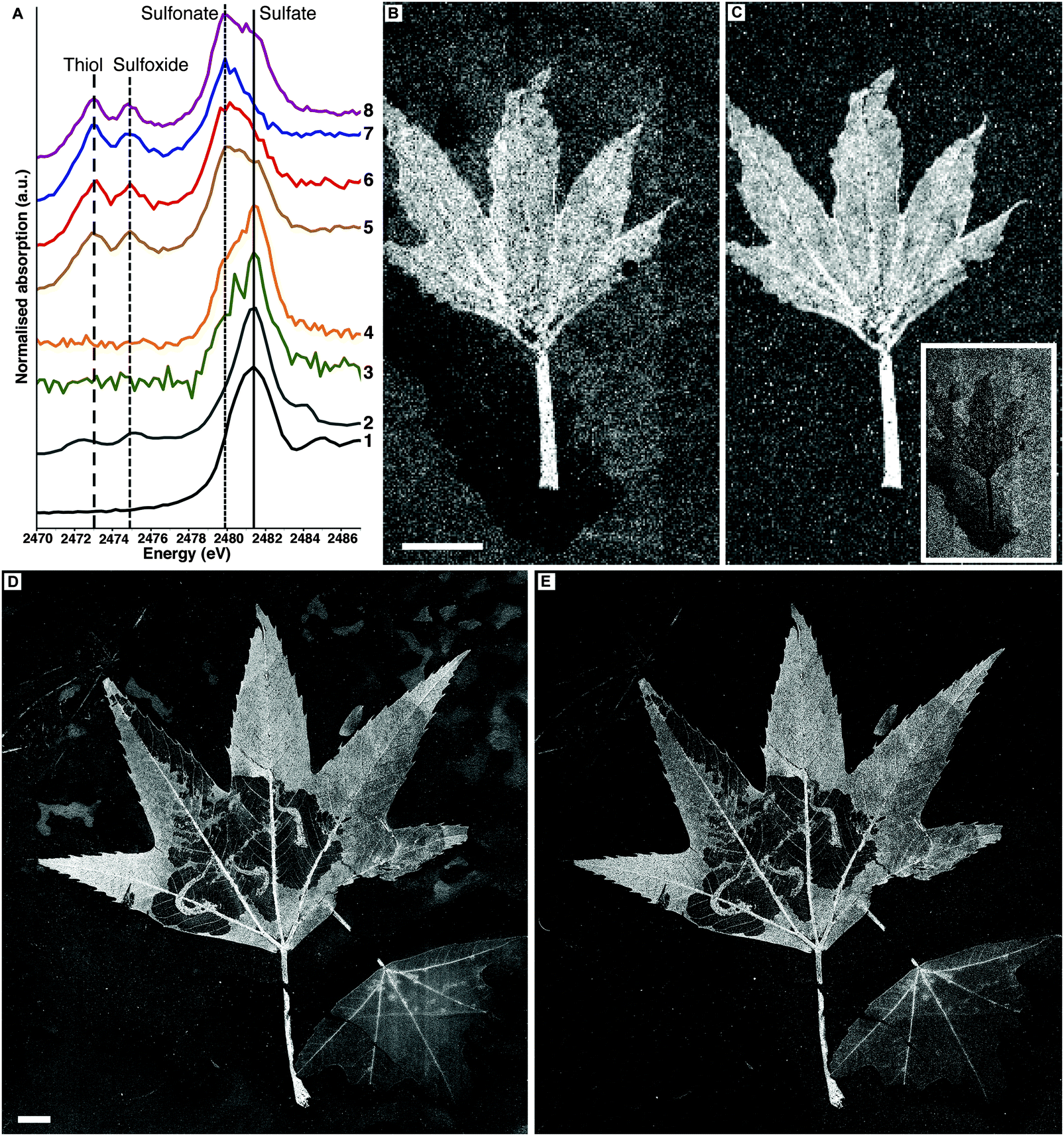 | ||
| Fig. 4 Sulfur spectroscopy and imaging of BHI-7032 and BHI-3113. (A) X-ray absorption spectra for S in fossil leaves. (1) Sulfate peak position as determined on a K2SO4 standard, vertical solid line (2481.4 eV). (2) Extant L. styraciflua. (3 and 4) Matrix of BHI-3113 and BHI-7032 respectively. (5) BHI-3113 insect larvae frass tubes. (6) BHI-3113 bulk leaf tissue. (7 and 8) Stems of BHI-3113 and BHI-7032 respectively. Organic sulfonate (2479.9 eV), sulfoxide (2474.6 eV) and thiol (2472.9 eV) assigned by comparison to literature values. S maps of BHI-7032 and BHI-3113 obtained with beam energies of (B and D) 3150 eV (equivalent to total S) and (C and E) 2472.9 eV (essentially thiol only). Inset of C shows image of sulfate only (total S with all organic S subtracted, see methods for details). Scale bars B = 5 mm, D = 1 cm, black = low relative concentrations, white = high relative concentrations. | ||
Sulfur speciation imaging
SRS-XRF imaging is uniquely able to map the distribution of specific oxidation states of an element within a sample by tuning the energy of the incident X-ray beam to specific XANES resonances. This ability allows sulfur speciation and distribution to be better resolved, as data are not restricted to a few limited point spectra. Sulfur maps of BHI-7032 and BHI-3113 (Fig. 4B and D) were obtained with an incident X-ray beam energy of 3150 eV, inducing X-ray fluorescence of all oxidation states of S (i.e., total S). These maps reveal that S is highly concentrated in the leaves but is also present in the matrices (note the mottling in top right of 4D). The maps in Fig. 4C and E were obtained using an incident energy of 2472.9 eV, inducing X-ray fluorescence primarily from thiol and almost completely removing the contribution from higher oxidation states such as inorganic sulfate. At this energy, matrix features are no longer resolved (the mottling in top right of 4D disappears) confirming that almost all reduced organic S is unequivocally confined within the fossil leaves. Maps of other oxidation states are presented in Fig. S4 (ESI†).![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretch at 1560 cm−1 and C–H antisymmetric stretch at 2930 cm−1) shows that these organic functional groups are confined within the fossil leaf tissue, comparable to the synchrotron maps of organic sulfur (FTIR absorption band assignments in Table S3, ESI†).
O stretch at 1560 cm−1 and C–H antisymmetric stretch at 2930 cm−1) shows that these organic functional groups are confined within the fossil leaf tissue, comparable to the synchrotron maps of organic sulfur (FTIR absorption band assignments in Table S3, ESI†).
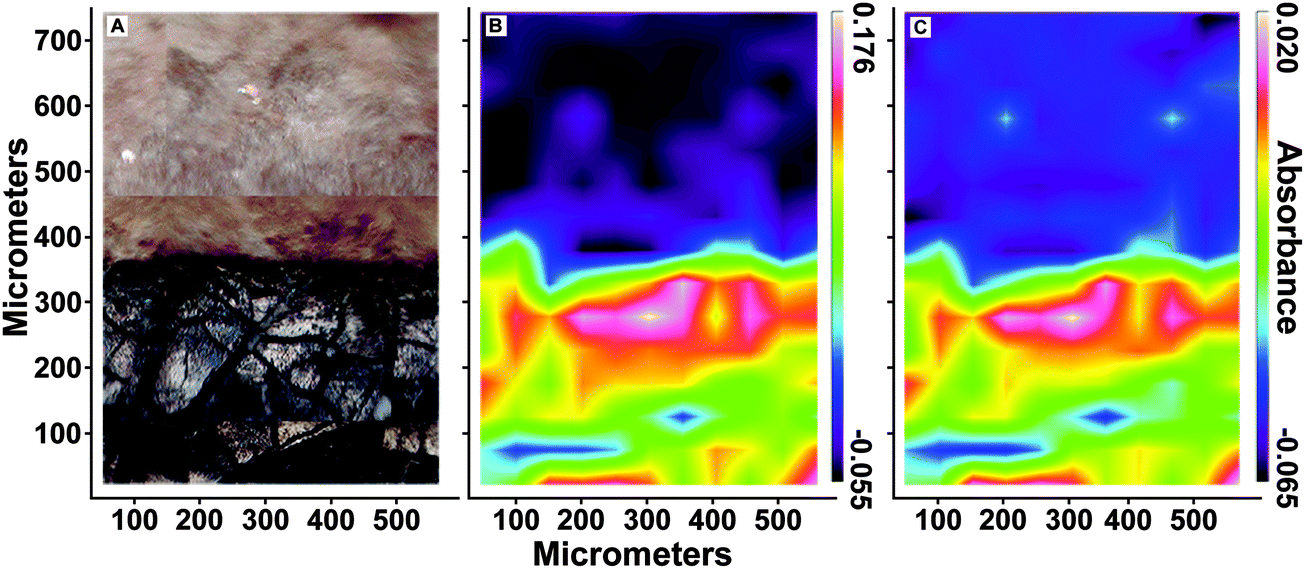 | ||
| Fig. 5 (A) Optical microscope image of fossilized leaf material [stem, bottom] and sedimentary matrix [top]. Infrared absorption maps of same area at (B) 1560 cm−1 and (C) 2930 cm−1 in leaf material (attenuated total reflectance mode). Absorbance is scaled from low (purple) to high (red/white). These maps show that organic functional groups are highly constrained within the fossil residue. | ||
Discussion
These results not only resolve important and previously unknown details of the chemistry of fossilized leaf material but also lead us to conclude that a significant fraction of this chemistry is derived from the biology of the ancient plants. Post-depositional addition of material is a distinct possibility however for P. wyomingensis (BHI-7032, BHI-3113) the data do not support the argument that non-biogenic processes such as pyritisation, sulfurisation and phosphatisation are contributors to the observed chemistry for the following reasons: X-ray diffraction (Fig. S6, ESI†) did not identify pyrite; S XANES and Py-GC/MS (Fig. S7, ESI†) are inconsistent with either sulfide precipitation or sulfurisation; and phosphorus is below XRF detection limits (<100 ppm). Chemical mapping and Py-GC/MS also clearly show that trace metals and organic moieties are constrained within the fossil residues and map tightly with discrete biological structures, not a pattern that may be attributed to random indiscriminate binding of mobile species to organic ligands. FTIR does not show enrichment in amides or phospholipids that could potentially be contributed from exogenous sources such as bacteria22 though this does not entirely rule out their presence. Also, unlike the diffuse Mn precipitates (Fig. S2, ESI†), there is virtually no evidence of Cu or Zn-enriched precipitates from fluid transport within any of the fossil leaf specimens. Most importantly, Cu EXAFS is inconsistent with inorganic precipitates.SRS-XRF mapping reveals that elemental distributions in the fossil leaves are directly comparable to extant leaves and XAS indicates that Cu is coordinated as an organometallic complex. The organometallic and organosulfur compounds that we detect and map within the fossil material may be inherited from several different functional roles within the ancient organism. For example the majority of metal binding occurs within either the cell wall (apoplast) or to specific metal binding molecules23 such as plastocyanin24 (a cofactor in photosynthesis), phytochelatins25 (specific sulfur rich metal detoxifying proteins), organic acids (citrate and malate),26 nitric oxide27 and nicotianamine28 to name a few. We now consider whether elemental distributions and spectroscopy can be combined to provide clues as to the likely biochemical roles of Cu in the original plant.
First, we note that organic S is only detectable within the fossil residues and is present in forms comparable to extant leaves. Sulfur is a major macronutrient for plants and is known to be present in both inorganic and organic forms (sulfate and carbon-bound sulfonate/cysteine etc., respectively).13,29 Although a clear spatial correlation exists between Cu and organic S in the fossil leaves [Cu vs. thiol in BHI-7032 (Fig. S4, ESI†) R2 = 0.86], XAS indicates that the bulk of Cu in these leaves is not S-coordinated. This suggests two possibilities. (1) Sulfur-rich metal chelates such as phytochelatins were not abundant in the living leaf matter at the time of burial and therefore the Cu was originally dominantly bound to carboxylate functional groups in cell walls. Despite volatile loss and condensation reactions, Cu has remained complexed in this configuration as observed by XAS. (2) Originally much of the Cu inventory was coordinated by sulfur-rich chelates, but post-depositional replacement of S by O in the inner coordination sphere of Cu occurred as sulfur compounds degraded, consistent with the strong affinity that Cu has for carboxylate-type complexes during soil formation.19
Our XRF maps which show the pervasive distribution of Cu within all of the leaves, both fossil and extant, supports the first possibility, with Cu being kept in reserve throughout the active leaf tissue by attachment to exposed carboxylate-type functional groups on cell walls within veins, parenchymal cells or in vacuoles. This is also consistent with other work,30 which predicts that the bulk of Cu within the leaf should be bound within the parenchyma through attachment to oxygen terminated carboxyl groups.
From all the data collected, we therefore conclude that organometallic Cu compounds in the fossils are derived from the original organism, either as remnants of original O- or N-coordinated Cu (in particular malate or malate-type moieties such as α-hydroxy fatty acids which are abundantly present both in extant plant tissues26,31 and in these fossils [see Fig. S7, ESI†]) or as Cu scavenged by carboxylate terminated molecules during breakdown reactions of S-bearing compounds such as phytochelatins or glutathione.25 Further compelling evidence supporting the endogeneity of the observed chemistry is provided by the preserved insect frass tubes (BHI-3113, Fig. 1I and J). It is clear that insect larvae (cf. Catastega aceriella) fed upon the leaf, subsequently excreting leaf-derived material to form the frass tubes. The trace metal inventory of the tubes reflects that of the host leaf, which is consistent with the tube material being derived from un-metabolized host leaf chemistry. It is difficult to postulate a post-depositional mechanism by which this and other detailed chemical relationships might be taphonomically reproduced.
Details of the preservation mechanism are yet to be determined, but the results suggest that bioaccumulated trace metals play a role in the overall reaction pathway and the exceptional morphological preservation of leaves in the Green River Formation. It has been shown that trace metals (especially Cu) participate in a range of geochemical reactions during degradation of plant tissue and the formation of stable molecules in sedimentary organic matter.32–37 Such reactions could promote exceptional preservation of plant tissue and result in trace metals retaining an original distribution within fossils. A parallel or alternate process is that bioaccumulated Cu may function as a biocide which may have inhibited microbial degradation. This is a property of Cu that is commonly exploited industrially in wood preservative products (e.g. Cu naphthenate). It is also possible that mechanisms proposed in other studies could enhance the preservation of metal distributions, such as the formation of a recalcitrant organic geopolymer e.g.,8,10 or ternary complexation with mineral surfaces.3 However, the presence of Cu has been observed in a range of exceptionally preserved fossil tissues and archaeological artifacts1–5,38 which have various original compositions and taphonomic pathways. This indicates that the observed positive correlation between the presence of trace metals, Cu especially, and the enhanced preservation of organic material is probably not coincidental.
Conclusion
The innovative application of the non-destructive imaging techniques employed here provides a powerful method for investigating fossil material and reveals that the fidelity of biochemical preservation in fossil plants is much higher than has previously been shown. The data presented show how state-of-the-art synchrotron chemical imaging and spectroscopy can uniquely and accurately resolve the details of chemical-structural relationships in fossilized plant material from the angstrom to decimeter length-scales and can discriminate among contributions from fossils, the embedding sedimentary matrices, and post-depositional geochemical precipitates. Results strongly indicate that, as has been observed in fossil animals, the chemical inventory in Green River Formation fossil leaves is derived from the original biochemistry of the organisms. The chemical distributions seen in the fossil specimens are comparable to those seen in extant plant material and it is this comparison that gives a tantalizing glimpse as to how ancient plants might have regulated their metal inventories, i.e., a first look at the metallome of extinct plants. Not only may this type of analysis inform us about ancient plant biochemistry but it may also inform us about local environmental conditions, degradation of organic matter and the formation of hydrocarbon deposits.Author contributions
All authors actively participated in synchrotron X-ray data acquisition. B.E.v.D and H.E.B performed and processed Py-GC/MS analysis. N.P.E, H.E.B, R.A.W performed FTIR analyses. N.P.E, R.A.W., and P.L.M. co-wrote the manuscript and all other authors contributed to the manuscript. N.P.E composed all the figures.Acknowledgements
Fossil specimens were provided by the Black Hills Institute Museum. Portions of this research were carried out at the Stanford Synchrotron Radiation Lightsource (SSRL), a national user facility operated by Stanford University on behalf of the U.S. Department of Energy, Office of Basic Energy Sciences. The authors would also like to thank Diamond Light Source (DLS) for beamtime awarded under proposal SP-8597. We thank support staff at both SSRL and DLS for patience with difficult samples. Funding was provided in part by a UK Natural Environment Research Council grant NE/J023426/1.References
- R. A. Wogelius, P. L. Manning, H. E. Barden, N. P. Edwards, S. M. Webb, W. I. Sellers, K. G. Taylor, P. L. Larson, P. Dodson, H. You, L. Da-qing and U. Bergmann, Science, 2011, 333, 1622–1626 CrossRef CAS PubMed.
- N. P. Edwards, R. A. Wogelius, U. Bergmann, P. L. Larson, W. I. Sellers and P. L. Manning, Appl. Phys. A: Mater. Sci. Process., 2013, 111, 147–155 CrossRef CAS.
- N. P. Edwards, H. E. Barden, B. E. van Dongen, P. L. Manning, P. L. Larson, U. Bergmann, W. I. Sellers and R. A. Wogelius, Proc. R. Soc. B, 2011, 278(1722), 3209–3218 CrossRef CAS PubMed.
- P. L. Manning, N. P. Edwards, R. A. Wogelius, U. Bergmann, H. E. Barden, P. L. Larson, D. Schwarz-Wings, V. M. Egerton, D. Sokaras, R. A. Mori and W. I. Sellers, J. Anal. At. Spectrom., 2013, 28, 1024–1030 RSC.
- U. Bergmann, R. W. Morton, P. L. Manning, W. I. Sellers, S. Farrar, K. G. Huntley, R. A. Wogelius and P. L. Larson, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 9060–9065 CrossRef CAS PubMed.
- R. Pal and J. Rai, Appl. Biochem. Biotechnol., 2010, 160, 945–963 CrossRef CAS PubMed.
- I. Raskin, P. B. A. Nanda Kumar, S. Dushenkov and D. E. Salt, Curr. Opin. Biotechnol., 1994, 5, 285–290 CrossRef CAS.
- N. S. Gupta, D. E. G. Briggs, M. E. Collinson, R. P. Evershed, R. Michels, K. S. Jack and R. D. Pancost, Org. Geochem., 2007, 38, 499–522 CrossRef CAS.
- S. Auras, V. Wilde, S. Hoernes, K. Scheffler and W. Püttmann, Palaeogeogr., Palaeoclimatol., Palaeoecol., 2006, 240, 305–317 CrossRef.
- M. E. Collinson, in Taphonomy: Process and Bias Through Time, ed. P. A. Allison and D. J. Bottjer, Springer, Dordrecht, 2nd edn, 2011, pp. 223–247 Search PubMed.
- G. Eglinton, P. M. Scott, T. Belsky, A. L. Burlingame and M. Calvin, Science, 1964, 145, 263–264 CAS.
- L. Grande, Paleontology of the Green River Formation, with a review of the Fish Fauna, The Geological Survey of Wyoming, 2nd edn, 1993 Search PubMed.
- F. Jalilehvand, in Sulfur Transport and Assimilation in Plants in the Post Genomic Era, ed. K. Saito, Backhuys Publishers, Leiden, The Netherlands, 2005 Search PubMed.
- M. Sandström, F. Jalilehvand, E. Damian, Y. Fors, U. Gelius, M. Jones and M. Salomé, Proc. Natl. Acad. Sci. U. S. A., 2005, 102(40), 14165–14170 CrossRef PubMed.
- V. A. Solé, E. Papillon, M. Cotte, Ph. Walter and J. Susini, Spectrochim. Acta, Part B, 2007, 62, 63–68 CrossRef.
- E. Harada, A. Hokura, I. Nakai, Y. Terada, K. Baba, K. Yazaki, M. Shiono, N. Mizuno and T. Mizuno, Metallomics, 2011, 3, 1340–1346 RSC.
- W. A. Côté and D. C. Allen, Can. Entomol., 1973, 105(3), 463–470 CrossRef.
- R. L. Brown, J. Lepid. Soc., 1986, 40(4), 327–346 Search PubMed.
- A. Manceau and A. Matynia, Geochim. Cosmochim. Acta, 2010, 74, 2556–2580 CrossRef CAS.
- D. H. McNear Jr., R. L. Chaney and D. L. Sparks, Phytochemistry, 2010, 71, 188–200 CrossRef PubMed.
- T. J. Strathmann and S. C. B. Myneni, Geochim. Cosmochim. Acta, 2004, 68(17), 3441–3458 CrossRef CAS.
- V. Erukhimovitch, V. Pavlov, M. Talyshinsky, Y. Souprun and M. Huleihel, J. Pharm. Biomed. Anal., 2005, 37, 1105–1108 CrossRef CAS PubMed.
- W. E. Rauser, Cell Biochem. Biophys., 1999, 31, 19–48 CrossRef CAS PubMed.
- M. R. Redinbo, T. O. Yeates and S. Merchant, J. Bioenerg. Biomembr., 1994, 26, 49–66 CrossRef CAS PubMed.
- C. S. Cobbett, Plant Physiol., 2000, 123, 825–832 CrossRef CAS PubMed.
- J. Schulze, M. Tesfaye, R. G. M. G. Litjens, B. Bucciarelly, G. Trepp, S. Miller, D. Samac, D. Allan and C. P. Vance, Plant Soil, 2002, 247, 133–139 CrossRef CAS.
- A. Besson-Bard, A. Pugin and D. Wendehenne, Annu. Rev. Plant Biol., 2008, 59, 21–39 CrossRef CAS PubMed.
- M. Takahashi, Y. Terada, I. Nakai, H. Nakanishi, E. Yoshimura, S. Mori and N. K. Nishizawa, Plant Cell, 2003, 15(6), 1263–1280 CrossRef CAS.
- S. Kopriva, eLS, John Wiley & Sons, Ltd, Chicester, 2001 Search PubMed.
- S. V. Sahi, M. Israr, A. K. Srivastava, J. L. Gardea-Torresdey and J. G. Parsons, Chemosphere, 2007, 67(11), 2257–2266 CrossRef CAS PubMed.
- B. Mösle, M. E. Collinson, P. Finch, B. A. Stankiewicz, A. C. Scott and R. Wilson, Org. Geochem., 1998, 29, 1369–1380 CrossRef.
- J. W. Bunting and K. M. Thong, Can. J. Chem., 1970, 48, 1654–1656 CrossRef CAS.
- J. H. Fitzpatrick and D. Hopgood, Inorg. Chem., 1974, 13, 568–574 CrossRef CAS.
- M. Eddaoudi, D. B. Moler, H. Li, B. Chen, T. M. Reineke, M. O'Keefe and O. M. Yaghi, Acc. Chem. Res., 2001, 34, 319–330 CrossRef CAS PubMed.
- D. L. Reger, J. J. Horger and M. D. Smith, Chem. Commun., 2011, 47, 2805–2807 RSC.
- V. V. Annenkov, E. N. Danilovtseva, V. V. Saraev and A. I. Mikhaleva, J. Polym. Sci., Part A: Polym. Chem., 2003, 41, 2256–2263 CrossRef CAS.
- J. G. Lawless and N. Levi, J. Mol. Evol., 1979, 13, 281–286 CrossRef CAS PubMed.
- K. Anheuser and M. Roumeliotou, The Conservator, 2003, 27, 23–33 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3mt00242j |
| This journal is © The Royal Society of Chemistry 2014 |