DOI:
10.1039/C4RA10654G
(Paper)
RSC Adv., 2014,
4, 60342-60348
Electronic and electrochemical properties as well as flowerlike supramolecular assemblies of fulleropyrrolidines bearing ester substituents with different alkyl chain lengths†
Received
17th September 2014
, Accepted 3rd November 2014
First published on 6th November 2014
Abstract
A series of alkyl (methyl, ethyl, propyl, butyl) benzoate ester substituted fulleropyrrolidine derivatives (FP1–FP4) were synthesized and their electronic and electrochemical properties were investigated by means of absorption spectra, electronic structure calculation, and cyclic voltammetry (CV), respectively. The LUMO–HOMO energies and energy gaps of fullerene derivatives were estimated by the first reduction potential measured with CV combined with absorption spectra, which are consistent with those obtained from density functional theory (DFT) calculations. It was found that all fulleropyrrolidines showed very similar absorption spectra, orbital energies and redox behaviors, which are comparable with those of well-known phenyl-C61-butyric acid methyl ester (PCBM). The flowerlike supramolecular architectures obtained from the self-assembly of FP1–FP4 in chloroform–alcohol mixture solvents were characterized by scanning electron microscopy (SEM) and X-ray diffraction (XRD). A lamellar structure with a d-spacing of 1.92–2.02 nm that depends on the molecular size, corresponding to the thickness of a bilayer structure, suggested a face-to-face conformation of the substituent of C60 and an interdigitation of the bare C60 side packing. These fulleropyrrolidines have high C60 content, are energetically PCBM-like, and are capable of forming complex flowerlike architectures, which provide fundamental insights into molecular design toward advanced fullerene materials.
1. Introduction
Fullerene (C60) and its derivatives have been employed to a considerable extent as excellent electron-acceptor materials in various optoelectronic devices (such as organic solar cells and field effect transistors).1–6 On the other hand, as typical π-conjugated molecules, fullerenes are also important building blocks for fabrication of functional supramolecular assemblies.7–10 It has been recognized that the performance of molecular device could be improved by fine molecular-tailoring and directed self-organization of π-conjugated units. In this regard, many efforts have been devoted to self-assembly of fullerenes to manipulating properties of fullerene-based materials.11–19 For example, several well-defined organized nano/microstructures such as spheres,20 rods,21 wires,22 belts,23,24 whiskers,25,26 and sheets,27,28 have been fabricated from the pristine C60, which retain the intrinsic optoelectronic properties of fullerenes but the numbers of solvents owning enough solubility for C60 are very limited (usually aromatic solvents and carbon disulfide). Alternatively, molecular-tailoring of fullerenes via attachment of proper substituent groups on periphery of C60 cage not only enhances its solubility in common organic solvents, which is benefit for solution processing in device fabrication, but also enables the regulation of self-assembly.29–44 For example, various superstructures such as nanofibers, vesicles, flowerlike architectures have been achieved by fulleropyrrolidines bearing multiple alkyl chains.41–44 However, the attachment of bulky and insulating appends largely decreased C60 content (usually lower than 50%), leading to these chemically-modified fullerenes are less electronically active that is actually undesirable for their optoelectronic applications. Therefore, grafting small substituent groups on fullerene surface should be more promising due to high C60 content will be retained in these fullerene derivatives.45,46 Recently, a small pyridine group equipped fulleropyrrolidine was reported to be capable of organizing into C60-rich and photoconductive flowerlike architectures, which retain high C60 content (84%) and photoinduced carrier-transporting properties comparable with pristine C60.45 Furthermore, it was also found that the morphology of self-organized objects could also be tuned by the position of N-atom in pyridine group. This example provides a useful molecular design concept for construction of C60-rich complex supramolecular assemblies via grafting a small substituent group on periphery of fullerene cage. Thus the design and synthesis of other small substituent attached fullerene derivatives to investigate their electronic properties and self-assembly would further provide fundamental insights into fullerene-containing materials.
In this work, by considering the molecular structure of well-known fullerene derivative, phenyl-C61-butyric acid methyl ester (PCBM), a small benzoate ester substituent group was then grafted on C60 to synthesize a series of alkyl (methyl, ethyl, propyl, butyl) benzoate ester-substituted fulleropyrrolidines (FP1–FP4, Scheme 1), and their electronic and electrochemical properties as well as self-assembly were investigated by means of absorption spectra, electronic structure calculation, cyclic voltammetry (CV), scanning electron microscopy (SEM) and X-ray diffraction (XRD) analysis, respectively. It was found that the LUMO–HOMO levels of these fullerene derivatives estimated by the experiments and theoretical calculations, are energetically PCBM-like. All of these fulleropyrrolidines could organize into flowerlike architectures of lamellar structures with alkyl chain length dependent d-spacing. These small ester substituent attached fullerenes are high C60 content (76–79%), energetically PCBM-like, and capable of forming flowerlike assemblies, which provide fundamental insights into molecular design toward advanced fullerene materials.
 |
| | Scheme 1 Chemical structures of fulleropyrrolidine derivatives FP1–FP4. | |
2. Experimental details
Absorption, 1H NMR and mass spectra were recorded on a TU-1901 (Persee), Brucker Avance III (400 MHz) and MALDI-TOF-MS (AB Sciex 4700) spectrometer, respectively. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) were performed on a HITACHI TM-1000 and JEOL-2100 microscopes, respectively. X-Ray diffraction (XRD) patterns were obtained using a RIGAKU D/Max-2550 PC X-ray diffractometer. Cyclic voltammetry (CV) and differential pulse voltammetry (DPV) curves were measured using a CHI 660D electrochemical workstation (Chenhua, Shanghai). The CV and DPV curves were collected at room temperature under N2 using a conventional three-electrode system (glassy carbon working electrode, Pt wire counter-electrode and Ag/Ag+ quasi-reference electrode) in 0.1 M tetrabutylammonium tetrafluoroborate (n-Bu4NBF4) solution in 1,2-dichlorobenzene (o-DCB) at a potential scan rate of 100 mV s−1, where the reduction potentials were calibrated using ferrocene/ferrocenium (Fc/Fc+) redox couple as internal standard. The geometries of fulleropyrrolidine derivatives were optimized by density functional theory (DFT) at the B3LYP/6-31G(d) level, and the orbital energy and orbital analysis were calculated at the PBEPBE/6-311G(d,p) level, with the Gaussian 09 program package.47
Fullerene C60 (99%) was purchased from XFNANO (Nanjing) and other reagents as well as solvents (AR) were obtained from Sinopharm Chemical Reagents Co. (Shanghai). Alkyl (methyl, ethyl, propyl, butyl) 4-formylbenzoates were prepared by esterification reaction from 4-formylbenzoic acid with the corresponding alcohols in the presence of thionyl chloride. Fulleropyrrolidine derivatives (FP1–FP4, Scheme 1) were synthesized by refluxing the toluene solution of C60, sarcosine and the corresponding benzaldehyde according to the standard Prato reaction.48
1, 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.12 (d, 2H, J = 8.0 Hz), 7.94 (br, 2H), 5.07 (br, 2H), 4.33 (d, 1H, J = 8.0 Hz), 3.91 (s, 3H), 2.85 (s, 3H). MALDI-TOF-MS [DCTB] m/z calcd, C71H13NO2 911.23; found, 911.12.
2, 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.13 (d, 2H, J = 8.0 Hz), 7.96 (br, 2H), 5.11 (br, 2H), 4.40–4.35 (m, 3H), 2.88 (s, 3H), 1.39 (t, 3H, J = 8.0 Hz). MALDI-TOF-MS [DCTB] m/z calcd, C72H15NO2 925.25; found, 925.76.
3, 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.12 (d, 2H, J = 8.0 Hz), 7.92 (br, 2H), 5.05 (br, 2H), 4.33–4.25 (m, 3H), 2.84 (s, 3H), 1.82–1.76 (m, 2H), 1.03 (t, 3H, J = 8.0 Hz). MALDI-TOF-MS [DCTB] m/z calcd, C73H17NO2 939.28; found, 939.86.
4, 1H NMR (400 MHz, CDCl3): δ (ppm) = 8.13 (d, 2H, J = 8.0 Hz), 7.97 (br, 2H), 5.12 (br, 2H), 4.37–4.30 (m, 3H), 2.88 (s, 3H), 1.79–1.71 (m, 2H), 1.51–1.45 (m, 2H), 0.98 (t, 3H, J = 8.0 Hz). MALDI-TOF-MS [DCTB] m/z calcd, C74H19NO2 953.31; found, 953.87.
3. Results and discussion
3.1. Photophysical properties
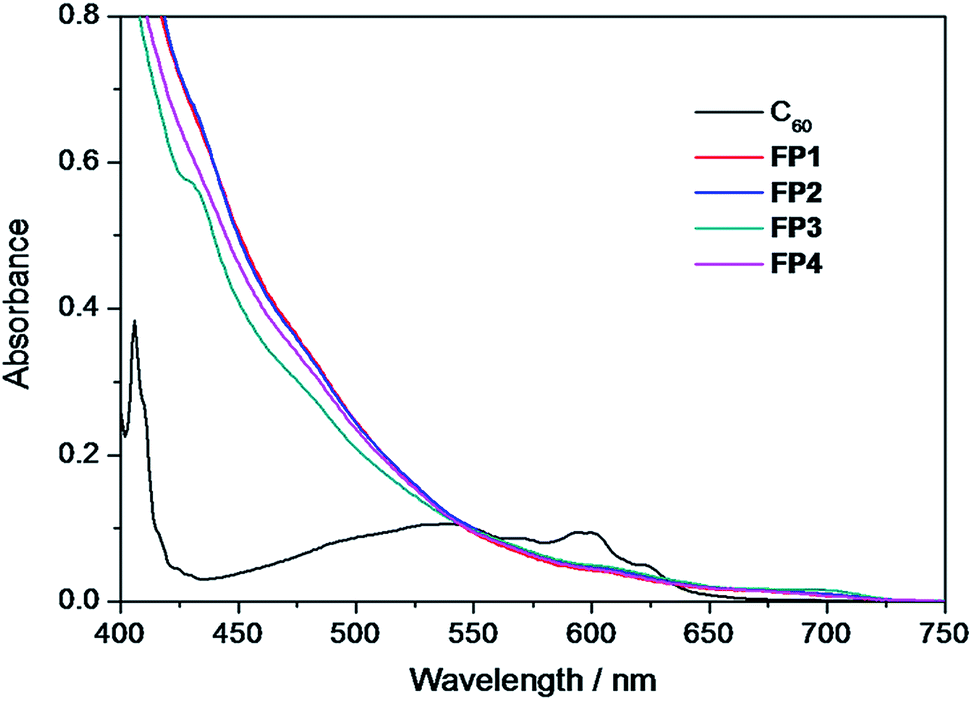
The absorption spectra (400–750 nm) of fulleropyrrolidine derivatives FP1–FP4 in chloroform (1.3 × 10−4 M) were shown in Fig. 1, together with the spectrum of pristine C60 (6.5 × 10−5 M) for comparison. All of fulleropyrrolidine derivatives exhibit nearly the same broad absorption bands from 400 to 750 nm those extend to longer wavelength range than that of the pristine C60 (Fig. 1). The difference of photophysical properties between FP1–FP4 and the pristine C60 could be explained from the symmetry breaking effect in the former that enhanced the forbidden transitions to some extent.49 It was noted that the fulleropyrrolidine derivatives have stronger visible light absorption in the red-shifted absorption edge above 650 nm with the longer λonset around 724 nm (Table 1), which is similar to that of PCBM and important to contribute the light harvesting in photovoltaics.31 The similar absorption spectra observed among fulleropyrrolidine derivatives FP1–FP4 indicated that the alkyl chain length scarcely affects their photophysical properties.
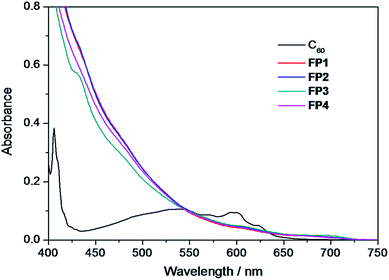 |
| | Fig. 1 Absorption spectra of fulleropyrrolidines FP1–FP4 and the pristine C60 in chloroform. | |
Table 1 Redox properties, energy gaps, LUMO and HOMO energy levels of fullerenes FP1–FP4, PCBM and the pristine C60
| Parameters |
C60 |
PCBM |
FP1 |
FP2 |
FP3 |
FP4 |
| Half-wave potential. Onset reduction potential. LUMO = −e(Eredonset + 4.60). Values from the DFT calculation. Onset absorption wavelength. Band gap = hc/λonset, converted [J] to [eV]; h, Planks constant; c, speed of light. HOMO = LUMO − Egap [eV]. Taken from ref. 55. |
| E1a [V] |
−0.75 |
−0.90h |
−0.84 |
−0.85 |
−0.85 |
−0.83 |
| E2a [V] |
−1.14 |
−1.31h |
−1.24 |
−1.24 |
−1.25 |
−1.23 |
| E3a [V] |
−1.60 |
−1.85h |
−1.77 |
−1.77 |
−1.78 |
−1.76 |
| Eredonsetb [V] |
−0.68 |
−0.77h |
−0.77 |
−0.78 |
−0.78 |
−0.78 |
| LUMOc [eV] |
−3.92 |
−3.83 |
−3.83 |
−3.82 |
−3.82 |
−3.82 |
| LUMOd [eV] |
−4.18 |
−4.00 |
−4.02 |
−4.01 |
−4.01 |
−4.01 |
| λonsete [nm] |
693 |
723 |
724 |
724 |
724 |
724 |
| Egapf [eV] |
1.79 |
1.72 |
1.72 |
1.72 |
1.72 |
1.72 |
| Egapd [eV] |
1.69 |
1.52 |
1.50 |
1.50 |
1.50 |
1.50 |
| HOMOg [eV] |
−5.71 |
−5.55 |
−5.55 |
−5.54 |
−5.54 |
−5.54 |
| HOMOd [eV] |
−5.87 |
−5.52 |
−5.52 |
−5.51 |
−5.51 |
−5.51 |
3.2. Electrochemical properties
It is well-known that fullerenes have rich electrochemical property and can accept up to six electrons in solution.50,51 The first reduction potentials of fullerene acceptors were usually used to estimate their lowest unoccupied molecular orbital (LUMO) energy, which determines the open circuit voltage performance of polymer solar cells together with the highest occupied molecular orbital (HOMO) energy levels of electron donor.52–54 We therefore measured the cyclic voltammograms of FP1–FP4 and the pristine C60 for a comparison. As shown in Fig. 2, a series of cyclic voltammograms shows that three well-defined and reversible redox waves are retained in the functionalized fullerenes ranging from 0 to −2.6 V vs. Ag/Ag+, under room temperature and anhydrous air-free conditions, relative to the ferrocene/ferrocenium (Fc/Fc+) internal standard. As shown in Fig. 3, three sharp and equal intensity peaks together with the noticeable fourth peaks were clearly observed from differential pulse voltammetry (DPV), further revealing that FP1–FP4 are typical redox-reversible fullerene derivatives. From the CV, it can be noted that three reversible redox peaks of all derivatives FP1–FP4 are all shifted to negative potentials by ca. 100 mV as compared to those of C60, an indication of the stronger electron acceptor. The measured half-wave potentials are listed in Table 1 together with the LUMO energy levels of the fullerene derivatives, estimated from the their onset reduction potentials (Eredonset) according to the equation, LUMO (eV) = −e(Eredonset + 4.60).55 The Eredonset of FP1–FP4 were −0.77, −0.78, −0.78, and −0.78 V, corresponding to the LUMO energy levels of −3.83, −3.82, −3.82, and −3.82 eV, respectively (Table 1), indicating the alkyl chain length has a little effect on their electrochemical properties. The LUMO energy levels of FP1–FP4 are ca. 0.1 eV higher than that of C60 (−3.92 eV), which is desirable for an electron acceptor in polymer solar cells to get higher open-circuit voltage.52–54 The redox properties, LUMO, HOMO and energy gap (Egap) of all derivatives are similar and also very close to those of PCBM (Table 1),55 an indicative of FP1–FP4 are PCBM-like electron acceptors.
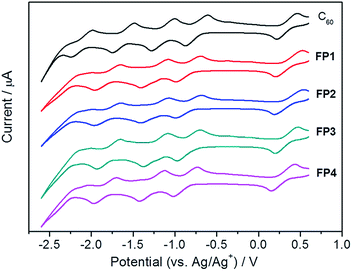 |
| | Fig. 2 CV curves of fulleropyrrolidine derivatives FP1–FP4 and the pristine C60 in o-DCB. | |
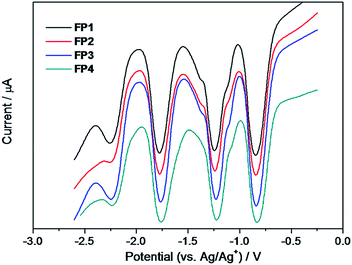 |
| | Fig. 3 DPV curves of fulleropyrrolidine derivatives FP1–FP4 in o-DCB. The parameters of DPV: amplitude, 0.05 V; pulse width, 0.2 s; sample width, 0.02 s; pulse period, 0.5 s. | |
3.3. DFT calculations
To further understand the substituent effect on the LUMO–HOMO energy levels of fullerene acceptors, DFT calculations were performed by using the Gaussian 09 program package.47 Ground state geometrical structures of four fulleropyrrolidine derivatives FP1–FP4, PCBM and C60 were optimized at the B3LYP/6-31G(d) level, and the molecular orbital energies were calculated at the more reliable PBEPBE/6-311G(d,p) level based on above optimized geometries.46,49,56,57 The calculated results are summarized in Table 1, and the schematic diagrams of HOMO and LUMO levels are shown in Fig. 4. It can be noted that the HOMO–LUMO energy gaps of fulleropyrrolidine derivatives FP1–FP4 reduced to 1.72 eV by comparison with 1.79 eV of C60, while their LUMO and HOMO energies are 0.17 and 0.35 eV higher than those of C60 (−4.18 and −5.87 eV), respectively (Fig. 4 and Table 1), which may explain the red-shifted absorption of substituted fullerenes. The calculated orbital energies of derivatives FP1–FP4 are nearly same as those of PCBM and also close to the experimental values (Table 1), confirming the PBE method a reliable in orbital energy calculation for fullerene systems. In addition, other molecular properties, such as molecular hardness, electrophilicity, electronegativity, etc., have also been provided (Table S2, ESI†). Considering the molecular hardness, large value means a hard molecule with less global reactivity. It was noted that the fulleropyrrolidine derivatives FP1–FP4 have smaller hardness values (ca. 0.75 eV) than that of C60 (ca. 0.84 eV), revealing they are more reactive. Orbital analysis of HOMO and LUMO were also carried out for FP1–FP4, PCBM and C60 at the PBEPBE/6-311G(d,p) level (Fig. 5). The fulleropyrrolidine derivatives FP1–FP4, PCBM and C60 showed very similar orbital distributions, in which both the LUMO and HOMO are mainly located on the fullerene cage, respectively. But it can still be noted that the electron delocalization for fullerene derivatives are slightly weakened relative to C60, which has been attributed to symmetry breaking of C60 molecule by functionalization.49,56 It should be noted that C60 has 3-fold degenerated LUMOs and 5-fold degenerated HOMOs (Fig. S1, ESI†), but the LUMO and HOMO of fulleropyrrolidine derivatives FP1–FP4 become non-degenerated (Table S3 and Fig. S2, ESI†). Further orbital analysis for other occupied and vacant MOs showed that the highest occupied and lowest vacant MOs localized on phenyl substituent are energetically far from the HOMO and LUMO of FP1–FP4 (Fig. S3–S6 and Table S3, ESI†), suggesting a little contribution to the frontier MOs. These findings suggest that the hybridization change (from sp2 to sp3) of C60 carbon atoms involved in connection with substituent, which modified the localization and the nature of HOMO and LUMO, also contribute to the observed substituent effect. The similar orbital distribution of fullerene derivatives may explain the similar electronic and electrochemical properties observed in absorption spectra and CV.
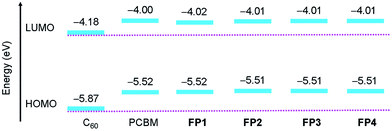 |
| | Fig. 4 Schematic diagrams of HOMO and LUMO energy levels of C60, PCBM, and fulleropyrrolidine derivatives FP1–FP4 calculated at PBEPBE/6-311G(d,p) level. | |
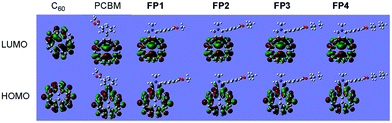 |
| | Fig. 5 LUMO (top) and HOMO (bottom) contours for C60, PCBM, and fulleropyrrolidine derivatives FP1–FP4 calculated at PBEPBE/6-311G(d,p) level. | |
3.4. Self-assembly
To demonstrate how the small alkyl benzoate ester substituent groups affect the packing of C60 molecules in solid state, the self-organized objects of FP1–FP4 were further investigated by SEM, TEM and XRD. The self-organized objects were prepared by mixing the chloroform solution of FP1–FP4 (1 mg ml−1) with 2-propanol or ethanol (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, v/v), then dropping on Si wafers and evaporating the solvent in a capped petri dish up to 1 h until dryness at room temperature. The assembled objects on Si wafers were directly used for SEM or XRD characterization, while they were scratched off and dispersed in EtOH for a TEM. Fig. 6 shows SEM and TEM images of self-organized architectures formed from FP1–FP4 derivatives. It can be noted that flowerlike objects with size distribution around 2–8 μm were consisted of plate nanostructures (Fig. 6a–e), which was also confirmed by TEM (Fig. 6f). To get information on molecular packing, the bulk flowerlike objects obtained from FP1–FP4 on Si wafer were further investigated by XRD (Fig. 7). As shown in Fig. 7, one intense diffraction peak appeared at around 5° accompanied by several weaker peaks at higher angle region in FP1–FP4 derivatives, resembling those previously observed in others fulleropyrrolidines.42–45,58 These peaks are therefore similarly attributed to the reflections of (001), (002) and (003) planes with a d-spacing value of 1.92, 1.95, 1.98 and 2.02 nm for FP1, FP2, FP3, and FP4, respectively, suggesting a lamellar structure. In addition, by comparison with the patterns obtained from the pristine C60, the residue peaks can be readily assigned to the reflections of (111), (220), (311) and (222) planes of a fcc structure with the average spacing of 0.8 nm (as indicated by asterisks in Fig. 7), suggesting a π–π (C60/C60) interaction in the assemblies. By taking the molecular size of 1.3–1.5 nm of FP1–FP4 obtained from the DFT calculation, the d-spacing of 1.92–2.02 nm that depends their molecular sizes, corresponds well to the thickness of a bilayer structure, where a face-to-face conformation of the substituent of C60 and an interdigitation of the bare C60 side packed as suggested before.7,13,45,58 These observations suggest that the smaller grafted group insufficiently disturbing the strong π–π interactions between adjacent C60 molecules,13 especially the strongest peaks from fcc packing were clearly noticed in the case of FP1 (Fig. 7).
:1, v/v), then dropping on Si wafers and evaporating the solvent in a capped petri dish up to 1 h until dryness at room temperature. The assembled objects on Si wafers were directly used for SEM or XRD characterization, while they were scratched off and dispersed in EtOH for a TEM. Fig. 6 shows SEM and TEM images of self-organized architectures formed from FP1–FP4 derivatives. It can be noted that flowerlike objects with size distribution around 2–8 μm were consisted of plate nanostructures (Fig. 6a–e), which was also confirmed by TEM (Fig. 6f). To get information on molecular packing, the bulk flowerlike objects obtained from FP1–FP4 on Si wafer were further investigated by XRD (Fig. 7). As shown in Fig. 7, one intense diffraction peak appeared at around 5° accompanied by several weaker peaks at higher angle region in FP1–FP4 derivatives, resembling those previously observed in others fulleropyrrolidines.42–45,58 These peaks are therefore similarly attributed to the reflections of (001), (002) and (003) planes with a d-spacing value of 1.92, 1.95, 1.98 and 2.02 nm for FP1, FP2, FP3, and FP4, respectively, suggesting a lamellar structure. In addition, by comparison with the patterns obtained from the pristine C60, the residue peaks can be readily assigned to the reflections of (111), (220), (311) and (222) planes of a fcc structure with the average spacing of 0.8 nm (as indicated by asterisks in Fig. 7), suggesting a π–π (C60/C60) interaction in the assemblies. By taking the molecular size of 1.3–1.5 nm of FP1–FP4 obtained from the DFT calculation, the d-spacing of 1.92–2.02 nm that depends their molecular sizes, corresponds well to the thickness of a bilayer structure, where a face-to-face conformation of the substituent of C60 and an interdigitation of the bare C60 side packed as suggested before.7,13,45,58 These observations suggest that the smaller grafted group insufficiently disturbing the strong π–π interactions between adjacent C60 molecules,13 especially the strongest peaks from fcc packing were clearly noticed in the case of FP1 (Fig. 7).
 |
| | Fig. 6 SEM (a–e) and TEM (f) images of self-organized architectures of FP1–FP4 obtained from the mixture of chloroform–alcohol. (a) FP1 from chloroform–2-propanol; (b) FP2 from chloroform–2-propanol; (c) FP3 from chloroform–2-propanol; (d) FP4 from chloroform–ethanol; (e) and (f) FP4 from chloroform–2-propanol. | |
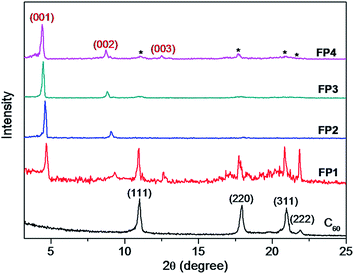 |
| | Fig. 7 XRD patterns of flowerlike objects of FP1–FP4 obtained from the mixture of chloroform–2-propanol. | |
4. Conclusions
In conclusion, a series of small alkyl (methyl, ethyl, propyl, butyl) benzoate ester group substituted fulleropyrrolidine derivatives (FP1–FP4) were synthesized and their electronic and electrochemical properties as well as self-assembly were investigated by means of absorption spectra, DFT calculation, CV, SEM, TEM and XRD, respectively. The LUMO–HOMO energies of fullerene derivatives were estimated by the first reduction potential measured with CV combined with absorption spectra, which are in consistent with those obtained from DFT calculations. All fulleropyrrolidines FP1–FP4 showed the very similar absorption spectra, orbital energies and redox behaviors, which are comparable with those of well-known PCBM, indicating alkyl chain length has little effect on their electronic and electrochemical properties. The SEM investigation of self-assembly of FP1–FP4 in chloroform–alcohol solution mixture solvents showed that flowerlike supramolcular architectures can be facilely achieved. XRD analysis revealed that these flowerlike objects were lamellar structures with the d-spacing of 1.92–2.02 nm that depends on the molecular size of FP1–FP4, corresponding well to the thickness of a bilayer structure. This suggested a face-to-face conformation of the substituent of C60 and an interdigitation of the bare C60 side packing fashion. The fulleropyrrolidines FP1–FP4 are high C60 content (76–79%), energetically PCBM-like, and capable of forming complex flowerlike architectures, which provide fundamental insights into molecular design toward advanced fullerene materials and may find potential application in optoelectronics.
Acknowledgements
This work was financially supported by Innovation Program of Shanghai Municipal Education Commission (12ZZ067), Shanghai Pujiang Program (11PJ1400200), the Research Fund for the Doctoral Program of Higher Education of China (20120075120018), and the Fundamental Research Funds for the Central Universities.
Notes and references
- C. Yang, J. Y. Kim, S. Cho, J. K. Lee, A. J. Heeger and F. Wudl, J. Am. Chem. Soc., 2008, 130, 6444–6450 CrossRef CAS PubMed.
- J. L. Segura, N. Martín and D. M. Guldi, Chem. Soc. Rev., 2005, 34, 31–47 RSC.
- D. M. Guldi, B. M. Illescas, C. M. Atienza, M. Wielopolskia and N. Martín, Chem. Soc. Rev., 2009, 38, 1587–1597 RSC.
- C.-Z. Li, H. L. Yip and A. K. Y. Jen, J. Mater. Chem., 2012, 22, 4161–4177 RSC.
- Y. J. He and Y. F. Li, Phys. Chem. Chem. Phys., 2011, 13, 1970–1983 RSC.
- Y.-Y. Lai, Y.-J. Cheng and C. S. Hsu, Energy Environ. Sci., 2014, 7, 1866–1883 CAS.
- M. J. Hollamby, M. Karny, P. H. H. Bomans, N. A. J. M. Sommerdjik, A. Saeki, S. Seki, H. Minamikawa, I. Grillo, B. R. Pauw, P. Brown, J. Eastoe, H. Möhwald and T. Nakanishi, Nat. Chem., 2014, 6, 690–696 CrossRef CAS.
- H.-G. Li, J. Choi and T. Nakanishi, Langmuir, 2013, 29, 5394–5406 CrossRef CAS PubMed.
- Y. Yamamoto, G. Zhang, W. Jin, T. Fukushima, N. Ishii, A. Saeki, S. Seki, S. Tagawa, T. Minari, K. Tsukagoshi and T. Aida, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 21051–21056 CrossRef CAS PubMed.
- V. Georgakilas, F. Pellarini, M. Prato, D. M. Guldi, M. Melle-Franco and F. Zerbetto, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 5075–5080 CrossRef CAS PubMed.
- L. Sánchez, R. Otero, J. María Gallego, R. Miranda and N. Martín, Chem. Rev., 2009, 109, 2081–2091 CrossRef PubMed.
- T. Nakanishi, Chem. Commun., 2010, 46, 3425–3436 RSC.
- H.-G. Li, S. S. Babu, S. T. Turner, D. Neher, M. J. Hollamby, T. Seki, S. Yagai, Y. Deguchi, H. Möhwald and T. Nakanishi, J. Mater. Chem. C, 2013, 1, 1943–1951 RSC.
- H. Asanuma, H.-G. Li, T. Nakanishi and H. Möhwald, Chem.–Eur. J., 2010, 16, 9330–9338 CrossRef CAS PubMed.
- P. Heremans, D. Cheyns and B. P. Rand, Acc. Chem. Res., 2009, 42, 1740–1747 CrossRef CAS PubMed.
- A. J. Moulé and K. Meerholz, Adv. Funct. Mater., 2009, 19, 3028–3036 CrossRef.
- L. K. Shrestha, Q. M. Ji, T. Mori, K. Miyazawa, Y. Yamauchi, J. P. Hill and K. Ariga, Chem.–Asian J, 2013, 8, 1662–1679 CrossRef CAS PubMed.
- S. S. Babu, H. Möhwald and T. Nakanishi, Chem. Soc. Rev., 2010, 39, 4021–4035 RSC.
- Y.-F. Shen and T. Nakanishi, Phys. Chem. Chem. Phys., 2014, 16, 7199–7204 RSC.
- X. Zhang and M. Takeuchi, Angew. Chem., Int. Ed., 2009, 48, 9646–9651 CrossRef CAS PubMed.
- L. Wang, B. Liu, D. Liu, M. Yao, Y. Hou, S. Yu, T. Cui, D. Li, G. Zou, A. Iwasiewicz and B. Sundqvist, Adv. Mater., 2006, 18, 1883–1886 CrossRef CAS.
- J. Geng, W. Zhou, P. Skelton, W. Yue, I. A. Kinloch, A. H. Windle and B. F. G. Johnson, J. Am. Chem. Soc., 2008, 130, 2527–2534 CrossRef CAS PubMed.
- X. Zhang and X. D. Li, Chin. Chem. Lett., 2014, 25, 912–914 CrossRef CAS PubMed.
- L. Wei, J. Yao and H. Fu, ACS Nano, 2013, 7, 7573–7582 CrossRef CAS PubMed.
- K. Miyazawa, Y. Kuwasaki, A. Obayashi and M. Kuwabara, J. Mater. Res., 2002, 17, 83–88 CrossRef CAS.
- M. H. Nurmawati, P. K. Ajikumar, R. Renu, C. H. Sow and S. Valiyaveettil, ACS Nano, 2008, 2, 1429–1436 CrossRef CAS PubMed.
- M. Sathish, K. Miyazawa, J. P. Hill and K. Ariga, J. Am. Chem. Soc., 2009, 131, 6372–6373 CrossRef CAS PubMed.
- C. Park, H. J. Song and H. C. Choi, Chem. Commun., 2009, 4803–4805 RSC.
- D. Mi, H.-U. Kim, J.-H. Kim, F. Xu, S.-H. Jin and D. H. Hwang, Synth. Met., 2012, 162, 483–489 CrossRef CAS PubMed.
- H. Yu, H.-H. Cho, C.-H. Cho, K.-H. Kim, D. Y. Kim, B. J. Kim and J. H. Oh, ACS Appl. Mater. Interfaces, 2013, 5, 4865–4871 CAS.
- Y. J. He, H.-Y. Chen, J. H. Hou and Y. F. Li, J. Am. Chem. Soc., 2010, 132, 1377–1382 CrossRef CAS PubMed.
- C.-Z. Li, S.-C. Chien, H.-L. Yip, C.-C. Chueh, F.-C. Chen, Y. Matsuo, E. Nakamura and A. K. Y. Jen, Chem. Commun., 2011, 47, 10082–10084 RSC.
- K.-H. Kim, H. Kang, S. Y. Nam, J. Jung, P. S. Kim, C.-H. Cho, C. Lee, S. C. Yoon and B. J. Kim, Chem. Mater., 2011, 23, 5090–5095 CrossRef CAS.
- I. Riedel, E. von Hauff, J. Parisi, N. Martin, F. Giacalone and V. Dyakonov, Adv. Funct. Mater., 2005, 15, 1979–1987 CrossRef CAS.
- N. Nakashima, T. Ishii, M. Shirakusa, T. Nakanishi, H. Murakami and T. Sagara, Chem.–Eur. J., 2001, 7, 1766–1772 CrossRef CAS.
- A. M. Cassell, C. L. Asplund and J. M. Tour, Angew. Chem., Int. Ed., 1999, 38, 2403–2405 CrossRef CAS.
- J.-F. Nierengarten, New J. Chem., 2004, 28, 1177–1191 RSC.
- X. Y. Meng, Q. Xu, W. Q. Zhang, Z. A. Tan, Y. F. Li, Z. X. Zhang, L. Jiang, C. Y. Shu and C. R. Wang, ACS Appl. Mater. Interfaces, 2012, 4, 5966–5973 CAS.
- A. L. Rosa, K. Gillemot, E. Leary, C. Evangeli, M. T. González, S. Filippone, G. Rubio-Bollinger, N. Agraït, C. J. Lambert and N. Martín, J. Org. Chem., 2014, 79, 4871–4877 CrossRef PubMed.
- X. Y. Meng, G. Y. Zhao, Q. Xu, Z. A. Tan, Z. X. Zhang, L. Jiang, C. Y. Shu, C. R. Wang and Y. F. Li, Adv. Funct. Mater., 2014, 24, 158–163 CrossRef CAS.
- T. Homma, K. Harano, H. Isobe and E. Nakamura, J. Am. Chem. Soc., 2011, 133, 6364–6370 CrossRef CAS PubMed.
- T. Nakanishi, W. Schmitt, T. Michinobu, D. G. Kurth and K. Ariga, Chem. Commun., 2005, 5982–5984 RSC.
- T. Nakanishi, Y.-F. Shen, J.-B. Wang, H.-G. Li, P. Fernandes, K. Yoshida, S. Yagai, M. Takeuchi, K. Ariga, D. G. Kurth and H. Möhwald, J. Mater. Chem., 2010, 20, 1253–1260 RSC.
- T. Nakanishi, K. Ariga, T. Michinobu, K. Yoshida, H. Takahashi, T. Teranishi, H. Möhwald and D. G. Kurth, Small, 2007, 3, 2019–2023 CrossRef CAS PubMed.
- X. Zhang, T. Nakanishi, T. Ogawa, A. Saeki, S. Seki, Y. F. Shen, Y. Yamauchi and M. Takeuchi, Chem. Commun., 2010, 46, 8752–8754 RSC.
- X. Zhang, L. X. Ma and X. D. Li, Synth. Met., 2014 DOI:10.1016/j.synthmet.2014.09.030.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, B. Mennucci, G. A. Petersson, H. Nakatsuji, M. Caricato, X. Li, H. P. Hratchian, A. F. Izmaylov, J. Bloino, G. Zheng, J. L. Sonnenberg, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, J. A. Montgomery Jr, J. E. Peralta, F. Ogliaro, M. Bearpark, J. J. Heyd, E. Brothers, K. N. Kudin, V. N. Staroverov, T. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, N. Rega, J. M. Millam, M. Klene, J. E. Knox, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, R. L. Martin, K. Morokuma, V. G. Zakrzewski, G. A. Voth, P. Salvador, J. J. Dannenberg, S. Dapprich, A. D. Daniels, O. Farkas, J. B. Foresman, J. V. Ortiz, J. Cioslowski and D. J. Fox, Gaussian 09, Revision C.01, Gaussian, Inc., Wallingford CT, 2010 Search PubMed.
- M. Maggini, G. Scorrano and M. Prato, J. Am. Chem. Soc., 1993, 115, 9798–9799 CrossRef CAS.
- H. Wang, Y. J. He, Y. F. Li and H. M. Su, J. Phys. Chem. A, 2012, 116, 255–262 CrossRef CAS PubMed.
- T. Suzuki, Y. Maruyama, T. Akasaka, W. Ando, K. Kobayashi and S. Nagasel, J. Am. Chem. Soc., 1994, 116, 1359–1363 CrossRef CAS.
- M. Carano, T. D. Ros, M. Fanti, K. Kordatos, M. Marcaccio, F. Paolucci, M. Prato, S. Roffia and F. Zerbetto, J. Am. Chem. Soc., 2003, 125, 7139–7144 CrossRef CAS PubMed.
- C. J. Brabec, A. Cravino, D. Meissner, N. S. Sariciftci, T. Fromherz, M. T. Rispens, L. Sanchez and J. C. Hummelen, Adv. Funct. Mater., 2001, 11, 374–380 CrossRef CAS.
- M. C. Scharber, D. Mühlbacher, M. Koppe, P. Denk, C. Waldauf, A. J. Heeger and C. J. Brabec, Adv. Mater., 2006, 18, 789–794 CrossRef CAS.
- F. B. Kooistra, J. Knol, F. Kastenberg, L. M. Popescu, W. Verhees, J. H. Kroon and J. M. Hummelen, Org. Lett., 2007, 9, 551–554 CrossRef CAS PubMed.
- G. D. Han, W. R. Collins, T. L. Andrew, V. Bulović and T. M. Swager, Adv. Funct. Mater., 2013, 23, 3061–3069 CrossRef CAS.
- X. Zhang and X. D. Li, Chin. Chem. Lett., 2014, 25, 501–504 CrossRef CAS PubMed.
- The comparison of the HOMO and LUMO energies as well as energy gaps of C60 and PCBM calculated with various basis sets and experimental values were listed in Table S1 (ESI†). It showed that the PBEPBE/6-311G(d,p) basis set is more reliable than B3LYP/6–311G(d,p) for energy calculation based on geometry optimized with B3LYP/6–31G(d). Energies calculated from PBEPBE/6-311G(d,p) based on geometry optimized at the same level and at B3LYP/6–31G(d) level exhibited the comparable value but the former is more cost in computation time. Therefore, we selected the B3LYP/6-31G(d) level for geometry optimization but the PBEPBE/6-311G(d,p) for energy calculation by considering both reliability and computation cost.
- S. S. Babu, A. Saeki, S. Seki, H. Möhwald and T. Nakanishi, Phys. Chem. Chem. Phys., 2011, 13, 4830–4834 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Data of additional computation results for C60 and PCBM at various basis sets, other molecular properties (ionization potential, electro affinity, electronegativity, global hardness, etc.) and MOs for fullerenes are available. See DOI: 10.1039/c4ra10654g |
|
| This journal is © The Royal Society of Chemistry 2014 |