Nanoconjugation: a materials approach to enhance epidermal growth factor induced apoptosis†
Linxi
Wu
,
Xinwei
Yu
,
Amin
Feizpour
and
Björn M.
Reinhard
*
Department of Chemistry and the Photonics Center, Boston University, Boston, MA 02215, USA. E-mail: bmr@bu.edu; Fax: +16173536466; Tel: +16173538669
First published on 9th October 2013
Abstract
Apoptosis evasion is a hallmark of cancer that motivates the development of novel strategies for inducing cell death in a controlled fashion. The size-compatibility of nanoparticles (NPs) with cellular components provides new opportunities for regulating cellular processes, potentially including apoptosis. We investigated the impact of the covalent attachment of epidermal growth factor (EGF) to 40 nm diameter Au NPs on cellular apoptosis levels, quantified as caspase-3 activity, in two in vitro cancer cell lines: A431 and HeLa. Our studies show that nanoconjugation enhances EGF-induced apoptosis in EGF receptor (EGFR) overexpressing A431 and triggers a quantifiable increase in apoptosis in HeLa. The latter has physiological receptor expression levels and does not show apoptosis in response to free EGF. Endocytosis and trafficking are involved in key EGFR regulation processes, most prominently signal termination. Our experimental findings indicate that these processes can be manipulated through nanoconjugation to induce apoptosis.
1. Introduction
Nanoconjugation, i.e. the covalent attachment of a biological functionality to a nanoparticle (NP), has been shown to influence endocytosis and trafficking of the tethered species.1 Given the role of these two cellular mechanisms in many signaling processes, it is important to understand the extent to which the function of signaling molecules can be modulated through nanoconjugation. We investigate this question in the case of the epidermal growth factor (EGF). The EGF receptor (EGFR) is a receptor tyrosine kinase (RTK) that plays a crucial role in normal cell and tissue growth but whose dysregulation can cause cancer.2 The EGFR signaling cascade is initiated by a ligand binding induced conformational change of the receptor, which results in the formation of dimers and potentially larger assemblies of the transmembrane protein.3–6 The kinase activity located in the cytoplasmic tail of EGFR facilitates a cross-phosphorylation of tyrosine residues in receptor dimers and can, thus, trigger a variety of intracellular signaling pathways after receptor activation. An integral part of the normal EGFR signaling process is the growth factor induced down-regulation of the receptor due to elevated endocytosis and degradation of the activated EGFR.2,7–11 Under physiological conditions (low ligand concentrations and low/moderate expression levels of the receptor), EGF and EGFR accumulate in early endosomal compartments after 2–5 min of continuous endocytosis.8,12–14 The early endosomes are highly dynamic and fuse into larger endosomes as part of their maturation process.15 After 15–20 min of continuous EGF-induced endocytosis, EGF and EGFR accumulate in the intraluminal membranes of multivesicular membrane bodies that are mostly located in the perinuclear area of the cell. The sequestration of the activated EGF*EGFR complex into intraluminal vesicles has been shown to represent a key step in signal termination since it inhibits interactions of the carboxyl terminus of the transmembrane protein with signal transducers in the cytoplasm.16 There is strong experimental evidence that a disruption of the temporal and spatial regulation of EGFR trafficking has dramatic consequences for the signaling outcome.7,17–19 In fact, the puzzling observation that EGFR signaling is not exclusively linked to promoting cell proliferation and differentiation but can also induce apoptosis in EGFR overexpressing cells20–23 has been linked to the enrichment of activated EGFR in the limiting membrane of early endosomes where the receptor can continue to signal.16,24The mechanism of endocytosis and subsequent trafficking of nanoconjugated biomolecules is expected to depend on both the nature of the biomolecule and NP related parameters (size, shape, surface composition and charge, and ligand presentation). Consequently, careful design of the NPs provides some leverage for manipulating the cellular response to the tethered functionality.25–28 In the case of an anti-EGFR antibody (AB), nanoconjugation has been shown to impact the uptake rate and route of the AB*EGFR complex and to achieve an enrichment of the NP-EGFR complex in early endosomes and the Golgi apparatus.1 We hypothesized that a similar enrichment of activated EGF*EGFR in the endosomal pathway through nanoconjugation of EGF would effectively delay the downregulation of EGFR signaling through lysosomal degradation and, thus, result in an enhanced induction of apoptosis. In this manuscript we, therefore, quantified the impact of nanoconjugation on the apoptotic efficacy of EGF. Our findings confirm an increase in apoptosis after EGF nanoconjugation in human epithelial carcinoma cell lines with both high (A431) and physiological (HeLa) EGFR expression levels. The experimental finding that covalent nanoconjugation enhances the apoptotic efficacy of the tethered ligand, in this case EGF, emphasizes the value of nanoparticle-based strategies not only for achieving an efficient delivery of small molecules to the tumor site29–32 but also for controlling their biological function.
2. Experimental section
2.1 Materials
5 nm and 40 nm citrate stabilized Au colloid (Ted Pella); HS–(CH2)11–(CH2CH2O)6–OCH2–COOH (PEG1) (ProChimia); HS–CH2CH2–(CH2CH2O)77–N3 (PEG2) (NANOCS); HS–CH2CH2–(CH2CH2O)54–N3 (PEG3) (NANOCS); HS–CH2CH2–(CH2CH2O)16–Biotin (PEG4) (NANOCS); propargyl dPEG–NHS ester (Quanta Biodesign); recombinant human epidermal growth factor (Life Technologies); β-nicotinamide adenine dinucleotide 2′-phosphate reduced tetrasodium salt hydrate (Life Technologies); NeutrAvidin (Thermo Scientific); L-ascorbic acid (Aldrich); copper(II) sulfate pentahydrate (Aldrich); nitric acid (EMD); lysis buffer (Cell Signaling Technology) and hydrochloric acid (Fisher) for ICP-MS. Zeba™ spin desalting columns (7 K MWCO) from ThermoScientific and D-Tube dialyzers (3.5 K MWCO) from EMD were used for protein purification. Amicon Ultra centrifugal filter units (100 kDa MWCO) from Millipore were used to purify 5 nm pegylated Au NPs. An EGF human ELISA kit and an EGFR [pY1068] ELISA kit from Life Technologies were used for EGF surface density and EGFR phosphorylation determination. An EnzChek® Caspase-3 Assay Kit and a Vybrant™ Apoptosis Kit #2 from Life Technologies were used as apoptosis assays. A LIVE/DEAD® Viability/Cytotoxicity Kit from Life Technologies was used to show cell viability. NucBlue® Live Ready Probe™ Reagent and Lyso Tracker Deep Red from Life Technologies were used to stain lysosomes and cell nuclei, respectively.2.2 Particle functionalization with EGF
PEG1 and PEG2 were added to 4 mL of commercial 40 nm Au NP (∼9 × 1010 particles per mL) colloid to yield a concentration of 25 μM for each PEG and then incubated overnight at room temperature. The pegylated Au NPs were washed three times by centrifugation (5.500 × 103 rpm, 10 min) and resuspension in distilled de-ionized (ddi) water. After the last centrifugation step, the NPs were resuspended in 20 μL of 0.5× PBS (∼4.5 × 1012 particles per mL). 2 μL of a 100 mg mL−1 propargyl-PEG-NHS ester solution in DMSO were added to 100 μL of a 1 mg mL−1 solution of EGF in 1× PBS, pH 7.4, and incubated at 4 °C for 6 h. This mixture was then dialyzed against 0.5× PBS for at least 24 h. 100 μL of the obtained propargyl-PEG-EGF were then incubated overnight at 4 °C with 80 μL of the pegylated AuNPs in 0.5× PBS buffer with a total volume of 800 μL that also contained 500 μM ascorbic acid and 100 μM CuSO4. The resulting NP-EGF particles were washed five times by centrifugation (4.600 × 103 rpm, 10 min) with ddi water. After the final centrifugation step, the cleaned NP-EGF constructs were resuspended in 0.5× PBS.2.3 EGF quantification with ELISA
PEG1 (20 μL, 10 mM) was mixed with NP-EGF (20 μL, 6 × 1011 particles per mL) in 1× PBS, pH 7.4 at 45 °C for 2 days and the mixture was sonicated every 12 h to achieve efficient replacement of surface bound ligands by PEG1. After that, the particles were spun down and the supernatant was collected. Zeba™ spin desalting columns were applied to clean the collected EGF and to remove excess PEG1. The purified EGF was diluted by a factor of 50 with 1× PBS. The ELISA quantification of EGF requires a standard of known concentration. We used PEG2–EGF conjugates for that purpose. Solutions of propargyl-PEG-EGF (10 μL, 66 μM) and PEG2 (90 μL, 7.3 mM), both in 0.5× PBS, were mixed in the presence of ascorbic acid (500 μM) and CuSO4 (100 μM) and then incubated overnight at 4 °C, resulting in a solution containing PEG2-EGF at a concentration of 0.4 mg mL−1 (66 μM). Zeba™ spin desalting columns were then applied to clean the PEG2-EGF construct. After that, the human EGF ELISA kit was used to determine the concentration of PEG2-EGF according to the manufacturer's protocol through the colorimetric reaction between peroxidase and tetramethylbenzidine.2.4 EGF labeling with 5 nm Au NPs
PEG3 and PEG4 were added to 1 mL of commercial 5 nm Au NP (∼5 × 1013 particles per mL) colloid to yield a concentration of 220 μM for PEG3 and 24 μM for PEG4 and then incubated overnight at room temperature. The pegylated 5 nm Au NPs were purified through 5 times filtration with 100 kDa centrifugal filters and then transferred into 1× PBS.A NP-EGF solution was incubated on a carbon type B coated gold TEM grid for 30 min and then removed. The surface was blocked with 10% bovine serum albumin (BSA) in 1× PBS on the grid for 2 h at 4 °C. After removal of the BSA solution, the surface was washed 5 times with 1× PBS. Then, a solution of a biotinylated anti-EGF antibody was incubated on the grid overnight at 4 °C. The grid was then washed 5 times with 1× PBS and a 10 μg mL−1 solution of Neutravidin containing 1% BSA in 1× PBS was incubated with the sample for 1 hour at room temperature. The sample was washed 5 times and the solution of biotinylated 5 nm GNPs (1015 particles per mL) in 1× PBS was incubated with the sample for 2 h at room temperature. A control sample was made using the same procedure, but without the anti-EGF antibody incubation step. The samples were then washed 3 times with 1× PBS (15 min each), finally thoroughly rinsed with ddi water and subsequently air-dried. The samples were imaged using a JEOL JEM 2010 high-resolution transmission electron microscope (HRTEM) with 200 kV acceleration voltage.
2.5 Cell culture
The A431 cell line was obtained from ATCC® (CRL-1555™) and was cultured in DMEM with 10% fetal bovine serum. The HeLa cell line was obtained from ATCC® (CCL-2™) and was cultured in DMEM with 5% fetal bovine serum. Cells were grown in a 5% CO2 containing atmosphere with 95% relative humidity in an incubator at 37 °C. For all experiments, cells were subcultured at a density of 1.1 × 104 cells per mL.2.6 ICP-MS quantification of cellular uptake of nanoparticles
A431 cells were plated in 6-well dishes as described above. After 24 h, the old growth medium was replaced with a fresh medium containing NP-EGF or pegylated NPs at a concentration of 0.06 nM. The cells were further incubated for 24 h before trypsinization with 1 mL of 0.25% trypsin-EDTA solution and subsequent quenching of the enzyme by the addition of 1 mL of complete medium in each well. Cells were collected by centrifugation (200g, 5 min) and were washed twice with 1× PBS. The cell densities of the samples were determined through a hemacytometer (no less than 200 cells). Aqua regia (1 mL) was added to the cells in a total volume of 10 μL to dissolve the Au NPs. The mixture was then dried overnight at 65 °C and re-dissolved in HCl solution (1 mL, 2%). After an additional 1000 fold dilution through ddi water, the samples were inserted into a VG Plasma Quad ExCell ICP-MS.2.7 EGFR phosphorylation assay
HeLa cells were cultured for 54 h to almost 100% confluency in 6-well plates and subsequently starved for 20 h before they were transferred into fresh complete growth medium containing 0.05 nM NP-EGF or pegylated NPs (no EGF). The cells were subsequently incubated under a 5% CO2 containing atmosphere with 95% relative humidity at 37 °C for 2 h or 8 h. Excess NPs were then removed through washing with copious amounts of ice-cold 1× PBS. The cells were scraped off the plates and washed twice by centrifugation with 1× PBS (300g, 5 min). 50 μL of lysis buffer with freshly added 1 mM phenylmethanesulfonyl-fluoride were then added to each cell pellet (1 × 106 cells) and incubated on ice for 30 min. The samples were vortexed every 10 min. The EGFR containing supernatant was then collected through decantation via centrifugation. The clear solutions were aliquoted to 20 μL per tube and stored at −80 °C until EGFR phosphorylation was quantified. Cell background and 1.6 nM EGF treated cells were used as controls. The EGFR [pY1068]-ELISA kit from Life Technologies was used to measure phosphorylation following the manufacturer's protocol. For each sample, the assay was performed in duplicate.2.8 Apoptosis activity quantification with the caspase-3 assay
A431 and HeLa cells were plated in 12-well dishes. After 24 h, the growth medium was replaced by fresh medium containing additional EGF or different NP preparations. After that the cells were further incubated for 1–3 days before they were trypsinized with 0.5 mL of a 0.25% trypsin-EDTA solution. After quenching the trypsinization by adding 0.5 mL of complete medium in each well, the cells were collected by centrifugation (200g, 5 min) and were washed once with 1× PBS. The cells were resuspended in 1× PBS (20 μL). After the cell density was determined using a hemacytometer (no less than 200 cells), the EnzChek® Caspase-3 assay was applied to determine apoptosis activity according to the manufacturer's protocol in a 96-well plate. Fluorescence was quantified with a Perkin Elmer 1420 Victor-3 multilabel counter using excitation and emission wavelengths of λexc = 485 nm and λem = 535 nm, respectively.2.9 Mapping of the NP-EGF spatial distribution
HeLa cells were cultured for 36 h to reach a confluency of 30%. The old medium was replaced with fresh complete growth medium containing 0.06 nM NP-EGF or pegylated NPs and further incubated under a 5% CO2 containing atmosphere with 95% relative humidity at 37 °C. Then cells were thoroughly washed with prewarmed 1× PBS buffer and 1 mL of 100 nM LysoTracker Deep Red in DMEM and 1 drop of NucBlue were added and incubated for 30 min to stain lysosomes and nuclei. Excess dyes were washed away with 1× PBS buffer and cells were then fixed with 2.5% glutaraldehyde for 15 min at room temperature before imaging in a dark-field microscope.2.10 Annexin V apoptosis assay
A431 cells were cultured in 6-well dishes as described above. After 24 h, the cells were treated with 0.06 nM NP-EGF in fresh complete growth medium for 24 h, 48 h and 72 h. 0.5 mL of 0.25% trypsin-EDTA solution was used to harvest the cells. Subsequently, the cells were washed with ice cold Dulbecco's PBS buffer and stained using the Vybrant™ Apoptosis Assay Kit #2 according to the manufacturer's protocol. Then cells were cytospun (500 rpm, 5 min) onto glass slides before inspection in the optical microscope.2.11 Characterizing the viability of detached cells
A431 cells were cultured in cell culture dishes as described above. After 24 h, the old growth medium was replaced either by fresh complete growth medium or by complete growth medium containing NP-EGF at a concentration of 0.06 nM. The cells were then incubated for 72 h. Cells that had detached from the dish bottom during that time were collected by centrifugation (500g, 5 min). Then the LIVE/DEAD® Viability/Cytotoxicity Kit from Life Technologies was used to determine the cell viability of the detached cells according to the manufacturer's protocol.2.12 Dark-field microscopy
We used an Olympus IX71 inverted microscope with a high numerical aperture (NA) oil condenser (NA = 1.2–1.4) and 10× air (NA = 0.25) or 60× oil (NA = 0.65) objective. The samples were illuminated with a 100 W tungsten lamp or a 100 W xenon lamp. Color images were taken using a Nikon D5100 digital camera attached to the microscope through an eyepiece adaptor. The relative intensities detected on the red, green, and blue color channels provided a rough spectral characterization of the cell images before and after NP uptake.2.13 Fluorescence microscopy
Fluorescence images were acquired on an Olympus IX71 inverted microscope using a 60× oil objective (NA = 1.25) in combination with appropriate filter sets for various dyes. NucBlue® (λexc = 340 nm; λem = 473 nm); LysoTracker (λexc = 647 nm; λem = 670 nm); Alexa488-labeled annexin V, calcein AM (λexc = 480 nm; λem = 510 nm); PI, EthD-1 (λexc = 530 nm; λem = 610 nm). The samples were illuminated with a 100 W mercury burner.2.14 Statistical analysis
A two-tailed Mann–Whitney U test was used to compare the two distributions. This non-parametric method compares medians and does not assume normality. U values were calculated based on non-parametric 0.95 and 0.99 confidence intervals.3. Results
3.1 Nanoconjugation of EGF
The design of the NPs used in this work is guided by the need for a NP platform that combines stability with a high degree of bioavailability for covalently attached EGF ligands. We chose Au NPs 40 nm in diameter for the EGF nanoconjugation in our studies since Au NPs show excellent biocompatibility,33–36 have exceptional photophysical properties,37–45 and are easy to quantify in biological tissue using inductively coupled mass spectrometry (ICP-MS).46,47 Furthermore, NPs of this diameter are close to the predicted optimum size for receptor mediated endocytosis.25–27,48,49 We grafted two different polyethylene glycol (PEG) molecules (HS–(CH2)11–(C2H4O)6–COOH, PEG1) and (HS–CH2CH2–(C2H4O)77–N3, PEG2) onto the NP surface (Fig. 1). Our goal was to create a brush of – under physiological conditions – negatively charged PEG1 molecules interspersed with azide-containing PEG2 molecules, which can be crosslinked with EGF peptides through the CuI catalyzed 1,3-dipolar cycloaddition reaction.50 We chose PEG2 to be much longer than PEG1 to ensure that the tethered EGF was readily bioavailable and not covered by the PEG1 brush. Furthermore, the long tether length provides a high degree of configurational flexibility and is, therefore, expected to limit the detrimental effect of nanoconjugation on the binding affinity of EGF to its cellular receptor. To generate EGF-functionalized NPs, PEG1 and PEG2 were first bound to the NPs by incubating citrate stabilized 40 nm diameter Au NPs with PEG1 and PEG2 (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 molar ratio of PEG1 and PEG2). Alkyne-functionalized EGF was then crosslinked to NP-tethered PEG2 through the click reaction. The hydrodynamic diameter (dhyd) increased in this step from 48.4 ± 0.6 nm for the pegylated NPs to 56.1 ± 2.5 nm for the EGF functionalized NPs, which we will refer to as NP-EGF in the following. For simple mixtures of pegylated NPs and EGF (no covalent attachment) the measured hydrodynamic diameter (dhyd = 47.8 ± 1.3 nm) was nearly identical to that of the pegylated NPs. We determined the average number of EGF molecules bound per NP through an ELISA assay. To this end, we first removed the PEG2-EGF construct from the purified NP-EGF particles by incubation in a large excess of PEG1 at 45 °C for 48 h with sonicating every 12 h and then quantified its concentration relative to a PEG2-EGF calibration solution colorimetrically. We found that on average each NP carries ∼4 EGF peptides under the chosen experimental conditions. The ELISA results were consistent with TEM studies, in which we visualized the EGF molecules on individual NP-EGF particles after labeling with biotinylated anti-EGF antibodies and Neutravidin functionalized 5 nm diameter Au NPs (Fig. 2a–f). The average number of 5 nm NP labels detected per 40 nm core was 2.7 (maximum number 12) in the case of NP-EGF, while the binding to the control was negligible (Fig. 2g). Approximately 90 Au cores were evaluated for each condition. Based on geometric considerations, we estimate that approximately 45% of all bound 5 nm NP labels are detectable in conventional TEM. The remaining NP labels are located in areas where the diameter of the smaller NP is entirely blocked by the larger core so that the contrast is insufficient for their detection. Taking this effect into account, one can anticipate that the average number of bound 5 nm Au labels is around 6, which is in good agreement with the ELISA assay.
:1 molar ratio of PEG1 and PEG2). Alkyne-functionalized EGF was then crosslinked to NP-tethered PEG2 through the click reaction. The hydrodynamic diameter (dhyd) increased in this step from 48.4 ± 0.6 nm for the pegylated NPs to 56.1 ± 2.5 nm for the EGF functionalized NPs, which we will refer to as NP-EGF in the following. For simple mixtures of pegylated NPs and EGF (no covalent attachment) the measured hydrodynamic diameter (dhyd = 47.8 ± 1.3 nm) was nearly identical to that of the pegylated NPs. We determined the average number of EGF molecules bound per NP through an ELISA assay. To this end, we first removed the PEG2-EGF construct from the purified NP-EGF particles by incubation in a large excess of PEG1 at 45 °C for 48 h with sonicating every 12 h and then quantified its concentration relative to a PEG2-EGF calibration solution colorimetrically. We found that on average each NP carries ∼4 EGF peptides under the chosen experimental conditions. The ELISA results were consistent with TEM studies, in which we visualized the EGF molecules on individual NP-EGF particles after labeling with biotinylated anti-EGF antibodies and Neutravidin functionalized 5 nm diameter Au NPs (Fig. 2a–f). The average number of 5 nm NP labels detected per 40 nm core was 2.7 (maximum number 12) in the case of NP-EGF, while the binding to the control was negligible (Fig. 2g). Approximately 90 Au cores were evaluated for each condition. Based on geometric considerations, we estimate that approximately 45% of all bound 5 nm NP labels are detectable in conventional TEM. The remaining NP labels are located in areas where the diameter of the smaller NP is entirely blocked by the larger core so that the contrast is insufficient for their detection. Taking this effect into account, one can anticipate that the average number of bound 5 nm Au labels is around 6, which is in good agreement with the ELISA assay.
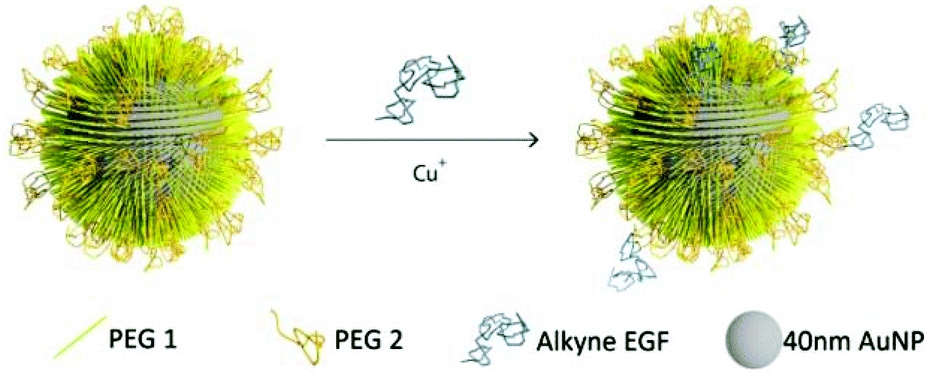 | ||
| Fig. 1 Schematics of NP-EGF preparation. 40 nm gold nanoparticles were functionalized with PEG1 and PEG2 to stabilize the NPs and to introduce binding sites (see text). Human EGF was then tethered to these binding sites through the CuI catalyzed 1,3-dipolar cycloaddition reaction. | ||
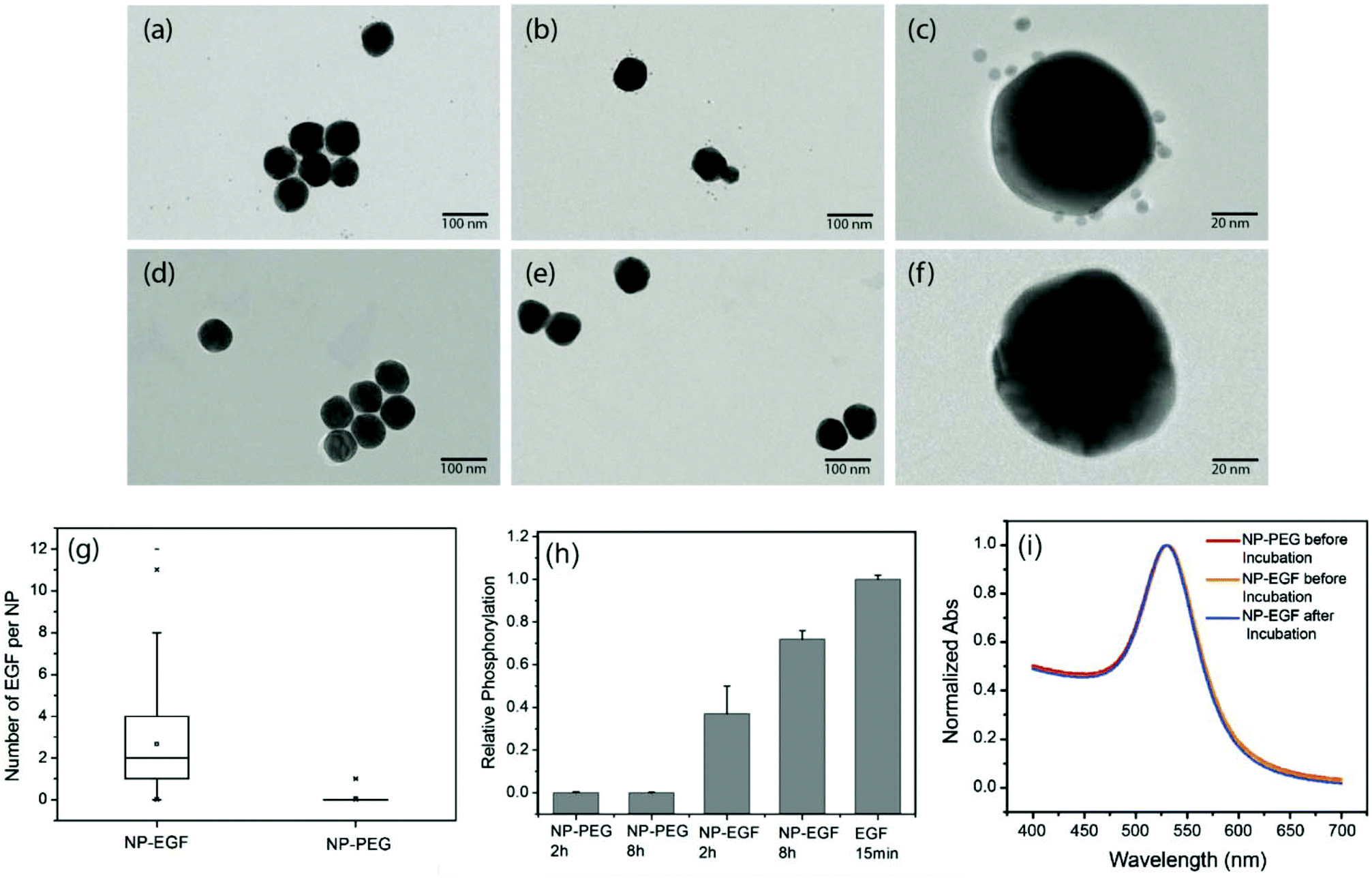 | ||
| Fig. 2 Characterization of NP-EGF conjugates. (a–c) TEM image of NP-EGF conjugates after incubation with biotinylated anti-EGF antibodies and Neutravidin and subsequent labeling with biotinylated 5 nm Au NPs. (d–f) TEM images of control experiments using the same particles but without the anti-EGF antibody. (g) Box plot of the number of 5 nm NP labels detected on NP-EGF (left) and control (right). The TEM data confirm a selective binding of 5 nm NP labels to NP-EGF. (h) EGFR phosphorylation in HeLa after treatment with NP-EGF and pegylated NPs for 2 h and 8 h. All phosphorylation levels are normalized to the reference condition: 15 min of incubation with 1.6 nM free EGF (last column). The measured phosphorylation levels confirm EGFR activation through nanoconjugated EGF. (i) UV-Vis spectra of NP-PEG (red) and NP-EGF before (yellow) and after (blue) incubation with A431 cells in serum containing medium for 4 h. The superposition of the graphs confirms the stability of NP-EGF against agglomeration. | ||
To further validate the bioavailability of the nanoconjugated EGF, we performed EGFR phosphorylation assays. These tests confirmed that NP-EGF conjugates but not the pegylated NPs induce EGFR activation (Fig. 2h).
In the next step, we characterized the stability of NP-EGF under physiological conditions. We incubated NP-EGF (3.6 × 1010 particles per mL) with A431 cells in serum-containing DMEM medium for 4 h at 37 °C and recovered the particles from the solution. The measured UV-Vis spectrum (Fig. 2i) superimposes almost perfectly with that of NP-EGF recorded prior to incubation with the cells. We also imaged the recovered NPs after random deposition on a silicon substrate. Consistent with the UV-Vis data, the SEM images show predominantly monomers before (94.1%) and after (93.5%) incubation with the cell (Fig. S1†), confirming a high degree of stability for NP-EGF in serum-containing DMEM. EGF functionalization did not affect the stability of the NPs under our experimental conditions, which was essentially identical to that of the pegylated NPs.
3.2 Nanoparticle panel and quantification of uptake
Our experimental strategy for validating a potential gain in apoptosis efficacy in A431 cells due to EGF nanoconjugation involves a quantitative comparison of apoptosis obtained with pegylated NPs (no EGF), simple mixtures of pegylated NPs and free EGF, and NP-EGF. In particular, we considered the following three different NP preparations: (P1) 0.06 nM pegylated NPs; (P2) 0.06 nM pegylated NPs + 1.6 nM EGF; (P3) 0.06 nM NP-EGF. The NP concentrations in these preparations were verified via the optical density measured at 520 nm. The effective EGF concentration for P3 was approximately 0.2 nM.Although the cytotoxicity of Au NPs with benign ligands has been shown to be, overall, low,51,52 large differences in the intracellular Au concentration are nevertheless to be avoided in comparative apoptosis studies to avoid spurious effects. We quantified the uptake of P1–P3 after incubating A431 cells with the respective NP preparations in serum containing DMEM for 24 h at 37 °C (Fig. 3a). The NP uptake was evident in all three cases “by eye” and was further confirmed through a characteristic red-shift of the cell spectra (Fig. 3b). We quantified the relative Au NP uptake for P1–P3 via ICP-MS. The results of eleven independent experiments are histogrammed in Fig. 3c. We find that under the chosen experimental conditions P1–P3 do not show systematic differences in uptake based on a two-tailed Mann–Whitney U test (95% confidence interval, p < 0.05), which we attribute to the formation of a protein corona under the chosen experimental conditions (vide infra). For the NP-EGF conjugate, P3, we characterized the intracellular spatial distribution of uptaken NPs through correlated dark-field/fluorescence imaging (Fig. 4). The high concentration of scatterers in the perinuclear area that colocalize with acidified compartments, as indicated by the LysoTracker stain, confirm an enrichment of the uptaken NPs in the lysosomal pathway. Controls without NPs do not show a similar concentration of scatterers (Fig. S2†).
 | ||
| Fig. 3 NP uptake in A431 cells. (a) Dark-field image of a cluster of A431 cells before (left) and after (right) incubation with NP-EGF. (b) The box plots (i–iii) show the relative pixel intensities on the red, green, and blue color channels for the cell areas marked accordingly. The detected spectral red-shift for the NP containing sample (iii) arises from the large scattering cross-section of clustered NPs in this spectral range. (c) Relative Au concentrations (normalized to maximum) as determined by ICP-MS after incubation of A431 cells with 0.06 nM solutions of (left to right) pegylated Au NPs (P1), pegylated Au NPs co-incubated with 1.6 nM EGF (P2), and NP-EGF (effective EGF concentration 0.2 nM) (P3) in serum containing DMEM for 24 h at 37 °C. Data were collected from at least 11 independent experiments. | ||
 | ||
| Fig. 4 Correlated dark-field and fluorescence microscopy of HeLa cells after 4 h of continuous incubation of the cells with NP-EGF. (a) The dark-field image shows the intracellular distribution of the NPs. (b) Lysosomes were stained with fluorescent LysoTracker. (c) Cell nuclei were stained with Hoechst 33342. (d) Overlay image of all three channels. The optical colocalization of NPs and LysoTracker confirms an enrichment of NPs in acidified membrane compartments in perinuclear regions. Similar distributions were obtained for A431 cells. | ||
3.3 Apoptosis quantification
Caspases play a central role in regulating apoptosis in mammalian cells53 and caspase-3, in particular, is frequently used to quantify apoptosis.24,54 We performed independent control experiments to validate that under our experimental conditions increased caspase-3 levels were indeed caused by apoptosis. Characteristic changes in the cell morphology (Fig. S3†), obvious cell nuclei contraction (Fig. S4†), and phosphatidylserine translocation in cells with intact plasma membrane (Fig. S5†) confirmed that caspase-3 activity was a useful metric to quantify apoptosis. We monitored the changes in the relative caspase-3 activity in A431 cells upon exposure to either (i) free EGF at concentrations of 1.6 and 33 nM, (ii) a mixture of 1.6 nM free EGF solution with pegylated Au NPs (P2), or (iii) nanoconjugated EGF (P3). We also included pegylated NPs (P1) and the supernatant of the NP-EGF crosslinking reactions in this study to determine the background apoptosis levels from NPs and residual free EGF or copper catalyst in P3. We chose the EGFR overexpressing cell line A431 for this systematic comparison, since it was shown before that nanomolar concentrations of free EGF induce apoptosis in this cell line.20–23 In Fig. 5 we plot, from left to right, the relative average caspase-3 activities (± standard deviation) of A431 cells without any additions (control), as well as of A431 cells incubated with P1, 1.6 nM free EGF, 33 nM free EGF, P2, P3, and the P3 supernatant. All cells were incubated in serum-containing DMEM at 37 °C and the experimental details followed in large part previous studies,55 which quantified apoptosis as the response to free EGF. | ||
| Fig. 5 Relative caspase-3 activity of A431 cells after 24 h, 48 h, and 72 h of incubation in serum containing DMEM at 37 °C with (left to right): blank controls; pegylated NPs (P1, 0.06 nM); free EGF (1.6 nM); free EGF (33 nM); pegylated NPs (0.06 nM) with 1.6 nM free EGF (P2); nanoconjugated EGF (P3, 0.06 nM); P3 supernatant. All caspase-3 activities are normalized to controls after 24 h. Measured activities that were significantly (p < 0.05) higher than controls are marked by *, and highly significant differences (p < 0.01) are marked by **. Data were collected from at least 6 independent experiments. | ||
We measured the caspase-3 activities after 24 h, 48 h, and 72 h of incubation for each condition and normalized the data with the caspase-3 activity for the A431 blank control obtained at the respective incubation times. The A431 controls show no significant (p < 0.05, two-tailed Mann–Whitney U test) change in caspase-3 activity as a function of time, as is the case for the pegylated NPs (P1). We conclude that the apoptotic effect of Au NPs by themselves is negligible under the chosen experimental conditions. In the next step we characterize the impact of free EGF at a concentration of 1.6 nM. Although the average caspase-3 activities are already higher (∼120%) than for controls, the data are broadly spread and we only measure a statistically significant increase in caspase-3 activity after 72 h of incubation. If the EGF concentration is, however, increased to 33 nM a highly significant (p < 0.01) increase in caspase-3 activity to ∼132% is already obtained after 24 h and 48 h, which further increases to ∼178% after 72 h. These observations are in very good agreement with previous studies that have reported apoptosis in A431 in response to free EGF concentrations in the low tens of nM range.55
Having analyzed the separate apoptotic effects of EGF and NPs, we can evaluate whether a simple mixing of EGF and NP solutions results in a synergistic increase in apoptosis. We find that for a 0.06 nM pegylated NP solution (P2), free EGF at a concentration of 1.6 nM yields a significant increase in the caspase-3 activity to ∼133% after 48 h, which slightly decreases to ∼125% after 72 h. Although the measured caspase-3 increase for P2 has a higher significance than that observed for 1.6 nM EGF alone, the magnitudes of average caspase-3 levels are similar. The data, therefore, do not support the presence of a strong synergistic effect between simple non-covalent mixtures of EGF and NPs. This changes upon covalent attachment of EGF to Au NPs. A highly significant increase in caspase-3 activity to ∼156% is obtained for the covalent NP-EGF conjugate, P3. The caspase-3 activity in the presence of P3 further increases to ∼185% after 72 h. Despite the low effective EGF concentration of only ∼0.2 nM, P3 achieves a higher average apoptosis level than free EGF at a concentration of 33 nM after 48 h. After 72 h, cells incubated with P3 and 33 nM free EGF still show comparable levels of apoptosis. According to these data, nanoconjugation strengthens the apoptotic efficacy, measured as caspase-3 activity, of EGF in the EGFR overexpressing cell line A431 by approximately two orders of magnitude.
In a second systematic study we validated whether nanoconjugation is also capable of inducing apoptosis in cells with physiological receptor expression levels. We chose HeLa for this experiment since this cell line does not undergo apoptosis in response to exposure to free EGF.16 The experimental details were otherwise identical to our studies for A431 described above. In Fig. 6 we show the relative caspase-3 activities as a function of time for HeLa blank controls (no EGF or NP addition) and after addition of pegylated NPs (P1), free EGF (33 nM), simple mixtures of pegylated NPs and EGF solutions (P2), or NP-EGF (P3). The data do not indicate P1- or free EGF-induced apoptosis in HeLa. Importantly, the non-covalent mixture of free EGF and pegylated NPs (P2) did also not result in enhanced apoptosis. Even increasing the concentration of free EGF in P2 to 33 nM did not yield a significant increase in caspase-3 activity (data not shown). While free EGF and simple mixtures of EGF and NPs do not suffice to induce apoptosis in HeLa, nanoconjugation changes the apoptotic efficacy of EGF and induces measurable apoptosis levels. For P3 a highly significant increase in caspase-3 activity to approximately 117% is obtained after 24 h, which further increases to ∼122% after 48 h and to ∼134% after 72 h.
 | ||
| Fig. 6 Relative caspase-3 activity in HeLa cells after 24 h, 48 h, and 72 h of incubation in serum containing DMEM at 37 °C with (left to right): blank controls; pegylated NPs (P1, 0.06 nM), free EGF (33 nM); pegylated NPs (0.06 nM) with 1.6 nM free EGF (P2); NP-EGF (P3, 0.06 nM). All caspase-3 activities are normalized to controls after 24 h. Measured activities that were significantly (p < 0.05) higher than controls are marked by *, and highly significant differences (p < 0.01) are marked by **. Data were collected from at least 6 independent experiments. | ||
The observation of a robust apoptosis increase even in HeLa demonstrates that the effect of EGF nanoconjugation is not limited to merely enhancing EGF-induced apoptosis in EGFR overexpressing cells. Instead, the ability to induce apoptosis in cells that do not normally experience apoptosis implies that nanoconjugation modulates fundamental cellular regulation processes associated with apoptosis.
In our analysis thus far, we have focused on the impact of EGF nanoconjugation on the ensemble level as a function of time. Additional insight can be obtained by characterizing the impact of EGF conjugation at the single cell level. To this end, we stained A431 cells after incubation with NP-EGF (P3) for 24–72 h with annexin V and propidium iodide (PI). Annexin V binds specifically to phosphatidylserine (PS) so that fluorescently labelled annexin V is commonly used to detect PS translocation from the inner to the outer leaflet, which is a characteristic event in apoptosis. PI increases its fluorescence when intercalated into DNA, and PI staining indicates ruptured plasma membranes in dead cells. After 24 h of incubation no staining with either annexin V or PI is observed (Fig. 7a), but this changes after 48 h (Fig. 7b) when up to 20% of the cells show strong staining with annexin V. A large fraction of the annexin V positive cells are PI negative, which is characteristic of early apoptosis. The number of annexin V labelled cells further increases after 72 h (Fig. 7c). Although the overwhelming majority of the cells in the ensemble is annexin V positive by 72 h, not all of the cells are stained by annexin V. Overall, Fig. 7a–c reveals a significant level of cell-to-cell variability in the outcome of P3 exposure. This finding is consistent with previous studies that have demonstrated apoptosis in general to be an intrinsically heterogeneous process even in genetically identical cell populations.56,57
 | ||
| Fig. 7 Analysis of apoptosis at the single cell level. Co-staining of A431 cells with fluorescently labelled annexin V (green) and propidium iodide, PI, (red) after (a) 24 h, (b) 48 h, and (c) 72 h of incubation with P3. The scattering images of the cells are included in black-and-white. Cells detached from the substrate after 72 h of culturing were recovered by cytospinning the growth medium supernatant. Representative images of the immobilized cells after incubation with EthD-1 (red) and calcein AM (green) for (d) the control sample cultured without NP-EGF for 72 h and (e) the sample cultured with P3 for 72 h. The magnification is 60× (a)–(c) and 10× in (d)–(e). | ||
Another important observation in Fig. 7c is that after 72 h of incubation many of the annexin V positive cells also show some PI staining indicative of the breakdown of the plasma membrane. In the absence of phagocytic cells, apoptosis has been demonstrated to terminate into secondary necrosis after extended culturing.58,59 We attribute the observed membrane rupturing to the presentation of necrotic features in late stage apoptotic cells. This interpretation is corroborated by the analysis of the growth medium supernatant after 72 h. Compared to the control (culturing without NP-EGF, Fig. 7d), the supernatant of the cell sample incubated with P3 shows a dramatic increase in the number of ethidium homodimer-1 (EthD-1) positive cells that have detached from the substrate (Fig. 7e). The cells were also treated with an acetomethoxy derivative of calcein (calcein AM) that is cleaved into the fluorescent calcein in living cells. Most cells are calcein negative, confirming that the detached cells are dead. Together, these observations suggest that after completing the NP-EGF induced apoptosis program, the cells finally die in a secondary necrosis process and subsequently detach from the substrate.
4. Discussion
Any use of NPs in a complex biological medium, such as the serum containing DMEM medium used in this work, raises the question of the actual charge and composition of the NP surface that interacts with the cells. The NPs used in this work have negative surface charges in aqueous solution under physiological pH conditions and will, consequently, attract positively charged components of the serum containing buffer.60,61 The attachment of proteins onto the NP surface will lead to the formation of a “corona” around the NPs with different physico-chemical properties.62 The composition of this corona, which is dynamic in nature,63 will influence the interactions between the NPs and the cells. In fact, the comparable uptake of P1–P3 observed in this work (Fig. 3) could result from the adsorption of proteins on the NPs. If surface receptors of the cell recognize fragments of the attached proteins, receptor-mediated binding and uptake occur even for those NPs that were initially lacking any ligands of cell surface receptors. We emphasize, however, that despite this corona formation, the EGF-functionalized NPs retain a distinct character that is evident in the significantly increased apoptosis levels. Both the length of the PEG2 spacer and the dynamic nature of the corona contribute to the biological availability of the tethered growth factor. Furthermore, due to the covalent attachment of the EGF, the growth factor molecules are expected to remain tethered to the NPs even under acidic conditions and in the presence of hydrolases, whereas components of the corona degrade. The verification of the relative contributions of these different factors requires additional experiments that go beyond the scope of this manuscript. One clear conclusion of the current work, however, is that corona formation does not prevent nanoconjugation from enhancing the apoptotic effect of EGF. Nanoconjugation has been shown before to support the apoptotic effect of some anticancer drugs due to targeted delivery, enhanced cell permeabilization or related mechanisms.64–68 EGF nanoconjugation induced apoptosis differs, however, fundamentally from the above cases since EGF is an endogenous growth factor that stimulates proliferation under native conditions. Especially, the observation of NP-EGF induced apoptosis in HeLa cells with normal EGFR expression levels underlines that nanoconjugation can change the cellular response to a specific signaling molecule, in this case EGF. Although an unambiguous elucidation of the underlying mechanisms requires additional systematic studies, we note that the observed apoptosis enhancement is consistent with an effective deceleration of intracellular EGF*EGFR signaling de-activation through EGF nanoconjugation.5. Conclusion
EGF is a growth factor that stimulates cell growth and differentiation under native conditions but that has some apoptotic efficacy in EGFR overexpressing cells. We have provided detailed experimental evidence in this manuscript that a covalent attachment of the EGF peptide to a pegylated 40 nm diameter Au nanoparticle results in an enhancement of its apoptotic efficacy even in cells that are not known to show apoptosis in response to free EGF. This discovery is important (1) because it indicates that nanoconjugation can modulate apoptosis associated cellular regulation mechanisms and (2) because it provides a new potential strategy for overcoming apoptosis evasion in cancer cells. The selective enrichment of circulating NPs in tumor tissue due to the well-known enhanced permeability and retardation (EPR) effect,69,70 together with the now demonstrated ability to enhance the apoptotic efficacy of EGF through nanoconjugation in vitro, warrants additional future studies into (i) the systematic optimization of the efficacy through variation of NP size, EGF surface concentration and presentation, and (ii) a validation of nanoconjugation as a potential strategy to induce apoptosis in a controlled fashion in vivo. An additional significant conclusion from this work is that, although the nanoconjugated EGF was incubated for extended times in complex media, corona formation did not prevent functional differences between NP-EGF and plain pegylated NPs. This observation is consistent with previous studies in which NPs with appropriately designed surfaces successfully targeted specific receptors even in complex media.71–73Acknowledgements
This work was supported by the National Institutes of Health (NIH/NCI) through grant 5R01CA138509 and the National Science Foundation through grants 0953121 and 1159552 to BMR. L.W. acknowledges a National Cancer Institute Cancer Nanotechnology Training Center (NCI-CNTC) Graduate Student Traineeship and financial support through NIH R25 CA153955.References
- S. Bhattacharyya, R. Bhattacharya, S. Curley, M. A. McNiven and P. Mukherjee, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 14541–14546 CrossRef CAS PubMed.
- M. A. Lemmon and J. Schlessinger, Cell, 2010, 141, 1117–1134 CrossRef CAS PubMed.
- Y. Yarden and J. Schlessinger, Biochemistry, 1987, 26, 1434–1442 CrossRef CAS PubMed.
- J. Schlessinger, Cell, 2000, 103, 211–225 CrossRef CAS PubMed.
- H. A. Clayton, S. G. Orchard, E. C. Nice, R. G. Posner and A. W. Burgess, Growth Factors, 2008, 26, 316–324 CrossRef PubMed.
- H. A. Clayton, F. Walker, S. G. Orchard, C. Henderson, D. Fuchs, J. Rothacker, E. C. Nice and A. W. Burgess, J. Biol. Chem., 2005, 280, 30392–30399 CrossRef PubMed.
- A. Sorkin and M. von Zastrow, Nat. Rev. Mol. Cell Biol., 2009, 10, 609–622 CrossRef CAS PubMed.
- A. Sorkin, in EGFR Signaling Networks in Cancer Therapy, Internalization and Degradation of the EGF receptor, ed. J. D. Haley, W. J. Gullick, Humana Press, New York, USA, 2008, pp. 47–61 Search PubMed.
- G. Carpenter and S. Cohen, Annu. Rev. Biochem., 1979, 48, 193–216 CrossRef CAS PubMed.
- A. Sorkin and L. K. Goh, Exp. Cell Res., 2009, 315, 683–696 CrossRef CAS PubMed.
- L. P. Sousa, I. Lax, H. Shen, S. M. Ferguson, P. De Camilli and J. Schlessinger, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 4419–4424 CrossRef CAS PubMed.
- H. Resat, J. A. Ewald, D. A. Dixon and H. S. Wiley, Biophys. J., 2003, 85, 730–743 CrossRef CAS PubMed.
- X. Jiang and A. Sorkin, Traffic, 2003, 4, 529–543 CrossRef CAS PubMed.
- J. A. Hanover, M. C. Willingham and I. Pastan, Cell, 1984, 39, 283–293 CrossRef CAS PubMed.
- M. R. Maxfield and T. E. McGraw, Nat. Rev. Mol. Cell Biol., 2004, 5, 121–132 CrossRef PubMed.
- J. S. Rush, L. M. Quinalty, L. Engelman, D. M. Sherry and B. P. Ceresa, J. Biol. Chem., 2012, 287, 712–722 CrossRef CAS PubMed.
- Y. Daaka, L. M. Luttrell, S. Ahn, G. J. Della Rocca, S. S. Ferguson, M. G. Caron and R. J. Lefkowitz, J. Biol. Chem., 1998, 273, 685–688 CrossRef CAS PubMed.
- R. J. Lefkowitz, J. Biol. Chem., 1998, 273, 18677–18680 CrossRef CAS PubMed.
- J. A. Lunn, H. Wong, E. Rozengurt and J. H. Walsh, Am. J. Physiol. Cell Physiol., 2000, 279, C2019–C2027 CAS.
- D. K. Armstrong, S. H. Kaufmann, Y. L. Ottaviano, Y. Furuya, J. A. Buckley, J. T. Isaacs and N. E. Davidson, Cancer Res., 1994, 54, 5280–5283 CAS.
- G. N. Gill and C. S. Lazar, Nature, 1981, 293, 305–307 CrossRef CAS PubMed.
- T. J. Kottke, A. L. Blajeski, L. M. Martins, P. W. Mesner Jr., N. E. Davidson, W. C. Earnshaw, D. K. Armstrong and S. H. Kaufmann, J. Biol. Chem., 1999, 274, 15927–15936 CrossRef CAS PubMed.
- D. W. Barnes, J. Cell Biol., 1982, 93, 1–4 CrossRef CAS PubMed.
- D. C. Hyatt and B. P. Ceresa, Exp. Cell Res., 2008, 314, 3415–3425 CrossRef CAS PubMed.
- W. Jiang, B. Y. Kim, J. T. Rutka and W. C. W. Chan, Nat. Nanotechnol., 2008, 3, 145–150 CrossRef CAS PubMed.
- D. Chithrani and W. C. W. Chan, Nano Lett., 2007, 7, 1542–1550 CrossRef PubMed.
- D. Chithrani, A. A. Ghazani and W. C. W. Chan, Nano Lett., 2006, 6, 662–668 CrossRef PubMed.
- A. Verma and F. Stellacci, Small, 2010, 6, 12–21 CrossRef CAS PubMed.
- Z. P. Xu, Q. H. Z, G. Q. Lu and A. B. Yu, Chem. Eng. Sci., 2006, 61, 1027–1040 CrossRef CAS.
- C. Dreaden, S. C. Mwakwari, Q. H. Sodji, A. K. Oyelere and M. A. El-Sayed, Bioconjugate Chem., 2009, 20, 2247–2253 CrossRef PubMed.
- C. Dreaden and M. A. El-Sayed, Acc. Chem. Res., 2012, 45, 1854–1865 CrossRef PubMed.
- S. Dhar, W. L. Daniel, D. A. Giljohann, C. A. Mirkin and S. J. Lippard, J. Am. Chem. Soc., 2009, 131, 14652–14653 CrossRef CAS PubMed.
- X. Huang, P. K. Jain, I. H. El-Sayed and M. A. El-Sayed, Nanomedicine, 2007, 2, 681–693 CrossRef CAS PubMed.
- N. Lewinski, V. Colvin and R. Drezek, Small, 2008, 4, 26–49 CrossRef CAS PubMed.
- G. von Maltzahn, J. H. Park, A. Agrawal, N. K. Bandaru, S. K. Das, M. J. Sailor and S. N. Bhatia, Cancer Res., 2009, 69, 3892–3900 CrossRef CAS PubMed.
- X. Zhang, H. Wu, D. Wu, Y. Wang, J. Chang, Z. Zhai, A. Meng, P. Liu, L. Zhang and F. Fan, Int. J. Nanomed., 2010, 5, 771–781 CrossRef CAS PubMed.
- J. Yguerabide and E. E. Yguerabide, Anal. Biochem., 1998, 262, 157–176 CrossRef CAS PubMed.
- E. C. Dreaden, A. M. Alkilany, X. Huang, C. J. Murphy and M. A. El-Sayed, Chem. Soc. Rev., 2012, 41, 2740–2779 RSC.
- J. Yguerabide and E. E. Yguerabide, Anal. Biochem., 1998, 262, 137–156 CrossRef CAS PubMed.
- W. Qian, X. Huang, B. Kang and M. A. El-Sayed, J. Biomed. Opt., 2010, 15, 046025 Search PubMed.
- B. Kang, M. A. Mackey and M. A. El-Sayed, J. Am. Chem. Soc., 2010, 132, 1517–1519 CrossRef CAS PubMed.
- R. A. Sperling, P. Rivera Gil, F. Zhang, M. Zanella and W. J. Parak, Chem. Soc. Rev., 2008, 37, 1896–1908 RSC.
- H. Wang, L. Wu and B. M. Reinhard, ACS Nano, 2012, 6, 7122–7132 CrossRef CAS PubMed.
- J. Wang, S. V. Boriskina, H. Wang and B. M. Reinhard, ACS Nano, 2011, 5, 6619–6628 CrossRef CAS PubMed.
- G. Rong, H. Wang, L. R. Skewis and B. M. Reinhard, Nano Lett., 2008, 8, 3386–3393 CrossRef CAS PubMed.
- F. Li, Q. Zhao, C. Wang, X. Lu, X. F. Li and X. C. Le, Anal. Chem., 2010, 82, 3399–3403 CrossRef CAS PubMed.
- V. I. Baranov, Z. Quinn, D. R. Bandura and S. D. Tanner, Anal. Chem., 2002, 74, 1629–1636 CrossRef CAS PubMed.
- S. Zhang, J. Li, G. Lykotrafitis, G. Bao and S. Suresh, Adv. Mater., 2009, 21, 419–424 CrossRef CAS PubMed.
- H. Gao, W. Shi and L. B. Freund, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 9469–9474 CrossRef CAS PubMed.
- C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef.
- M. Alkilany and C. J. Murphy, J. Nanopart. Res., 2010, 12, 2313–2333 CrossRef PubMed.
- N. Khlebtsov and L. Dykman, Chem. Soc. Rev., 2010, 40, 1647–1671 RSC.
- J. Li and J. Yuan, Oncogene, 2008, 27, 6194–6206 CrossRef CAS PubMed.
- G. M. Cohen, Biochem. J., 1997, 326, 1–16 CrossRef CAS PubMed.
- P. S. Grudinkin, V. V. Zenin, A. V. Kropotov, V. N. Dorosh and N. N. Nikolsky, Eur. J. Cell Biol., 2007, 86, 591–603 CrossRef CAS PubMed.
- J. C. Goldstein, R. M. Kluck and D. R. Green, Ann. N. Y. Acad. Sci., 2000, 926, 132–141 CrossRef CAS PubMed.
- S. L. Spencer, S. Gaudet, J. G. Albeck, J. M. Burke and P. K. Sorger, Nature, 2009, 459, 428–432 CrossRef CAS PubMed.
- M. T. Silva, FEBS Lett., 2010, 584, 4491–4499 CrossRef CAS PubMed.
- G. Majno and I. Joris, Am. J. Pathol., 1995, 146, 3–15 CAS.
- M. Lundqvist, J. Stigler, G. Elia, I. Lynch, T. Cedervall and K. A. Dawson, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 14265–14270 CrossRef CAS PubMed.
- A. Hung, S. Mwenifumbo, M. Mager, J. J. Kuna, F. Stellacci, I. Yarovsky and M. M. Stevens, J. Am. Chem. Soc., 2011, 133, 1438–1450 CrossRef CAS PubMed.
- E. Nel, L. Mädler, D. Velegol, T. Xia, E. M. V. Hoek, P. Somasundaran, F. Klassig, V. Castranova and M. Thompson, Nat. Mater., 2009, 8, 543–557 CrossRef PubMed.
- T. Cedervall, I. Lynch, S. Lindman, T. Berggard, E. Thulin, H. Nilsson, K. A. Dawson and S. Linse, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 2050–2055 CrossRef CAS PubMed.
- Y. Zhang, X. Li, Z. Huang, W. Zheng, C. Fan and T. Chen, Nanomed. Nanotech. Biol. Med., 2013, 9, 74–84 CrossRef CAS PubMed.
- K. Vandita, B. Shashi, K. G. Santosh and K. I. Pal, Mol. Pharm., 2012, 9, 3411–3421 CrossRef CAS PubMed.
- A. Khdair, D. Chen, Y. Patil, L. Ma, Q. P. Dou, M. P. V. Shekhar and J. Panyam, J. Controlled Release, 2010, 141, 137–144 CrossRef CAS PubMed.
- C. Wang, H. Zhang, B. Chen, H. Yin and W. Wang, Int. J. Nanomed., 2011, 6, 1929–1035 CAS.
- B. Chen, J. Cheng, M. Shen, F. Gao, W. Xu, H. Shen, J. Ding, C. Gao, Q. Sun, X. Sun, H. Cheng, G. Li, W. Chen, N. Chen, L. Liu, X. Li and X. Wang, Int. J. Nanomed., 2009, 4, 65–71 CrossRef CAS PubMed.
- K. Cho, X. Wang, S. Nie, Z. Chen and D. M. Shin, Clin. Cancer Res., 2008, 14, 1310–1316 CrossRef CAS PubMed.
- H. Maeda, Bioconjugate Chem., 2010, 21, 797–802 CrossRef CAS PubMed.
- E. C. Dreaden, L. A. Austin, M. A. Mackey and M. A. El-Sayed, Ther. Deliv., 2012, 3, 457–478 CrossRef CAS PubMed.
- M. P. Melancon, W. Lu, Z. Yang, R. Zhang, Z. Cheng, A. M. Elliot, J. Stafford, T. Olson, J. Z. Zhang and C. Li, Mol. Cancer Ther., 2008, 7, 1730–1739 CrossRef CAS PubMed.
- M. Eghtedari, A. V. Liopo, J. A. Copland, A. A. Oraevsky and M. Motamedi, Nano Lett., 2009, 9, 287–291 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: The ESI contains SEM images of NP-EGF before and after incubation with cells, dark-field and fluorescence images of HeLa cells with stained lysosomes, and apoptosis assays (cell morphology, nucleus condensation and phosphatidylserine translocation/plasma membrane integrity). See DOI: 10.1039/c3bm60142k |
| This journal is © The Royal Society of Chemistry 2014 |