 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
In situ-forming robust chitosan-poly(ethylene glycol) hydrogels prepared by copper-free azide–alkyne click reaction for tissue engineering†
Vinh X.
Truong
a,
Matthew P.
Ablett
b,
Hamish T. J.
Gilbert
b,
James
Bowen
c,
Stephen M.
Richardson
*b,
Judith A.
Hoyland
*b and
Andrew P.
Dove
*a
aDepartment of Chemistry, University of Warwick, Coventry, CV4 7AL, UK. E-mail: a.p.dove@warwick.ac.uk; Tel: +44 (0)24 7652 4107
bCentre for Tissue Engineering and Repair, Institute of Inflammation and Repair, Faculty of Medical and Human Sciences, The University of Manchester, Manchester, M13 9PT, UK. E-mail: Judith.hoyland@manchester.ac.uk; Tel: +44 (0)161 275 5425
cSchool of Chemical Engineering, University of Birmingham, Edgbaston, Birmingham, B15 2TT, UK
First published on 4th October 2013
Abstract
A water-soluble azide-functionalised chitosan was crosslinked with propiolic acid ester-functional poly(ethylene glycol) using copper-free click chemistry. The resultant hydrogel materials were formed within 5–60 min at 37 °C and resulted in mechanically robust materials with tuneable properties such as swelling, mechanical strength and degradation. Importantly, the hydrogels supported mesenchymal stem cell attachment and proliferation and were also non-toxic to encapsulated cells. As such these studies indicate that the hydrogels have potential to be used as injectable biomaterials for tissue engineering.
1. Introduction
Chitosan is a linear polysaccharide that can be readily obtained from natural sources such as shrimp and other crustacean shells.It displays excellent biocompatibility in addition to other unique properties such as being hemostatic, mucoadhesive and having high affinity to anionic biologically active molecules.1,2 Consequently, chitosan is an attractive precursor for the preparation of hydrogels for a range of biomedical applications such as wound healing,3,4 drug delivery5,6 and tissue engineering.7,8
Chitosan-based hydrogels have been widely studied, largely on account of their ability to form thermally-responsive, injectable gels that are capable of directing the differentiation of human mesenchymal stem cells (MSCs).9 Such gels are prepared from chitosan and glycerophosphate9–11 or PEG-grafted-chitosan,12–14 which remain liquid at ambient temperature and form gels within 10–15 minutes at 37 °C. Other injectable chitosan-based hydrogels have been reported using pH-responsive nanostructure transformations15,16 in which gelation occurs when the precursor solution is subjected to a change in pH. However, despite their attractive properties for biological applications, these hydrogels possess poor biomechanical properties and fast degradation rates that result from weak physical or ionic crosslinking interactions within the gel, thus limiting their potential for tissue engineering applications.
Several attempts to increase the strength of chitosan-based hydrogels have been reported by covalently crosslinking chitosan precursors using a range of chemistries such as thiol–ene Michael addition,17,18 enzyme-catalysed crosslinking (horseradish peroxidase),8,19 Schiff base formation,20 and by crosslinking with the natural crosslinker genipin.21,22 While in general, these processes led to hydrogels with increased mechanical properties in comparison to the ionic/physically crosslinked networks, some of the crosslinking methods resulted in a very fast gelation process (within seconds) which coupled with a low degree of control over the gelation time8,19 limits the potential application of these systems as injectable hydrogels. To this end, we sought to identify a system that was able to produce a wide range of chitosan-based hydrogels with tuneable mechanical properties and gelation times as well as demonstrate their ability to support MSC growth.
In recent years, so called ‘click’ reactions that are fast, selective and high yielding,23,24 have been used to prepare macrostructures with diverse and complex architecture in both organic and aqueous media. Notably, a selection of click reactions have even been shown to be carried out in complex biological media and in the presence of living cells. Such reactions are termed bioorthogonal click chemistry and have been successfully used in applications such as cell labelling25 and imaging26 and cell surface modification.27 Bioorthogonal click reactions are extremely attractive for fabrication of hydrogels to be used as cell culture scaffolds because they allow cell encapsulations and at the same time provide 3D environment which mimics the extracellular environment of natural tissues.28,29 Michael addition reactions are probably the most common bioorthogonal crosslinking chemistries utilised to prepare in situ-forming hydrogels on account of the mild reaction conditions, which require only basic buffer environment, and the availability of the functional precursors.28,30 More recently, several other click reactions have been introduced to the expanding bioorthogonal click toolkit for hydrogel preparation in biological media. They include reaction of an azide with a ring-strained alkyne (SPAAC),31,32 oxime click reaction with glutaraldehyde,33 tetrazine–norbonene reaction,34 and the tetrazole–alkene click reaction.35
Among these chemistries, SPAAC is gaining widespread attention for use in preparation of in situ-forming hydrogels31,32 as a consequence of the inert virtue of the functional groups which allow its use in conjunction with other bioorthogonal click chemistries to provide precise spatial and temporal manipulation of the microenvironment within the hydrogel scaffold. However, the synthetic challenges of both the multi-step preparation of the ring-strained alkyne and its incorporation in chitosan-based gels led us to focus on the more simple click reaction between organic azides and propiolic acid esters.23,36 In common with SPAAC, this reaction occurs readily at ambient temperature and does not require the addition of any catalytic species. Herein we demonstrate the preparation of robust chitosan-poly(ethylene glycol) (PEG) hydrogels by crosslinking an azide-functionalised chitosan with a water soluble 3-arm PEG containing an activated ester alkyne group. The tuneable nature of the gelation and gel properties are also demonstrated.
2. Materials and methods general considerations
5-Pentazoic acid was synthesized according to a previously reported method.37 Chitosan (low molecular weight, viscosity 20–300 cP, 1 wt% in 1% acetic acid) was obtained from Sigma-Aldrich. EDC·HCl was purchased from Carbosynth Limited. All other reagents were purchased from Sigma-Aldrich and used without further purification. NMR spectra were recorded on a Bruker DPX-300, DPX-400, AC400, or DRX-500 spectrometer at 293 K unless stated otherwise. Chemical shifts are reported as δ in parts per million (ppm) and referenced to the chemical shift of the residual solvent resonances (CDCl31H: δ = 7.26 ppm; 13C δ = 77.16 ppm). Size-exclusion chromatography (SEC) was used to determine the molar masses and molar mass distributions (dispersities, ÐM) of the synthesised polymers. SEC in tetrahydrofuran was conducted on a system comprised of a Varian 390-LCMulti detector suite fitted with differential refractive index (DRI), light scattering (LS) and ultra-violet (UV) detectors equipped with a guard column (Varian Polymer Laboratories PLGel 5 mM, 50 × 7.5 mm) and two mixed D columns (Varian Polymer Laboratories PLGel 5 mM, 300 × 7.5 mm). The mobile phase was tetrahydrofuran with 5% triethylamine eluent at a flow rate of 1.0 mL min−1, and samples were calibrated against Varian Polymer laboratories Easi-Vials linear poly(styrene) standards (2.4–162 × 105 g mol−1) using Cirrus v3.3.Synthesis of PEG-propiolate, 3-arm
Glycerol ethoxylate (10 g, 10 mmol) and propiolic acid (2.8 g, 40 mmol) were mixed in toluene (100 mL) before the solution was heated to 80 °C while stirring until a clear solution was obtained. To this solution, 2 drops of concentrated H2SO4 were added before the solution was heated to reflux under Dean–Stark conditions. After completion of water collection, the solvent was evaporated in vacuo and the oily mixture was taken up in CH2Cl2 (100 mL), washed with saturated aqueous NaHCO3 solution (20 mL), dried using MgSO4, and the solvent evaporated in vacuo. The resulting oil was dissolved in EtOH (50 mL) and charcoal (0.1 g) was added. The solution was stirred for 30 min at 40 °C after which the charcoal was removed by filtration. EtOH was evaporated in vacuo and the product was run through a column packed with silica gel using CH2Cl2 as the eluent. After removal of the solvent under vacuum, the product was collected as a clear oil (yield = 8.4 g, 73%). 1H NMR (CDCl3, 400) δ 4.35–4.37 (t, 3JHH = 6.8 Hz –CH2CH2OC(O)CH2–), 3.65–3.67 (m, –OCH2CH2O–) 3.01 (s, –CH![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CC(O)O–); IR (neat, cm−1) 3211, 2863, 2106, 1717, 1432, 1256, 1101, 967, 840. Mn = 1130 (Ð = 1.04). 1H NMR spectroscopy indicated ∼99% conversion of the hydroxyl group to propiolate group.
CC(O)O–); IR (neat, cm−1) 3211, 2863, 2106, 1717, 1432, 1256, 1101, 967, 840. Mn = 1130 (Ð = 1.04). 1H NMR spectroscopy indicated ∼99% conversion of the hydroxyl group to propiolate group.
Synthesis of azide functionalised chitosan
A mixture of 5-azidopentanoic acid (1 g, 7 mmol) and chitosan (1.56 g, degree of deacetylation (DD) = 84% from 1H NMR spectroscopy) was added to deionised water (200 mL) and stirred for 30 min to obtain a clear solution. N-Hydroxysuccinimide (NHS) (0.966 g, 8.2 mmol) and EDC (2 g, 10.5 mmol) were then added after which the solution was gently heated to 40 °C and stirred for 48 h. The solution was allowed to cool to room temperature before the pH was brought to 7.0 using NaOH 1 M aqueous solution. It was then dialysed in a dialysis membrane (MW cut off 3500 Da) against distilled water for 5 days with frequent changes of water. The purified solution was lyophilised and the resultant solid was Soxhlet extracted with ethanol for 1 day before drying to obtain product as a white fibre material (yield 78%, DD = 64.5 from 1H NMR spectroscopy). 1H NMR (D2O–CD3COOD), 400) δ 4.5 (chitosan anomeric proton), 3.5–3.8 (chitosan glucopyranose ring protons), 3.2 (m, CH2N3), 2.25 and 2.6 (m, CH2–CONH), 1.55 (m, –CH2CH2CH2–); IR (neat, cm−1) 3338, 2925, 2109, 1651, 1539, 1393, 1304, 1035, 1069.Hydrogel preparation
Hydrogels were prepared in 10 mL vials previously silanised with Sigmacote® to prevent gels adhering to the glassware. In a typical procedure for a hydrogel with [azide]![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :[alkyne] of 1:1, 25 μL of PEG-alkyne solution (80 wt%) was added to 200 μL of a 2.25 wt% solution of CS-azide and then mixed for 5 s using a vortex mixer before transferring into a water bath at 37 °C. The gelation time, i.e. the time when the solution stopped flowing, was determined using the vial tilt method.
:[alkyne] of 1:1, 25 μL of PEG-alkyne solution (80 wt%) was added to 200 μL of a 2.25 wt% solution of CS-azide and then mixed for 5 s using a vortex mixer before transferring into a water bath at 37 °C. The gelation time, i.e. the time when the solution stopped flowing, was determined using the vial tilt method.
Gel fraction and equilibrium water uptake
In order to determine gel fraction, the prepared gels were lyophilized and the weights (Wg) were recorded. The gels were then allowed to swell in deionised water for 3 days with frequent change of water to extract the unreacted polymers. The gels were then lyophilized and weights (Wr) recorded again. The gel fraction was expressed as Wg/Wr.In order to determine the water uptake in phosphate-buffered saline (PBS) the prepared gels were allowed to swell in PBS for 1 day so that the swelling reached equilibrium. The surface water was then removed with soft tissue paper and the weight was recorded (Ws). The gels were then lyophilized and the weight recorded (Wd). The equilibrium water uptake was expressed as:
| (Ws − Wd)/Wd × 100% |
Degradation studies
The prepared gels were placed in PBS solution pH 7.4 and incubated at 37 °C in an environmental-shaker incubator with a shaking speed of 90 rpm. In a separate set the gels were subjected to the same degradation conditions with addition of 0.1 wt% lysozyme from chicken egg white (≥40000 units per mg protein) in the PBS solution. The PBS solution was replaced after every 3 days to prevent build-up of solute concentration. At pre-set time intervals, the hydrogels were removed, gently blotted dry and the weight was recorded. The degradation was monitored by the percentage of weight remaining which is defined as:where Wt is the weight measured at specific time point and Wo is the weight at equilibrium swelling (after 24 h).
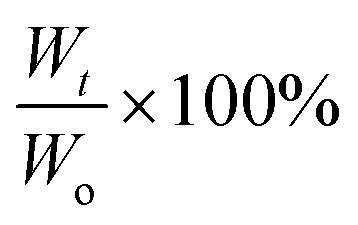
Rheological tests
Rheological tests were undertaken using an AR-G2 rheometer (TA Instruments, UK) in a parallel plate configuration, employing sandblasted stainless steel 40 mm diameter plates throughout and a Peltier plate for temperature control. In a typical rheological test for gelling kinetics, 800 μL of CS-azide 1.25 wt% solution in PBS was mixed with 200 μL of PEG-alkyne 80 wt% solution in PBS and the mixture was applied to the lower plate, preheated to 37 °C. The upper plate was immediately brought down to a plate separation of 0.5 mm and the measurement was started. A layer of high vacuum grease was applied around the gap to prevent water evaporation. A low frequency of 1 Hz and 0.5% strain was applied to minimise interference with the gelation process and keep the measurement within the linear viscoelastic regime. The gelation kinetics were monitored by the evolution of the storage (G′) and loss (G′′) moduli as a function of time. All measurements were repeated in triplicate.Unconfined compressive tests
Stress-strain measurements were performed using a Z030 mechanical tester (Zwick/Roell, UK). The hydrogel samples were prepared in a 10 mL vial previously silanised with Sigmacote® to prevent gels adhering to the glassware. After gelation at 37 °C the hydrogels were allowed to swell in PBS for 5 h before placing in between the parallel platens for measurement. Each specimen (approximately 5 g) had a thickness of approximately 25 mm and a diameter of 10 mm. The machine was operated at a compression velocity of 5 mm min−1 with a load cell of 100 N sensitivity and the force–displacement data were acquired at a frequency of 10 kHz. Each specimen was subjected to 90% strain in order to determine the compressive failure. The compressive modulus was defined as the slope of the initial linear modulus. All experiments were performed in triplicate.Mesenchymal stem cell culture and cell analysis
Human bone marrow mesenchymal stem cells (MSCs) (n = 3) were obtained, with ethical approval and written informed consent, and isolated following a previously published protocol.2 Adherent monolayer cells were cultured in Minimum Essential Medium (α-modification, Sigma), supplemented with 10% foetal calf serum, 10 μM ascorbic acid, 1× GlutaMAX™ (Gibco) and 1× Antibiotic Antimycotic solution (50000 units penicillin, 50 mg streptomycin and 125 μg amphotericin, Sigma). MSCs were incubated at 37 °C and 5% humidified CO2, and used when ∼80% confluent between passages 3–5 for all experiments.
For 2D MSC culture, hydrogels were prepared in tissue culture inserts (24-well plate inserts, BD Biosciences) as described above, but under sterile conditions and transferred to a 37 °C incubator until gelation had occurred. Cells were seeded at 5 × 104 cells per cm2 onto the gels with media changes every 3–4 days. For cell encapsulation (3D culture), MSCs were resuspended in CS-azide at 4 × 106 cells per mL before mixing with PEG-alkyne and pippetting into tissue culture inserts. Once gelation had occurred, media was added to the culture inserts and wells.
To assess cell viability both in 2D and 3D, gels were removed from tissue culture inserts after either 1 or 7 days in culture, washed in PBS and stained using Live/Dead® Viability/Cytotoxicity Kit for mammalian cells (Invitrogen) following the manufacturer's recommended protocol.
For morphological analysis, gels were removed from cell inserts after 7 days in culture, washed in PBS and fixed in 4% paraformaldehyde for 15 minutes at 4 °C. Cells were treated with 0.25% Triton-X in PBS (for 5 min) and then blocked in 1% bovine serum albumin (BSA) for 1 h at room temperature. Gels were incubated with 1:100 (in 1% BSA) mouse monoclonal vinculin primary antibody (Abcam) for 1 h at 4 °C followed by goat-anti mouse secondary antibody conjugated to Alexa Fluor® 488 (Invitrogen) at 1:300 dilution for 1 h, room temperature. 1 Unit of phalloidin per sample (Alexa Fluor 594, Invitrogen) was then added at room temperature for 20 min. Gels were then incubated with a 5 μM solution of DRAQ5™ (Biostatus) at 37 °C for 15 min to visualise the nuclei, after which the gels were washed and resuspended in PBS for imaging.
Images were collected on a Leica TCS SP5 AOBS upright confocal using a 20× dipping objective. ImageJ software was used to create maximum intensity projections of each 100 μm optical stack gathered using the confocal software.
3. Results and discussion
Chitosan presents many synthetic challenges, not least its inherent insolubility in aqueous solution at pH > 6.4 that results from the inter- and intra-molecular hydrogen bonds between the amine and the hydroxyl groups.38 To both overcome the limited water solubility of chitosan and introduce azide functional groups to the polysaccharide structure, 5-azidopentanoic was grafted onto the polysaccharide chain through amide formation using ethyl(dimethylaminopropyl) carbodiimide/N-hydroxysuccinimide coupling chemistry in water (Scheme S1A in ESI†). Following dialysis of the resultant material to remove small molecule side products and impurities, the presence of the azide functional group in the structure of the chitosan product was evidenced by the presence of a peak at 2150 cm−1 in the IR spectrum (Fig. 1A). 1H NMR spectroscopic analysis of the purified product showed new resonances at δ = 1.55, 2.26 and 3.27 ppm (Fig. 1B) which correspond to the protons within the azido-pentanoate side chain, which confirms the incorporation of the azido-pentanoate pendant group in the chitosan product. The degree of deacetylation of the chitosan before and after grafting were estimated from 1H NMR spectra as described elsewhere.39 These values were found to be 84% and 64.5% respectively which in turn enabled estimation of the degree of the substitution (i.e. the percentage of the glucosamine groups converted to glucosamide in the chitosan product) with azide groups calculated as 19.5%. This chitosan-azide precursor was found to be soluble in aqueous solution at the concentration of up to 3 wt% and pH up to 10. In a separate synthesis, a 3-arm PEG crosslinker with terminal propiolic acid ester groups was synthesised by Fisher esterification of the glycerol ethoxylate with propiolic acid (Scheme S1B in ESI†) in good yield (73%).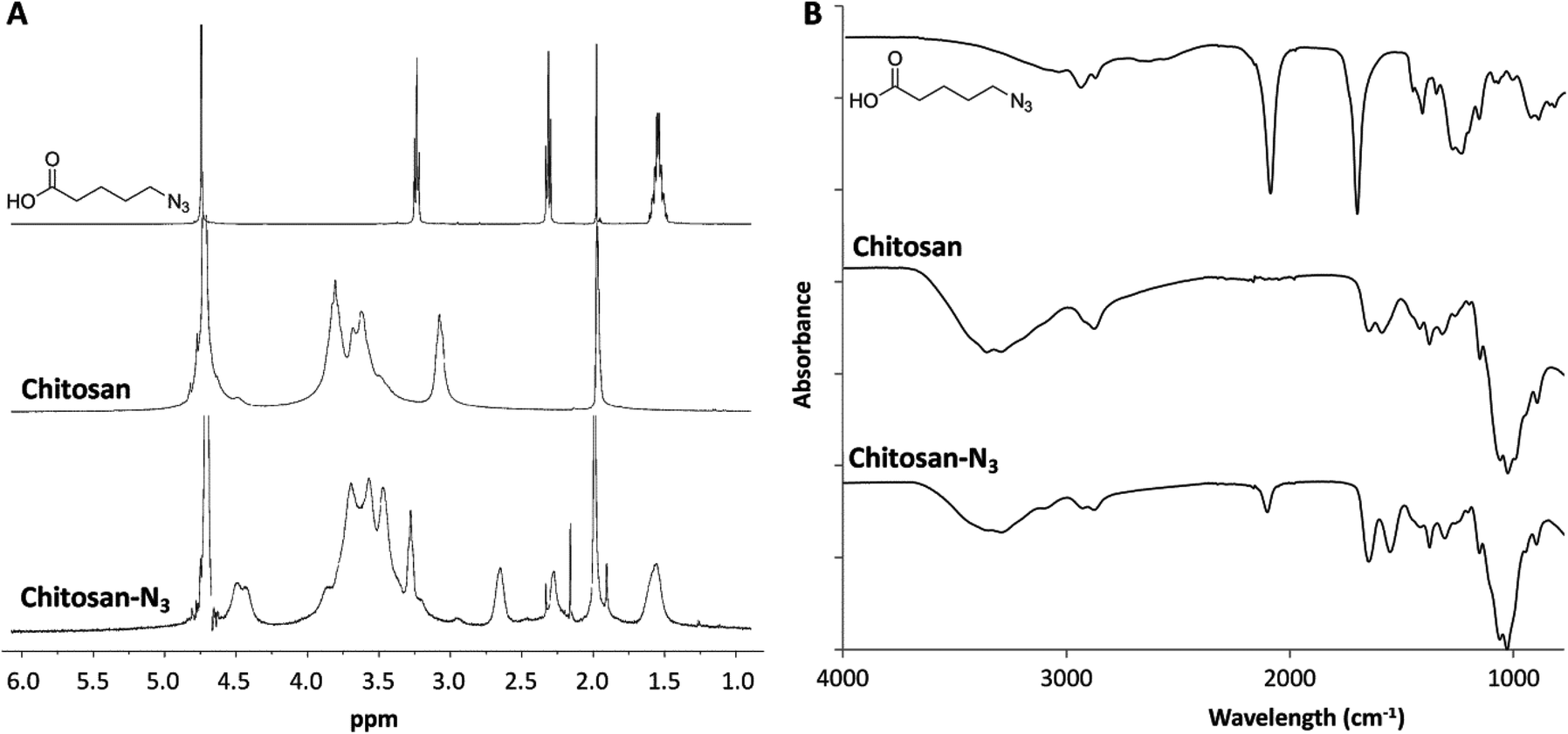 | ||
| Fig. 1 1H NMR spectra (in D2O–CD3COOD, 400 MHz) (A) and IR spectra (B) of the starting materials and chitosan-azide product. | ||
Initial hydrogel network formation was studied in nanopure water by crosslinking CS-azide (2 wt%) and PEG-alkyne (1:1 molar ratio to CS-azide) by metal-free, low temperature, alkyne–azide cycloaddition (Scheme 1). In contrast to the more commonly-applied copper catalysed alkyne–azide cycloaddition, this process does not require elevated temperatures or metal-based catalysts as a consequence of the activation of the alkyne by the adjacent electron withdrawing group.36 Gelation of these precursors was observed within 90 minutes at ambient temperature and 15 minutes at 37 °C as assessed by the vial tilt method. Such performance in pure water solutions demonstrates that gelation occurs in this system through the desired crosslinking reaction, eliminating the possibility of ionic crosslinking. To further demonstrate the biorthogonality of this reaction however, gelation in solutions that better mimic the cellular microenvironment i.e. those that include a range of components including salts, sugars, amino acids etc., we sought to evaluate the gelation process in PBS solution and cell culture media (MEM). Remarkably, in both solutions at 37 °C, gelation was complete within approximately the same time period, validating the hypothesis that the alkyne–azide cycloaddition remains bioorthogonal.
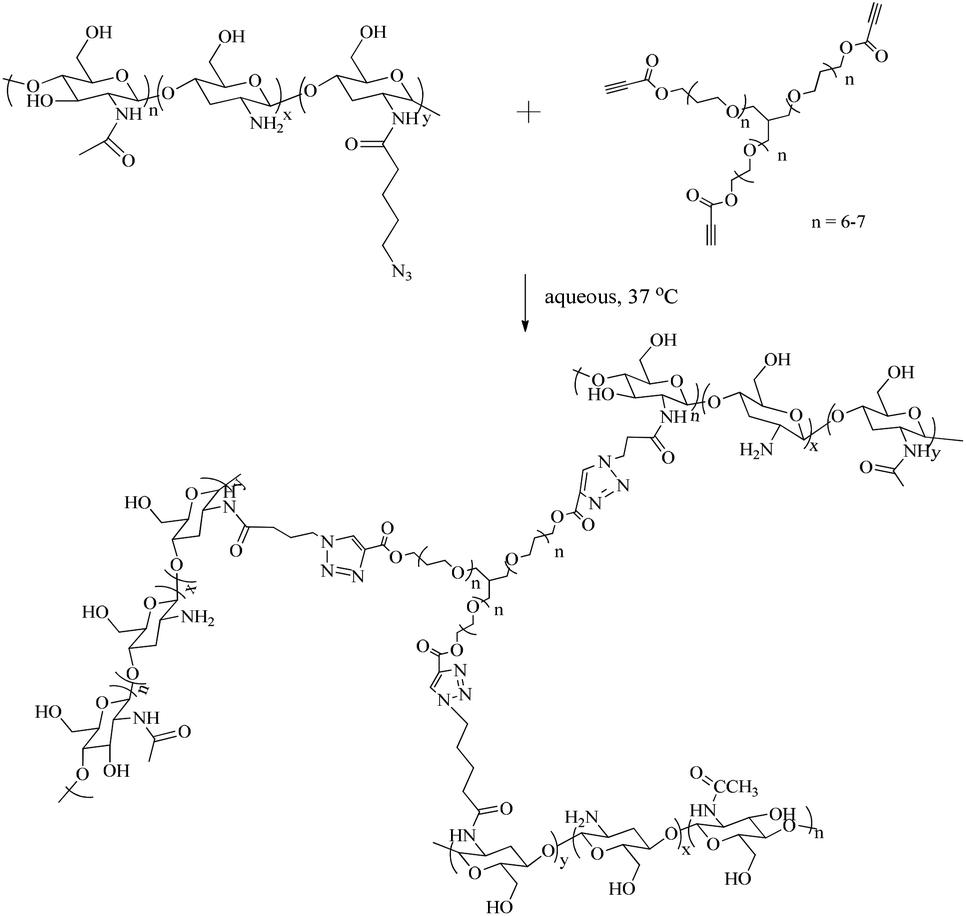 | ||
| Scheme 1 Crosslinking reaction of CS-azide with PEG-alkyne. | ||
The ability to control both gelation times and gel properties is important in the design of injectable hydrogel systems for tissue engineering and regenerative medicine applications. Ideally, mechanically-stable gels should form at a rate that enables mixing and uniform distribution of cells or proteins before injection into the body, but is sufficiently rapid to prevent leaching of the active biological components or cell sedimentation. A range of hydrogels, with the total weight percentage of polymer precursors in solution of 2–15 wt%, were prepared in PBS with increasing CS-azide concentrations from 1 to 3 wt% and [alkyne]:[azide] ratios between 0.25 to 2.5:1 with gelation times measured in triplicate using the vial tilt method (Fig. 2A). At the highest CS-azide and PEG-alkyne concentration and [alkyne]:[azide] ratio, gels were found to form within 4 minutes; however at lower concentrations of either component, gelation times were observed to be reduced significantly such that at 1 wt% CS-azide and a 0.25:1 molar ratio of [alkyne]:[azide], a period of 55 minutes was required for gelation to occur, most likely on account of the decrease in the concentration of the functional group of the gelation system.
 | ||
| Fig. 2 (A) Gelation time of CS-azide and PEG-alkyne mixture as a function of CS-azide concentration with different [azide]:[alkyne] molar ratios at 37 °C, and (B) rheological analysis of the gelation of CS-azide 1 wt% solution with the [alkyne]:[azide] ratio of 0.5 at 37 °C. | ||
The viscoelastic profiles of selected chitosan-PEG hydrogels during gelation were evaluated by rheological studies. The kinetics of gelation for a CS-azide 1 wt% solution with a [alkyne]:[azide] ratio of 0.5 was also followed by rheological analysis (Fig. 2B). Gelation was shown to have commenced within 1 minute at 37 °C, as characterized by the crossover point of the rheological storage (G′) and loss moduli (G′′).21 Complete gelation was observed after approximately 45 minutes which is in agreement with the gelation time observed by vial tilt method. Notably, the G′ value at the gel formation time was 44.2 ± 1.2 kPa, which indicates a robust gel network structure was formed. This value is higher than the G′ value of chitosan hydrogel formed by chitosan/glycerol phosphate system (G′ value of ca. 5 kPa)10 and other chemically crosslinked chitosan networks in which G′ values in the range of 10–20 kPa have been reported.8,17,21 Higher G′ values (87–103 kPa) at the gel point were observed with increasing CS-azide wt% (Fig. S3 in ESI†). The fast gelation time observed is the result of two factors: the elevated temperature (37 °C) used for crosslinking; and the high functionality of the CS-azide. By using the generalization of the Arrhenius’ equation, the reaction rate should be double at the elevated temperature compared to at ambient temperature which consequently increases the rate of the crosslinking. In addition, the average functionality of the CS-azide is ca. 19.5 (by estimation of the degree of substitution by 1H NMR spectroscopy), and thus gelation is proposed to occur rapidly because the critical conversion is likely to be low as a consequence of the high average functionality of the chitosan-azide.
When the viscoelastic properties of the mixture were monitored for a longer period of time, the storage modulus of the network continued to increase slowly to an upper limit before decreasing significantly with extreme fluctuation in the collected data. This phenomenon is related to the syneresis properties of the cross-linked network, which occur when the network pressure exceeds the osmotic pressure generated by water bound within the gel, which in turn leads to gel contraction and water expelled to the environment.21 In turn, the gel shrinks away from the cone of the oscillatory rheometer causing a reduction in the recorded values. Similar rheological profiles were also reported for other chitosan hydrogel systems.21,40
The gel fraction, which is the percentage of the weight of the dried gel over the total weight of the polymer precursors, was measured as an indication of the efficiency of the gelation process. In these systems, the gel fraction was found to vary from 0.6 to 0.95 (Table 1) and increase with increasing concentration of CS-azide in the precursor solution, which indicates that the crosslinking is more efficient at higher CS-azide concentrations. Moreover, the gel fraction was found to be highest with the [alkyne]:[azide] ratio of 1:1 at all CS-azide concentrations, which shows that an equimolar ratio of [alkyne]:[azide] produced the most efficient crosslinking. The gels all displayed a high water sorption capacity, measured by equilibrium water content (EWC) of the prepared CS-PEG hydrogels found to be in the range of 85–95% at ambient temperature (Fig. 3). The EWC was found to decrease with increasing CS-azide concentration which indicates that the gels formed from higher CS concentrations have a higher crosslinking density which reduces the water sorption capacity. These values are in agreement with the gel fraction values, which showed higher crosslinking efficiency at higher CS-azide concentration. In addition, the EWC was found to decrease with increasing [alkyne]:[azide] ratio, i.e. increasing the [PEG-alkyne] while keeping the [CS-azide] constant, which is also a result of the increase in crosslinking density.
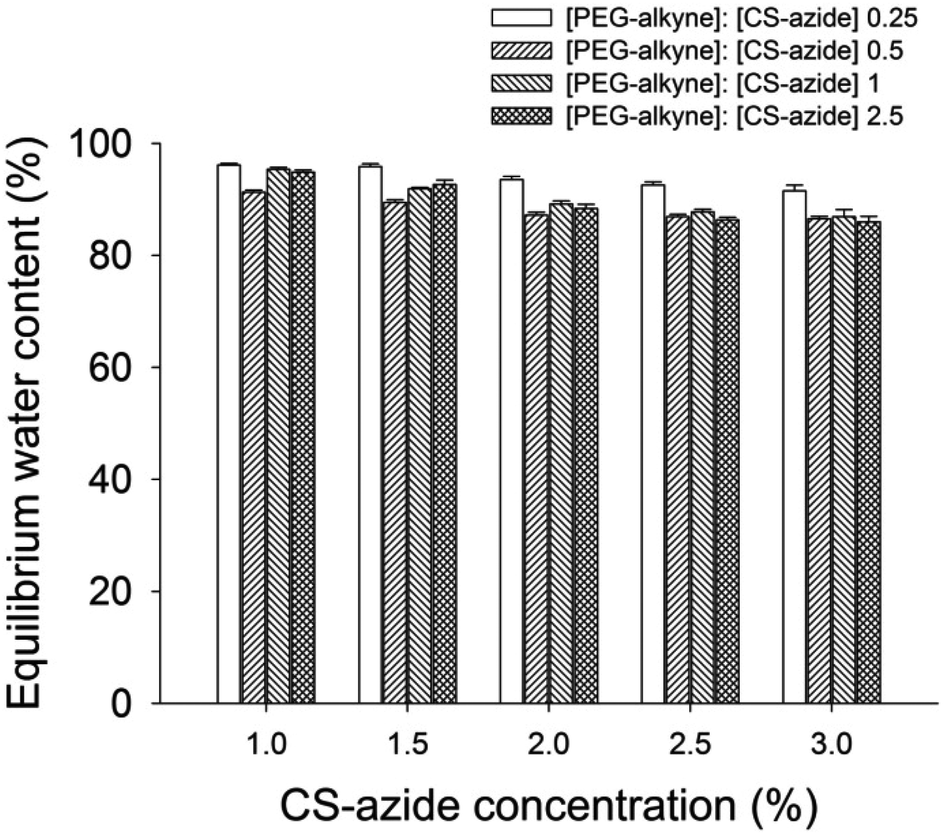 | ||
| Fig. 3 Equilibrium water content of CS hydrogel as a function of functionalized CS with different [alkyne]:[azide] ratios. | ||
| CS-azide wt% | [Azide] mM | PEG-alkyne wt% | [Alkyne] mM | Gel fraction |
|---|---|---|---|---|
| 1 | 53 | 0.5 | 13 | 0.68 ± 0.06 |
| 1 | 53 | 1 | 26 | 0.63 ± 0.06 |
| 1 | 53 | 2 | 53 | 0.84 ± 0.05 |
| 1 | 53 | 5 | 133 | 0.71 ± 0.13 |
| 2 | 106 | 1 | 26 | 0.81 ± 0.02 |
| 2 | 106 | 2 | 52 | 0.76 ± 0.05 |
| 2 | 106 | 4 | 106 | 0.90 ± 0.01 |
| 2 | 106 | 10 | 266 | 0.85 ± 0.09 |
| 3 | 159 | 1.5 | 39 | 0.89 ± 0.06 |
| 3 | 159 | 3 | 78 | 0.82 ± 0.01 |
| 3 | 159 | 6 | 159 | 0.96 ± 0.01 |
| 3 | 159 | 15 | 399 | 0.91 ± 0.07 |
Uniaxial compression tests were undertaken on the CS-PEG gels at concentrations of 1, 2 and 3 wt% of CS-azide (with [alkyne]:[azide] ratio of 1:1) after gelation was complete in order to evaluate their mechanical strength (Fig. 4). The gels were shown to display a rupture compression stress of 81 ± 11, 160 ± 15 and 192 ± 20 kPa and compressive modulus of 42 ± 4, 61 ± 7 and 77 ± 9 kPa respectively. The compressive strength of the CS-PEG hydrogels increased with increasing polymer precursor concentration, most likely a consequence of the CS-PEG gels formed from higher polymer concentration having higher crosslinking density, as evidenced in the swelling studies described above. It should be noted that these hydrogels, while being robust in terms of mechanical strength, have high water sorption capacity with the water contents of 97 wt%, 94 wt%, and 91 wt% respectively. These strengths and moduli values are higher than the values obtained for chitosan-PEG hydrogels by thiol–ene Michael reaction, which have the highest previously reported compressive moduli for chitosan-based hydrogels of 35–50 kPa,17,18 and comparable to the mechanical strength of the gels formed by step growth polymerization such as poly(carbonate)-PEG hydrogels formed by photo-catalyzed thiol–ene reaction, which has the compressive strength of 2–450 kPa.41 The high mechanical integrity observed from these gels is attributed to the step-growth crosslinking progress which results in highly heterogeneous network structure comprised of multiple network chains being able to distribute mechanical stress across the gel network.42
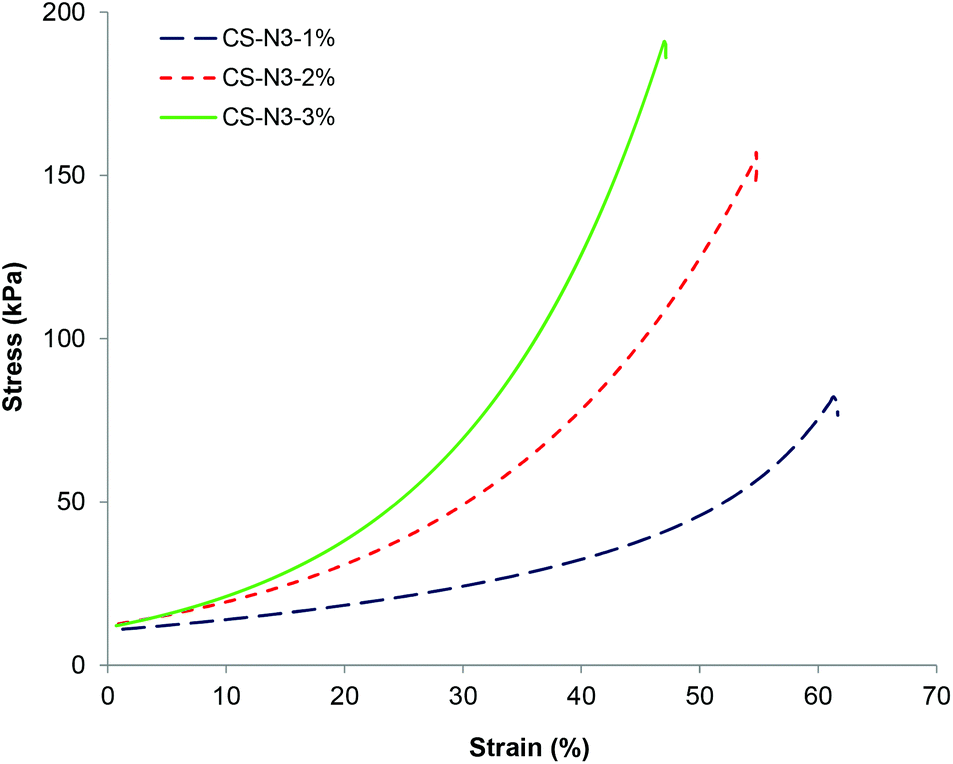 | ||
| Fig. 4 Stress–strain curves under uniaxial compression for CS gels formed from different CS-azide concentrations with the [azide]:[alkyne] ratio of 1:1. | ||
The in vitro stability of these hydrogels in PBS solution at pH 7.4 was also investigated by examining the mass of a range of hydrogels over time (Fig. 5). Specifically, gels comprised from CS-azide concentration of 1 wt%, 2 wt% and 3 wt% and PEG-alkyne with the [alkyne]:[azide] ratio of 1:1 were studied. The mass of the gels were observed to increase until it reached a critical point where sudden mass loss and collapse of the entire cleavage of the ester bonds, which in turn reduces the network occurred. The initial increase in mass is attributed to the crosslinking density and increase the internal volume of the network, allowing more water uptake. This degradation profile is consistent with bulk-degradation profile which results from hydrolysis of the ester linkage within hydrogel networks.43,44 CS gels formed from CS 1 wt% and 2 wt% solutions completely degraded in 50 days and 60 days respectively, while gels formed from CS 3 wt% solution were stable for more than 70 days. This slower degradation rate with increasing CS wt% is consistent with the higher crosslinking density of the hydrogel network. In the presence of lysozyme, an enzyme that can hydrolyze the glycosidic bond of the acetylated unit within the chitosan chain,45 faster degradation of hydrogels was observed where CS 3 wt% gel completely degraded in 70 days while the CS 1 wt% gel and CS 2 wt% gel degraded in 45 and 50 days respectively.
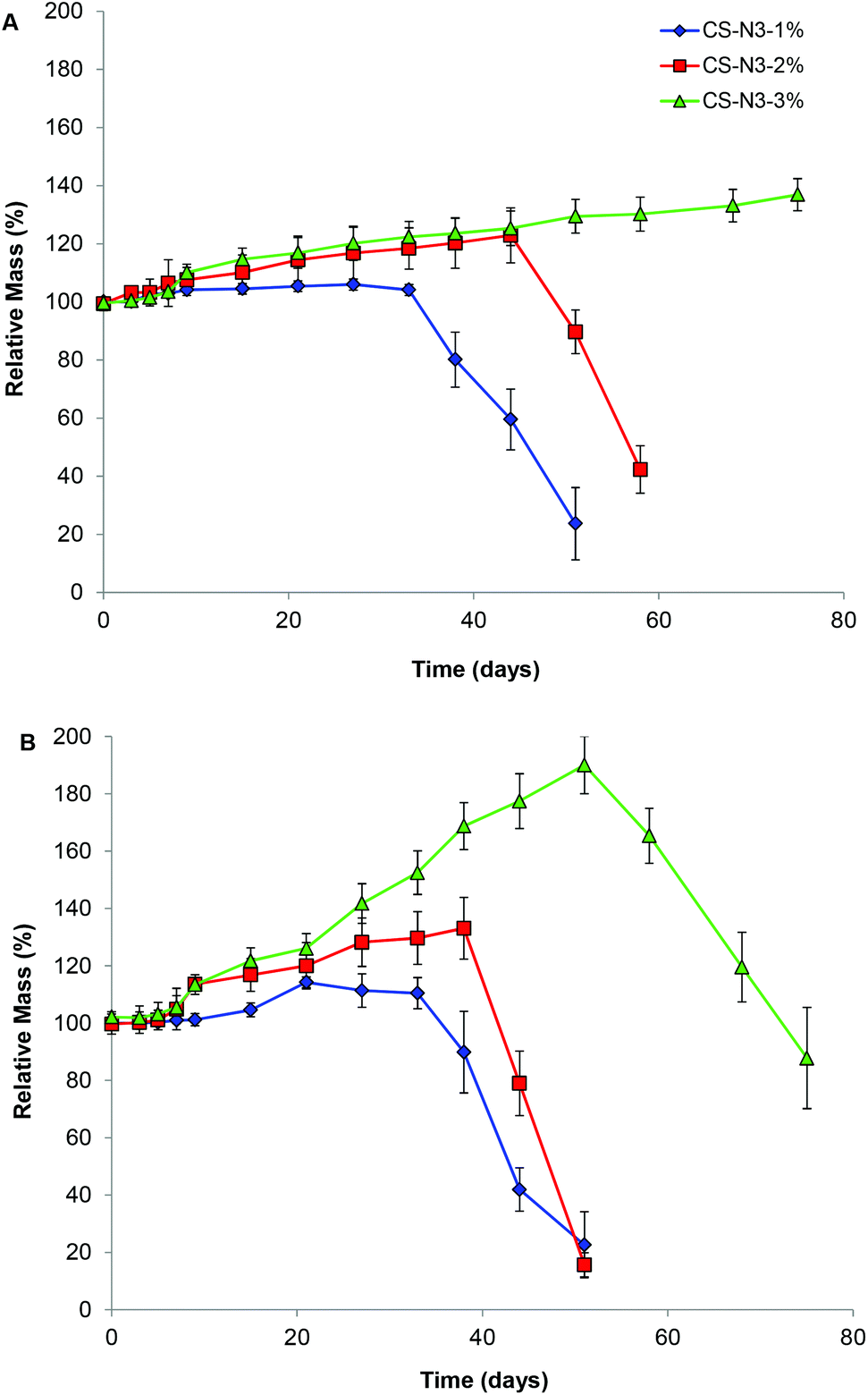 | ||
| Fig. 5 Degradation profiles of CS-PEG gels at different CS-azide concentrations in (A) PBS solution pH 7.4, and (B) PBS solution pH 7.4 with addition of lysozyme (0.1 mg mL−1). | ||
Tissue engineering scaffolds must also be able to support cellular function in vivo. As such, the ability of these hydrogels to support the growth of MSCs was assessed. MSCs were encapsulated in the gels in vitro and cell viability assessed after 24 h in culture. MSCs demonstrated high viability (>95%, Fig. 6A and B). MSCs were also seeded onto formed gels and viability and morphology assessed after 7 days in culture. The MSCs adhered to the surface of the gels and no cytotoxic response was observed, with the majority of cells remaining viable (Fig. 6C and D). Furthermore, morphological analysis of the MSCs revealed a typical fibroblastic morphology similar to that observed during in vitro culture on tissue culture polystyrene, with defined F-actin filaments and vinculin staining (important cytoskeletal components for cell adhesion and spreading) (Fig. 6E). These data confirm that the chitosan hydrogels prepared by the activated alkyne–azide cycloaddition chemistry are not cytotoxic and can support human MSCs.
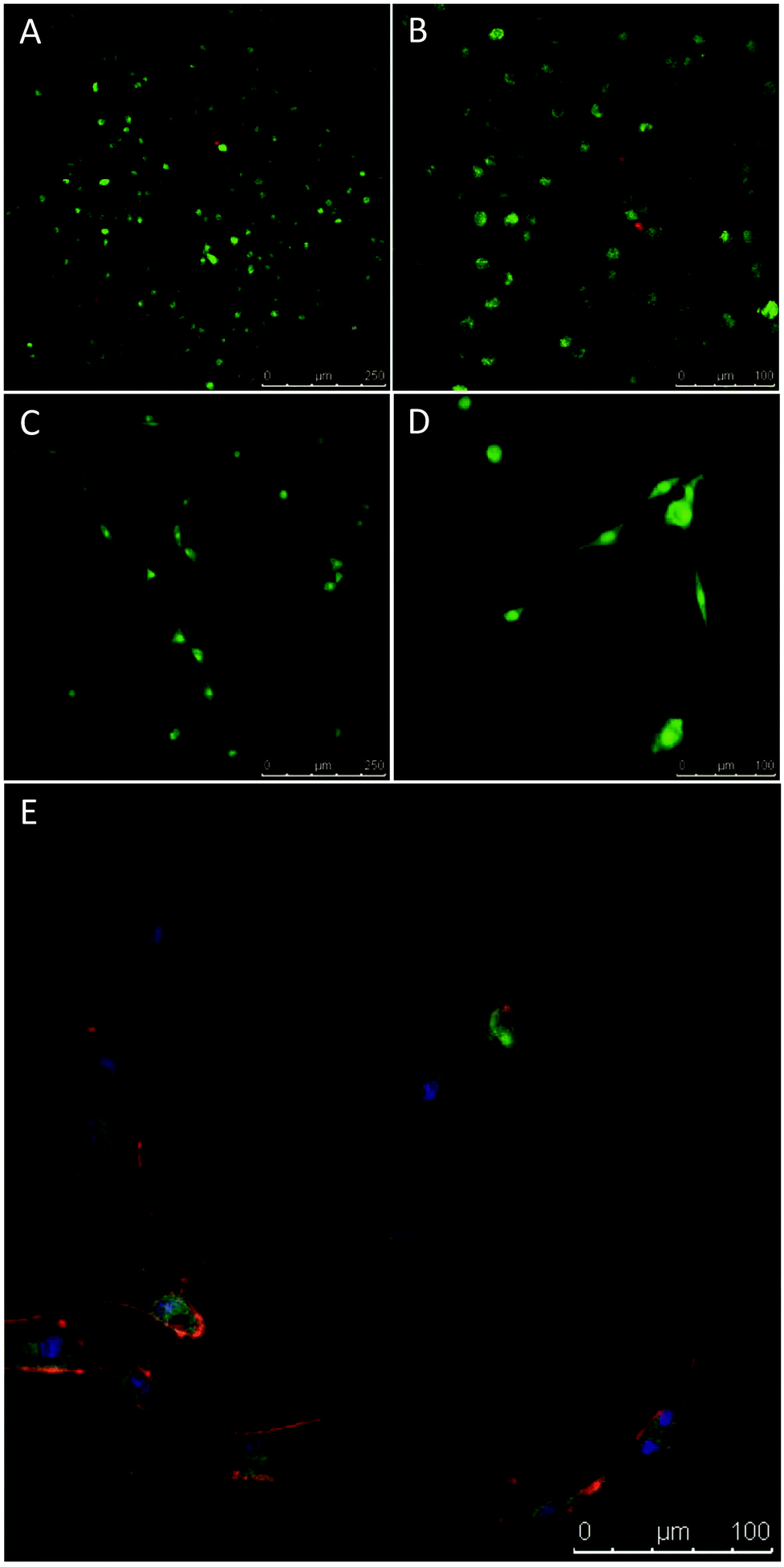 | ||
| Fig. 6 Novel chitosan-PEG gels supported MSC attachment and cell survival following encapsulation. (A & B) MSCs were encapsulated in PEG-chitosan gels and demonstrated high viability after 24 hours. Viability was assessed using calcein AM (green – viable cells) and ethidium homodimer-1 (red – dead cells). (C–E) MSCs were grown on the surface of PEG-chitosan gels for 7 days and cell viability and morphology assessed. Seeded cells demonstrated high viability after 7 days in culture (C & D). Viability in 2D was assessed as with 3D. Cell morphology was examined through staining of F-actin (red), vinculin (green) and the nucleus (blue). After 7 days, MSCs displayed a typical fibroblastic morphology (composite; E), with defined F-actin filaments and vinculin staining. | ||
4. Conclusions
In conclusion, we have developed a method for preparing in situ-forming chitosan-PEG hydrogels by copper free alkyne–azide click chemistry. The gelation time as well as gel properties and degradation rate can be tuned by varying the concentration of the chitosan and PEG precursors. These hydrogels have robust mechanical strength and are stable in PBS pH 7.4 solution for over 2 months. Preliminary cell studies showed that human MSCs can be encapsulated within the gel network with high viability, which indicates that the chemistry is bioorthogonal and the materials are nontoxic to cells. In addition, the hydrogels were able to support cell growth and attachment. As such, these studies indicate that the hydrogels have potential to be used as injectable biomaterials for tissue engineering.Acknowledgements
The Research Councils UK (RCUK) and The Royal Society are acknowledged for funding a fellowship to S.M.R. and A.P.D. respectively. We gratefully acknowledge financial support from BBSRC (BB/I002286/1) for funding postdoctoral fellowships to V.X.T., M.P.A. and H.T.G. as well as an ISIS award (BB/J014389/1) to A.P.D. The Z030 mechanical tester, GPC equipment and Cryo-SEM stage used in this research was obtained, through Birmingham Science City: Innovative Uses for Advanced Materials in the Modern World (West Midlands Centre for Advanced Materials Project 2), with support from Advantage West Midlands (AWM) and part funded by the European Regional Development Fund (ERDF). The University of Manchester Bioimaging Facility microscopes used in this study were purchased with grants from BBSRC, Wellcome Trust and The University of Manchester Strategic Fund.Notes and references
- I.-Y. Kim, S.-J. Seo, H.-S. Moon, M.-K. Yoo, I.-Y. Park, B.-C. Kim and C.-S. Cho, Biotechnol. Adv., 2008, 26, 1–21 CrossRef CAS PubMed.
- C. Shi, Y. Zhu, X. Ran, M. Wang, Y. Su and T. Cheng, J. Surg. Res., 2006, 133, 185–192 CrossRef CAS PubMed.
- M. P. Ribeiro, A. Espiga, D. Silva, P. Baptista, J. Henriques, C. Ferreira, J. C. Silva, J. P. Borges, E. Pires, P. Chaves and I. J. Correia, Wound Repair Regen., 2009, 17, 817–824 CrossRef PubMed.
- N. Q. Tran, Y. K. Joung, E. Lih and K. D. Park, Biomacromolecules, 2011, 12, 2872–2880 CrossRef CAS PubMed.
- M. Prabaharan, J. Biomater. Appl., 2008, 23, 5–36 CrossRef CAS PubMed.
- N. Bhattarai, J. Gunn and M. Zhang, Adv. Drug Delivery Rev., 2010, 62, 83–99 CrossRef CAS PubMed.
- Y. Hong, H. Song, Y. Gong, Z. Mao, C. Gao and J. Shen, Acta Biomater, 2007, 3, 23–31 CrossRef CAS PubMed.
- R. Jin, L. S. Moreira Teixeira, P. J. Dijkstra, M. Karperien, C. A. van Blitterswijk, Z. Y. Zhong and J. Feijen, Biomaterials, 2009, 30, 2544–2551 CrossRef CAS PubMed.
- S. M. Richardson, N. Hughes, J. A. Hunt, A. J. Freemont and J. A. Hoyland, Biomaterials, 2008, 29, 85–93 CrossRef CAS PubMed.
- A. Chenite, C. Chaput, D. Wang, C. Combes, M. D. Buschmann, C. D. Hoemann, J. C. Leroux, B. L. Atkinson, F. Binette and A. Selmani, Biomaterials, 2000, 21, 2155–2161 CrossRef CAS PubMed.
- L. Wang and J. P. Stegemann, Biomaterials, 2010, 31, 3976–3985 CrossRef CAS PubMed.
- N. Bhattarai, F. A. Matsen and M. Zhang, Macromol. Biosci., 2005, 5, 107–111 CrossRef CAS PubMed.
- N. Bhattarai, H. R. Ramay, J. Gunn, F. A. Matsen and M. Zhang, J. Controlled Release, 2005, 103, 609–624 CrossRef CAS PubMed.
- F. Ganji and M. J. Abdekhodaie, Carbohydr. Polym., 2008, 74, 435–441 CrossRef CAS.
- Y.-L. Chiu, M.-C. Chen, C.-Y. Chen, P.-W. Lee, F.-L. Mi, U. S. Jeng, H.-L. Chen and H.-W. Sung, Soft Matter, 2009, 5, 962–965 RSC.
- X. Qu, A. Wirsén and A. C. Albertsson, Polymer, 2000, 41, 4589–4598 CrossRef CAS.
- C. Chen, L. Wang, L. Deng, R. Hu and A. Dong, J. Biomed. Mater. Res., Part A, 2013, 101, 684–693 CrossRef PubMed.
- Y. Yu, C. Deng, F. Meng, Q. Shi, J. Feijen and Z. Zhong, J. Biomed. Mater. Res., Part A, 2011, 99, 316–326 CrossRef PubMed.
- S. Sakai, Y. Yamada, T. Zenke and K. Kawakami, J. Mater. Chem., 2009, 19, 230–235 RSC.
- H. Zhang, A. Qadeer and W. Chen, Biomacromolecules, 2011, 12, 1428–1437 CrossRef CAS PubMed.
- M. J. Moura, M. M. Figueiredo and M. H. Gil, Biomacromolecules, 2007, 8, 3823–3829 CrossRef CAS PubMed.
- R. A. A. Muzzarelli, Carbohydr. Polym., 2009, 77, 1–9 CrossRef CAS.
- H. C. Kolb, M. G. Finn and K. B. Sharpless, Angew. Chem., Int. Ed., 2001, 40, 2004–2021 CrossRef CAS.
- C. Barner-Kowollik, F. E. Du Prez, P. Espeel, C. J. Hawker, T. Junkers, H. Schlaad and W. Van Camp, Angew. Chem., Int. Ed., 2011, 50, 60–62 CrossRef CAS PubMed.
- M. Fernandez-Suarez, H. Baruah, L. Martinez-Hernandez, K. T. Xie, J. M. Baskin, C. R. Bertozzi and A. Y. Ting, Nat. Biotechnol., 2007, 25, 1483–1487 CrossRef CAS PubMed.
- S. T. Laughlin and C. R. Bertozzi, ACS Chem. Biol., 2009, 4, 1068–1072 CrossRef CAS PubMed.
- S. C. Hsiao, B. J. Shum, H. Onoe, E. S. Douglas, Z. J. Gartner, R. A. Mathies, C. R. Bertozzi and M. B. Francis, Langmuir, 2009, 25, 6985–6991 CrossRef CAS PubMed.
- M. A. Azagarsamy and K. S. Anseth, ACS Macro Lett., 2012, 2, 5–9 CrossRef PubMed.
- R. Langer and J. Vacanti, Science, 1993, 260, 920–926 CAS.
- A. B. Lowe, Polym. Chem., 2010, 1, 17–36 RSC.
- C. A. DeForest and K. S. Anseth, Nat. Chem., 2011, 3, 925–931 CrossRef CAS PubMed.
- J. Xu, T. M. Filion, F. Prifti and J. Song, Chem.–Asian J., 2011, 6, 2730–2737 CrossRef CAS PubMed.
- G. N. Grover, J. Lam, T. H. Nguyen, T. Segura and H. D. Maynard, Biomacromolecules, 2012, 13, 3013–3017 CrossRef CAS PubMed.
- D. L. Alge, M. A. Azagarsamy, D. F. Donohue and K. S. Anseth, Biomacromolecules, 2013, 14, 949–953 CrossRef CAS PubMed.
- Y. Fan, C. Deng, R. Cheng, F. Meng and Z. Zhong, Biomacromolecules, 2013, 2814–2821 CrossRef CAS PubMed.
- Z. Li, T. S. Seo and J. Ju, Tetrahedron Lett., 2004, 45, 3143–3146 CrossRef CAS.
- R. P. McGeary, Tetrahedron Lett., 1998, 39, 3319–3322 CrossRef CAS.
- I. A. Sogias, V. V. Khutoryanskiy and A. C. Williams, Macromol. Chem. Phys., 2010, 211, 426–433 CrossRef CAS.
- M. Lavertu, Z. Xia, A. N. Serreqi, M. Berrada, A. Rodrigues, D. Wang, M. D. Buschmann and A. Gupta, J. Pharma. Biomed., 2003, 32, 1149–1158 CrossRef CAS.
- S. Johnson, D. Dunstan and G. Franks, Colloid Polym. Sci., 2004, 282, 602–612 CAS.
- V. X. Truong, I. A. Barker, M. Tan, L. Mespouille, P. Dubois and A. P. Dove, J. Mater. Chem. B, 2013, 1, 221–229 RSC.
- M. W. Tibbitt, A. M. Kloxin, L. A. Sawicki and K. S. Anseth, Macromolecules, 2013, 46, 2785–2792 CrossRef CAS PubMed.
- H. Shih and C.-C. Lin, Biomacromolecules, 2012, 13, 2003–2012 CrossRef CAS PubMed.
- V. Truong, I. Blakey and A. K. Whittaker, Biomacromolecules, 2012, 13, 4012–4021 CrossRef CAS PubMed.
- K. M. Vårum, M. M. Myhr, R. J. N. Hjerde and O. Smidsrød, Carbohydr. Res., 1997, 299, 99–101 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3bm60159e |
| This journal is © The Royal Society of Chemistry 2014 |