 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Picomolar inhibition of cholera toxin by a pentavalent ganglioside GM1os-calix[5]arene†
Jaime
Garcia-Hartjes‡
a,
Silvia
Bernardi‡
b,
Carel A. G. M.
Weijers
a,
Tom
Wennekes
a,
Michel
Gilbert
c,
Francesco
Sansone
b,
Alessandro
Casnati
*b and
Han
Zuilhof
*ad
aLaboratory of Organic Chemistry, Wageningen University, Dreijenplein 8, 6703 HB Wageningen, The Netherlands. E-mail: Han.Zuilhof@wur.nl
bUniversità degli Studi di Parma, Dipartimento di Chimica, Parco Area delle Scienze 17/a, 43124 Parma, Italy. E-mail: Casnati@unipr.it
cInstitute for Biological Sciences, National Research Council Canada, 100 Sussex Drive, Ottawa, Ontario, Canada
dDepartment of Chemical and Materials Engineering, King Abdulaziz University, Jeddah, Saudi Arabia
First published on 20th May 2013
Abstract
Cholera toxin (CT), the causative agent of cholera, displays a pentavalent binding domain that targets the oligosaccharide of ganglioside GM1 (GM1os) on the periphery of human abdominal epithelial cells. Here, we report the first GM1os-based CT inhibitor that matches the valency of the CT binding domain (CTB). This pentavalent inhibitor contains five GM1os moieties linked to a calix[5]arene scaffold. When evaluated by an inhibition assay, it achieved a picomolar inhibition potency (IC50 = 450 pM) for CTB. This represents a significant multivalency effect, with a relative inhibitory potency of 100![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 compared to a monovalent GM1os derivative, making GM1os-calix[5]arene one of the most potent known CTB inhibitors.
000 compared to a monovalent GM1os derivative, making GM1os-calix[5]arene one of the most potent known CTB inhibitors.
Introduction
Cholera still represents a serious health problem in areas of the developing world where there is a lack of clean water and proper sanitation. In 2012, the World Health Organization estimated that annually 3–5 million cholera cases occur that result in more than 100000 deaths.1 Although several treatments exist for cholera,2 resistance development and mutations in the causative pathogen mean that efforts made to better understand the disease pathogenesis and develop new treatments are crucial.3,4 The symptoms of cholera are caused by cholera toxin (CT), which is produced by the Vibrio cholerae bacterium. CT is a member of the AB5 toxin family that contains a pentameric binding domain (CTB) for recognition and binding to cell surfaces.5 The natural target ligand for CTB is the glycosphingolipid ganglioside GM1, on cellular membranes of the infected hosts’ intestinal epithelial surface. CTB can bind five GM1 saccharide epitopes simultaneously with the terminal Gal- and the Neu5Ac carbohydrate units of the ganglioside as the major contributors to the binding.6,7 The adhesion of CTB to ganglioside GM1 on cell surfaces is the prerequisite for endocytosis of the toxic enzymatically active A subunit of CT, and the ensuing severe clinical symptoms.8 One avenue in cholera research is to study the binding of CTB to GM1 and to develop CTB inhibitors that might prevent CT from binding the hosts’ cell surface and thereby also the development of cholera. Here, we present the second of two examples of a pentavalent GM1os-based inhibitor for CTB, GM1os-calix[5]arene (1; Fig. 1). In the previous paper in this issue, we also reported on pentavalent inhibitors of CTB based on a GM1os-presenting corannulene scaffold. In the past, several studies have focused on the development of multivalent glycosylated inhibitors for CTB based on ganglioside GM1.9 It is noteworthy that in none of these studies, inhibitors were investigated with a pentavalent structure that matches the pentavalent structure of CTB. On the other hand, pentavalent cyclens10,11 and cyclic peptides12 have been described as CTB inhibitors, but those contained only the much simpler galactoseepitope likely to get around the difficulty to obtain sufficient tailor-made GM1os. Therefore, also GM1 mimics have been used, e.g. Thompson and Schengrund described poly(propylene) imine dendrimers that present the Galβ1–3GalNAcβ1–4(Neu5Acα2–3)Galβ-epitope of GM1,13 with IC50 values for the tetravalent and octavalent dendrimers of 7 and 3 nM, respectively. Bernardi et al. published a series of GM1-mimics (pseudo-GM1), in which the residues in the GM1os that are not essential for binding were replaced by a conformationally restricted cyclohexane-diol and the Neu5Ac-unit was substituted by various α-hydroxy acids.14,15 When attached to multivalent dendritic structures,16 the relative inhibitory potency (RIP) values per mimic unit of the tetravalent and octavalent inhibitors were 111 and 55, respectively. Interestingly, when the same mimic was linked to a divalent calix[4]arene scaffold,17 a 4000-fold enhancement in binding efficiency was achieved compared to the monovalent pseudo-GM1. These data suggested to us that the calixarenemacrocycle, from which the binding inhibitors are projected, could be a promising multivalent scaffold18–20 to design CTB inhibitors with improved efficiency. In collaboration with the group of Pieters, we previously published divalent, tetravalent, and octavalent dendritic structures decorated with GM1os.21,22 For the octavalent compound the unprecedentedly low IC50 value of 50 pM was observed with an RIP of 17500 per arm compared to its monovalent counterpart. However, its mismatched valency compared to CTB prompted us to investigate a pentavalent scaffold as core structure that when decorated with GM1os has the potential to form 1:1 inhibitor–CTB complexes. The current paper presents the convergent synthesis of the first, water-soluble, pentavalent CTB inhibitor (1), which was made by coupling five GM1os units to a calix[5]arene scaffold.
![Developed CTB inhibitor: penta-GM1os-calix[5]arene (1).](/image/article/2013/OB/c3ob40515j/c3ob40515j-f1.gif) | ||
| Fig. 1 Developed CTB inhibitor: penta-GM1os-calix[5]arene (1). | ||
Results and discussion
We designed a 5-fold symmetric calix[5]arene as a pentavalent scaffold structure. This calix[5]arene (Fig. 1) presents small methoxy groups at the lower rim, which confer a high conformational flexibility to the macrocyclic structure.23 The upper rim of the calixareneinhibitor is decorated with the GM1 pentasaccharide separated from the macrocyclic core by appropriate linkers. Fan and coworkers12 have demonstrated that an optimal linker length is vital for the potency of a synthetic multivalent inhibitor. For the calix[5]arene, described here, a 31 atom-containing linker was chosen. This should allow the simultaneous interaction of the five GM1os units with the five B-subunits of a single toxin.5The route towards our target (1) started with the synthesis of the pentavalent scaffold that began with the preparation of the known p-tert-butyl-calix[5]arene.24 This product was converted into p-H-calix[5]arene 225 by following literature procedures. Next, penta-aldehyde 3 was obtained in 57% yield from 2 by exploiting the Duff formylation reaction.26,27 Compound 3 was subsequently methylated at the lower rim by using CH3I and K2CO3 in acetonitrile affording the penta-methoxy-calix[5]arene 4 in 68% yield (Scheme 1).
![Synthesis of the penta-azido-calix[5]arene (8) scaffold. Reagents and conditions: (a) HMTA, CF3COOH, reflux, N2, 5 days, 57%; (b) CH3I, K2CO3 CH3CN, reflux, N2, 20 h, 68%; (c) NaClO2, NH2SO3H, (CH3)2CO, CHCl3, H2O, rt, 24 h, 79%; (d) (COCl)2, dry CH2Cl2, N2, rt, 18 h, quant.; (e) Et3N, dry CH2Cl2, N2, rt, 20 h, 44%.](/image/article/2013/OB/c3ob40515j/c3ob40515j-s1.gif) | ||
| Scheme 1 Synthesis of the penta-azido-calix[5]arene (8) scaffold. Reagents and conditions: (a) HMTA, CF3COOH, reflux, N2, 5 days, 57%; (b) CH3I, K2CO3 CH3CN, reflux, N2, 20 h, 68%; (c) NaClO2, NH2SO3H, (CH3)2CO, CHCl3, H2O, rt, 24 h, 79%; (d) (COCl)2, dry CH2Cl2, N2, rt, 18 h, quant.; (e) Et3N, dry CH2Cl2, N2, rt, 20 h, 44%. | ||
Oxidation of 4 with NaClO2 and NH2SO3H produced the penta-carboxylic acid 5. The unsymmetrically substituted azido-penta-(ethyleneglycol)-amine 7 was synthesized from hexa-ethylene glycol by ditosylation, substitution to the diazide, and finally selective Staudinger reduction of one azide.28,29 Initial attempts to condense amine7 with the carboxylic acids in 5 using HBTU resulted in difficult purification and low yields (∼20%) of 8. However, when this spacer (7) was attached to calix[5]arene 5via penta-acyl chloride intermediate 6 it provided penta-azido-calix[5]arene 8 in a 44% yield.
With the calix[5]arene (8) scaffold in hand we proceeded to the next stage, attaching five GM1 oligosaccharides. We chose the copper-catalyzed azide–alkynecycloaddition (CuAAC) reaction to achieve this, which meant a GM1os derivative with a terminal alkyne was required. This C11-alkyne-terminated GM1os 9 (Scheme 2) was made via a chemo-enzymatic procedure previously reported by us,30 which allowed the production of 9 on gram scale. Compound 9 was subsequently “clicked” to scaffold 8 under standard CuAAC conditions in H2O while exposed to microwave irradiation to successfully provide our crude target inhibitor1. With our target pentavalent GM1os-calix[5]arene 1 in hand, in order to properly assess the role of the GM1os in inhibitor1, we also set out to synthesize derivatives of 1 containing fragments of the GM1os to use for comparison in the biological assays. The first of these was penta-GM2os-calix[5]arene (11) that lacks the terminal galactoseepitope compared to the GM1os. We synthesized 11 using the same CuAAC reaction conditions from scaffold 8 and a chemo-enzymatically produced alkyne-terminated C11-linked GM2os sugar (10).
![Synthesis of GM1os-calix[5]arene (1) and GM2os-calix[5]arene (11); (a) CuSO4·5H2O, sodium ascorbate, Triton X-100, CH3OH, H2O, MW (150 W), 80 °C, 1 h; 51% 1, 59% 11.](/image/article/2013/OB/c3ob40515j/c3ob40515j-s2.gif) | ||
| Scheme 2 Synthesis of GM1os-calix[5]arene (1) and GM2os-calix[5]arene (11); (a) CuSO4·5H2O, sodium ascorbate, Triton X-100, CH3OH, H2O, MW (150 W), 80 °C, 1 h; 51% 1, 59% 11. | ||
With both our target 1 and its derivative 11 in hand as crude products, we investigated what purification method would be suitable for these large complex molecules. An initial purification by size exclusion chromatography (SEC) efficiently removed an excess of alkyne-terminated GM1os 9 and GM2os 10, respectively. However, the crude products both also contained a minor amount of tetravalent byproducts as could be clearly seen with mass spectrometry and their separation proved to be quite challenging. Initial attempts to separate these by aqueous HPLCGPC failed, but after extensive optimization, reversed phase HPLC purification proved the most successful for this final purification step (see experimental section for details). Fig. 2 shows a typical HPLC chromatogram for the separation of both the GM1os- and GM2os-calix[5]arenes. Despite attempts to improve the moderate resolution, the purification remained quite complicated because of long elution times (up to 40 minutes per run) and high affinity of the products with the column material. Collection of small fractions in a specific retention time window over multiple HPLC injections and subsequent lyophilization resulted indeed in pure pentavalent GM1os- and GM2os-calix[5]arenes (1 and 11) as shown by HR-MS and NMR analyses. The other impure fractions that also contained 1 or 11 were collected, pooled, lyophilized and re-injected to achieve optimal yields. Attempts were made to isolate the tetravalent byproducts, but insufficient amounts could be obtained for further analyses.
![LCMS trace of the purification of GM2-calix[5]arene 11. (left) Chromatogram of the purification on a reversed phase column (see experimental section for details). (right) Mass spectra for two fractions, the pentavalent product 11 at RT = 12 min, and the tetravalent byproduct at 21 min.](/image/article/2013/OB/c3ob40515j/c3ob40515j-f2.gif) | ||
| Fig. 2 LCMS trace of the purification of GM2-calix[5]arene 11. (left) Chromatogram of the purification on a reversed phase column (see experimental section for details). (right) Mass spectra for two fractions, the pentavalent product 11 at RT = 12 min, and the tetravalent byproduct at 21 min. | ||
Besides the GM2os containing calixarene (11), we also prepared two further derivatives of 1 that contained fragments of GM1os, a pentavalent β-galactoside- (16) and β-lactoside-calix[5]arene (17) (Scheme 3). These more simple carbohydrates enabled a modified synthesis procedure that circumvented the potentially challenging HPLC purification, as encountered for compounds 1 and 11. The coupling was also performed by employing the microwave-assisted CuAAC reaction on penta-azido-calixarene scaffold 8, but instead of using the deprotected carbohydrates, acetyl-protected galactoside12, and lactoside 13 were reacted. The resulting products could now be purified by normal phase silica gel chromatograpy. The acetyl-protected 14 and 15 were deprotected by employing standard Zemplén31 conditions to obtain galactoside-calix[5]arene 16, and lactoside-calix[5]arene 17, respectively, which did not require further purification after work-up.
![Synthesis of galactoside-calix[5]arene (16), and lactoside-calix[5]arene (17); (a) CuSO4·5H2O, sodium ascorbate, DMF, H2O, MW (150 W), 80 °C, 1 h; 67% 14, 57% 15; (b) NaOMe–MeOH, 4 h – 18 h, H+-resin; 90% 16, 72% 17.](/image/article/2013/OB/c3ob40515j/c3ob40515j-s3.gif) | ||
| Scheme 3 Synthesis of galactoside-calix[5]arene (16), and lactoside-calix[5]arene (17); (a) CuSO4·5H2O, sodium ascorbate, DMF, H2O, MW (150 W), 80 °C, 1 h; 67% 14, 57% 15; (b) NaOMe–MeOH, 4 h – 18 h, H+-resin; 90% 16, 72% 17. | ||
Finally, in order to properly determine the multivalency effect of the interaction of 1 with CTB in our biological assays, we also synthesized the monovalent GM1os derivative 20 (Scheme 4). This was achieved by first in situ generation of the acyl chloride of commercially available 4-methoxybenzoic acid with oxalyl chloride and, subsequently, reacting this with amino-azide 7, yielding azide19 in 20% over two steps. Again, by employing the microwave-assisted CuAAC reaction on alkyne-terminated 9 and azide 19, GM1os-monomer 20 was obtained in a reasonable yield (49%).
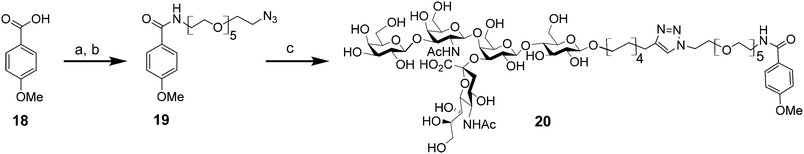 | ||
| Scheme 4 Synthesis of GM1-monomer 20; (a) (COCl)2, dry CH2Cl2, N2, rt, 18 h; (b) 7, Et3N, dry CH2Cl2, N2, rt, 20 h; 20% in two steps; (c) 9, CuSO4·5H2O, sodium ascorbate, Triton X-100, CH3OH, H2O, MW (150 W), 80 °C, 1 h; 49% 20. | ||
The inhibitory potency of the four pentavalent compounds (1, 11, 16, and 17) was determined by ELISA experiments. In the assays, the ability of 1, 11, 16 and 17 to inhibit the binding of HRP-labeled CTB was measured in competition with the natural ligandganglioside GM1, which was adsorbed to the well surface of the ELISA plate. GM1os-calix[5]arene 1 showed a high inhibition potency, i.e., a very low IC50 value of 450 pM (Fig. 3, Table 1). Comparing the IC50 value (44 μM) of the monovalent control compound (20) to that of 1 revealed a 100-thousand increase in inhibitory potency, and an RIP of 20-thousand per arm. Pentavalent inhibitors based on a more rigid corannulene scaffold, which we also report in this issue, inhibited CTB in the nanomolar range.32 Assay results for the GM2os-calix[5]arene 11 confirmed the importance of using GM1os. Lacking only the terminal galactose compared to 1, it produced an IC50 of 9 μM, which is 20-thousand fold worse compared to 1. The galactose-terminated (16) and lactose-terminated (17) calix[5]arenes displayed a higher inhibition concentration than their solubility in the assay medium, and their IC50 could therefore only be determined as being >1 mM (Table 1).
![GM1os-calix[5]arene GM2os-calix[5]arene GM1os-monomer; Fitted curves of the experimental ELISA inhibition data. For details of the inhibition assays see experimental section.](/image/article/2013/OB/c3ob40515j/c3ob40515j-f3.gif) | ||
Fig. 3
 GM1os-calix[5]arene GM1os-calix[5]arene  GM2os-calix[5]arene GM2os-calix[5]arene  GM1os-monomer; Fitted curves of the experimental ELISA inhibition data. For details of the inhibition assays see experimental section. GM1os-monomer; Fitted curves of the experimental ELISA inhibition data. For details of the inhibition assays see experimental section. | ||
| Entry | Saccharide | Valency | # | IC50 |
|---|---|---|---|---|
| 1 | GM1os | 5 | 1 | 450 pM |
| 2 | GM1os | 1 | 20 | 44 μM |
| 3 | GM2os | 5 | 11 | 9 μM |
| 4 | Galactose | 5 | 16 | >1 mM |
| 5 | Lactose | 5 | 17 | >1 mM |
Conclusions
In summary, we here report the synthesis and initial biological evaluation of the first known example of a pentavalent GM1os-based inhibitor of cholera toxin that matches the valency of the cholera toxin B-subunit. With an IC50 of 450 pM, the pentavalent GM1os-calix[5]arene (1) also displays the highest relative inhibitory potency, 20-thousand per arm (compared to 20), documented thus far for CT inhibitors. We are currently using the here reported convergent synthetic route to further explore the structure–activity-relationship of 1 and improve its potency. Among other issues we are interested in investigating the effect of the length, rigidity and hydrophobicity of the used spacer and restricting the flexibility of the calix[5]arene scaffold to a fixed cone structure.Experimental section
General experimental information
All moisture sensitive reactions were carried out under a nitrogen atmosphere, using previously oven-dried glassware. All dry solvents were prepared according to standard procedures, distilled before use and stored over 3 Å or 4 Å molecular sieves. Reagents were obtained from commercial sources and used without further purification unless stated otherwise. Analytical TLC was performed using prepared plates of silica gel (Merck 60 F-254 on aluminium) and then, according to the functional groups present on the molecules, revealed with UV light or using staining reagents: FeCl3 (1% in H2O–CH3OH 1:1), H2SO4 (5% in EtOH), ninhydrin (5% in EtOH), basic solution of KMnO4 (0.75% in H2O). Merck silica gel 60 (70–230 mesh) was used for flash chromatography and for preparative TLC plates. 1H NMR and 13C NMR spectra were recorded on Bruker AV300, Bruker AV400, Bruker DPX400, and Bruker AV600 equipped with cryoprobe spectrometers (observation of 1H nucleus at 300 MHz, 400 MHz, 600 MHz, respectively, and of 13C nucleus at 75 MHz, 100 MHz, and 151 MHz, respectively). Chemical shifts are reported in parts per million (ppm), calibrated on the residual peak of the solvent, whose values are referred to tetramethylsilane (TMS, δTMS = 0), as the internal standard. 13C NMR spectra were performed with proton decoupling. Electrospray ionization (ESI) mass analyses were performed with a Waters spectrometer, while high resolution ESI mass analyses were recorded on a Thermo Scientific Q Exactive spectrometer. Melting points were determined on an Electrothermal apparatus in closed capillaries. Microwave reactions were performed on a CEM Discovery System reactor running on Discover Application Chemdriver Software v3.6.0. HPLC was performed on an HP 1100 series with a DAP 190–600 nM detector, equipped with a Waters Xterra 100 × 4.6 mm C18 column eluted with isocratic iPrOH–H2O 35:65, and a flow of 0.4 mL min−1, unless stated otherwise. Materials for the ELISA experiments i.e. bovine serum albumin (BSA), bovine brain GM1, ortho-phenylenediamine (dihydrochloride salt) (OPD), cholera toxin horseradish peroxide (CTB-HRP) conjugate, Tween-20, 30% H2O2 solution, sodium citrate, and citric acid were purchased at Sigma Aldrich and used without further modification, phosphate-buffered saline (PBS) 10× concentrate was diluted ten times with demineralized water prior to use. Nunc F96 MaxisorpTM 96-well microtiter plates were used as purchased at Thermo Scientific. The microtiter plates were washed using an automated Denville® 2 Microplate Washer. Optical density (OD) was measured between 1.5 and 0.5 units on a Thermo Labsystems Multiskan Spectrum Reader running on Skanit software version 2.4.2. Data analysis and curve fitting of inhibition experiments were performed on Prism Graphpad software v5.04. Simplified nomenclature proposed by Gutsche33 is used to name the calix[5]arene compounds.
Compounds 31,32,33,34,35-pentahydroxycalix[5]arene 2,25 17-azide-3,6,9,12,15-pentaoxaheptadecane-1-amine 7,29 undec-10-ynyl-2,3,4,6-tetra-O-acetyl-β-D-galactopyranoside 12,30 undec-10-ynyl-2,3,4,6-tetra-O-acetyl-β-D-galactopyranosyl-(1→4)-2,3,6-tri-O-acetyl-β-D-glucopyranoside 1330 were prepared according to literature procedures.
GM1-calix[5]arene (1)
Calix[5]arene 8 (15.7 mg, 6.93 μmol) was dissolved in 0.5 mL of CH3OH in a microwave tube. Then the GM1os derivative 9 (59.8 mg, 52.1 μmol), previously synthesized by chemo-enzymatic procedures,30,34 was combined with CuSO4·5H2O (0.52 mg, 2.1 μmol), sodium ascorbate (0.82 mg, 4.2 μmol), 4 mL of H2O and a drop of Triton X-100. The mixture was heated at 80 °C by microwave irradiation (150 W) for 60 min. The reaction progression was monitored viaTLC (eluent: AcOEt–CH3OH–H2O–AcOH 4:2:1:0.1) and ESI-MS analyses. The crude mixture was purified via size exclusion column chromatography (Sephadex G-15, eluent: H2O 100%) and HPLC purification (see General information) giving product 1 as a white solid. Yield: 51%. 1H NMR (600 MHz, D2O): δ (ppm) 7.61 (s, 5H, H5 triazole); 7.50 (s, 10H, Ar); 4.69 (bs, 5H, H1-GalNAc); 4.43 (d, 5H, J = 8.0 Hz, H1-Gal′); 4.44–4.41 (m, 5H, H1-Gal); 4.38–4.37 (m, 5H); 4.32–4.31 (5H, d, J = 7.9 Hz, H1-Glc); 4.05–4.02 (m, 15H); 3.94–3.92 (m, 5H, H2-GalNAc); 3.86–3.60 (m, 105H); 3.60–3.45 (m, 80H); 3.46–3.33 (m, 75H); 3.26–3.24 (m, 5H, H2-Gal); 3.18–3.15 (m, 5H, H2-Glc); 3.10 (bs, 10H); 2.56–2.54 (m, 5H, H3a-Neu5Ac); 2.45–2.42 (m, 10H, triazole-CH2CH2CH2); 1.92 (s, 15H, NC(O)CH3-Neu5Ac); 1.89 (s, 15H, NC(O)CH3-GalNAc); 1.85–1.80 (m, 5H, H3b-Neu5Ac); 1.44–1.35 (m, 20H, CH2 aliphatic chain); 1.14–1.08 (m, 10H, CH2CH2OC1-Glc), 1.03 (m, 40H, CH2 aliphatic chain). 13C NMR (151 MHz, D2O): δ (ppm) 175.4, 175.1, 174.5 (C(O)); 169.1 (ArC(O)NH); 159.7 (Ar-ipso); 134.8 (Ar-ortho); 129.2 (Ar-para); 128.9 (Ar-meta); 123.7 (C5 triazole); 105.1 (C1-Gal′); 103.0 (C1-Gal); 102.8 (C1-GalNAc); 102.5 (C1-Glc); 102.0 (C2-Neu5Ac); 80.7 (C3-GalNAc); 79.0 (C4-Glc); 77.6 (C4-Gal); 75.2 (C5-Gal′); 75.1 (C5-Glc); 74.9 (C3-Gal); 74.7; 74.4 (C5-Gal); 73.4 (C6-Neu5Ac); 73.1 (C2-Glc); 72.8 (C3-Gal′); 72.6 (C7-Neu5Ac); 71.0 (C2-Gal′); 70.9 (β-COCH2); 70.4 (C2-Gal); 70.1–69.8; 69.4; 69.2; 69.0; 68.9; 68.4; 68.3 (C4-GalNAc); 63.1; 61.4; 61.3; 60.9; 60.5; 52.0; 51.5 (C2-GalNAc); 50.3; 40.0; 37.2; 31.1 (ArCH2Ar); 29.3, 29.1, 29.0, 28.9, 28.8 (CH2 aliphatic chain); 25.6 (CH2CH2OC1-Glc); 25.0 (triazole-CH2CH2CH2); 23.0 (NHC(O)CH3-GalNAc); 22.4 (NHC(O)CH3-Neu5Ac). HR-ESI-MS(−): m/z 1600.5013 [100% (M − 5H)5−] calcd: 1600.7112.
5,11,17,23,29-Pentaformyl-31,32,33,34,35-pentahydroxycalix[5]arene (3)
Calix[5]arene 225 (0.41 g, 0.78 mmol) was added to a solution of HMTA (2.5 g, 17.8 mmol) in 50 mL TFA and the mixture was refluxed for 5 days under N2. The solvent was then removed under reduced pressure and the residue dissolved in 12 mL of a 1:1 CH2Cl2–HCl 1 M solution. The mixture was stirred at room temperature for 24 h. The aqueous phase was extracted with CH2Cl2 (5 × 5 mL). The combined organic phases were washed with water (2 × 10 mL), dried over anhydrous Na2SO4, filtered and the solvent removed under vacuum. The residue was purified by trituration in CHCl3–hexane 1:1 to give the product 3 as a brownish solid. Yield: 57%. 1H NMR (300 MHz, CDCl3–CD3OD 9:1): δ (ppm) 9.75 (s, 5H, CHO); 7.70 (s, 5H, ArOH); 7.22 (s, 10H, ArH); 3.45 (s, 10H, ArCH2Ar). 13C NMR (75 MHz, CDCl3–CD3OD 9:1): δ (ppm) 192.1 (CHO); 150.6 (Ar-ipso); 131.9 (Ar-para); 128.1 (Ar-meta); 127.2 (Ar-ortho); 31.4 (ArCH2Ar). HR-ESI-MS(+): m/z 671.1921 [100% (M + H)+] calcd: 671.1917. M.p. > 300 °C.
5,11,17,23,29-Pentaformyl-31,32,33,34,35-pentamethoxy-calix[5]arene (4)
In a two-neck round-bottomed flask, pentaformylcalix[5]arene 3 (0.7 g, 1.1 mmol) was dissolved in 150 mL of dry CH3CN, then K2CO3 (4.5 g, 32 mmol) and CH3I (2 mL, 32 mmol) were added and the mixture was refluxed for 20 h under a nitrogen atmosphere. The solvent was removed under reduced pressure and the residue dissolved in 150 mL of a 1:1 solution CH2Cl2–HCl 1 M. The mixture was stirred for 2 h at room temperature. The organic layer was separated, and the aqueous phase extracted with CH2Cl2 (2 × 50 mL). The combined organic phases were washed with water (2 × 100 mL), dried over anhydrous Na2SO4, filtered and the solvent removed under reduced pressure. The product 4 was obtained as a brown solid. Yield: 68%. 1H NMR (300 MHz, CDCl3): δ (ppm) 9.72 (s, 5H, CHO); 7.50 (s, 10H, ArH); 3.92 (s, 10H, ArCH2Ar); 3.25 (s, 15H, OCH3). 13C NMR (75 MHz, CDCl3–CD3OD 9:1): δ (ppm) 191.4 (CHO); 161.8 (Ar-ipso); 134.8 (Ar-para); 132.0 (Ar-ortho); 130.8 (Ar-meta); 60.6 (OCH3); 30.7 (ArCH2Ar). HR-ESI-MS(+): m/z 741.2700 [100% (M + H)+] calcd: 741.2700. M.p.: 217–219 °C.
5,11,17,23,29-Pentacarboxy-31,32,33,34,35-pentamethoxycalix[5]arene (5)
Pentaformyl-pentamethoxycalix[5]arene 4 (0.25 g, 0.34 mmol) was dissolved in a two-neck round-bottomed flask in 100 mL of a mixture acetone–CHCl3 1:1, and cooled to 0 °C with an ice-water bath. In another flask, a solution of NaClO2 80% pure (0.47 g, 4.20 mmol) was dissolved in the minimum amount of water. Subsequently, sulfamic acid (0.49 mg, 5.04 mmol) was added. This solution was slowly poured into the reaction flask. The mixture was stirred at 0 °C for 15 min and gradually warmed to room temperature while it remained stirred for 24 h. The solvent was then removed under reduced pressure and the residue triturated with 1 M HCl. After filtration on a Büchner funnel, product 5 was obtained as a solid. Yield: 79%. 1H NMR (300 MHz, CD3OD): δ (ppm) 7.75 (s, 10H, ArH); 3.93 (s, 10H, ArCH2Ar); 3.30 (s, 15H, OCH3). 13C NMR (25 MHz, CD3OD): δ (ppm) 169.6 (CO); 162.1 (Ar-ipso); 135.8 (Ar-ortho); 132.0 (Ar-meta); 126.8 (Ar-para); 61.3 (–OCH3); 31.9 (ArCH2Ar). HR-ESI-MS(+): m/z 843.2269 [100% (M + Na)+] calcd: 843.2265.
5,11,17,23,29-Pentakis[(17-azide-3,6,9,12,15-pentaoxahepta-decane-1-amino)carbonyl]-31,32,33,34,35-pentamethoxy-calix[5]arene (8)
In a round-bottomed flask, 0.12 g of calix[5]arene 5 (0.14 mmol) and 0.51 mL of oxalyl chloride (5.82 mmol) were solubilized in 15 mL of dry CH2Cl2 under a nitrogen atmosphere. The solution was stirred for 18 h at room temperature and then the solvent evaporated to dryness. The residual compound 6 was dissolved again in 5 mL of dry CH2Cl2 and then added dropwise to a solution of amine compound 7 (0.29 g, 0.87 mmol) and NEt3 (0.12 mL, 0.87 mmol) in 5 mL of dry CH2Cl2. The mixture was stirred for 20 h at room temperature under a nitrogen atmosphere. The mixture was then washed with 1 M HCl, an aqueous solution of Na2CO3 and water till neutral pH was reached. The solvent was removed under vacuum and the crude purified by flash chromatography (eluent: CHCl3–CH3OH 95:5) to give the product 8 as a yellow oil. Yield: 44%. 1H NMR (400 MHz, CDCl3): δ (ppm) 7.50 (s, 10H, ArH); 7.07 (bs, 5H, CONH); 3.89 (s, 10H, ArCH2Ar); 3.65–3.50 (m, 110H, OCH2, CH2NHCO); 3.34 (t, 10H, J = 4.8 Hz, CH2N3); 3.28 (s, 15H, OCH3). 13C NMR (100 MHz, CDCl3): δ (ppm) 167.3 (CO); 159.2 (Ar-ipso); 134.2 (Ar-para); 129.8 (Ar-ortho); 128.2 (Ar-meta); 70.6, 70.5, 70.2, 70.0, (OCH2); 60.8 (OCH3); 50.6 (CH2N3); 39.7 (CH2NHCO); 30.9 (ArCH2Ar). HR-ESI-MS(+): m/z 1131.5698 [100% (M + 2H)2+]; calcd: 1131.5756.
GM2-calix[5]arene (11)
Calix[5]arene 8 (15.7 mg, 6.93 μmol) was dissolved in 0.5 mL of CH3OH in a microwave tube. Then the GM2os derivative 10 (50.5 mg, 52.1 μmol), previously synthesized by chemo-enzymatic procedures,30,34 was added together with samples of CuSO4·5H2O (0.52 mg, 2.1 μmol), sodium ascorbate (0.82 mg, 4.2 μmol), 4 mL of H2O and a drop of Triton X-100. The mixture was heated at 80 °C by microwave irradiation (150 W) for 60 min. The reaction progress was monitored viaTLC (eluent: AcOEt–CH3OH–H2O–AcOH 4:2:1:0.1) and ESI-MS analyses. The crude material was purified via size exclusion column chromatography (Sephadex G-15, eluent: H2O 100%), followed by HPLC purification (see General information) giving pure product 11 as a white solid. Yield: 59%. 1H NMR (400 MHz, D2O/CD3OD): δ (ppm) 7.65 (s, 5H, H5 triazole); 7.54 (s, 10H, ArH); 4.70 (m, 5H, H1-Gal (determined by HSQC)); 4.47–4.42 (m, 15H, H1-GalNAc); 4.36–4.35 (d, 5H, J = 7.6 Hz, H1-Glc); 4.08–4.06 (m, 10H); 3.89–3.66 (m, 95H, ArCH2Ar); 3.58–3.43 (m, 150H); 3.32–3.30 (m, 5H, H2-Gal); 3.23–3.19 (m, 5H, H2-Glc); 3.14 (bs, 10H); 2.61–2.58 (m, 5H, H3a-Neu5Ac); 2.48 (bs, 10H); 1.97 (s, 15H, NHC(O)CH3-Neu5Ac); 1.95 (s, 15H, NHC(O)CH3-GalNAc); 1.89–1.84 (m, 5H, H3b-Neu5Ac); 1.51–1.36 (bs, 20H, CH2 aliphatic chain); 1.23–0.95 (50H, m, CH2 aliphatic chain). HR-ESI-MS(−): m/z 1440.4670 [100% (M − 5H)5−] calcd: 1438.6588.
Peracetylated-galactosylcalix[5]arene (14)
Calix[5]arene 8 (32.0 mg, 14.1 μmol) and the β-galactoside derivative 12 (52.9 mg, 106 μmol) were dissolved in 2.5 mL of DMF in a microwave tube. CuSO4·5H2O (2.0 mg, 8.5 μmol), sodium ascorbate (3.3 mg, 16.9 μmol) and 0.5 mL H2O were then added. The mixture was heated at 80 °C by microwave irradiation (150 W) for 60 min. When the reaction was completed (checked viaTLC, eluent: CH2Cl2–CH3OH 20:1), it was quenched by addition of water (15 mL) and extracted with AcOEt (5 × 15 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and the solvent removed under vacuum. The crude material was purified on preparative TLC plates (eluent: CH2Cl2–CH3OH 9:1) giving product 14 as a yellow oil. Yield: 67%. 1H NMR (300 MHz, CD3OD): δ (ppm) 8.21 (bs, 5H, C(O)NH); 7.77 (s, 5H, H5 triazole); 7.61 (bs, 10H, ArH); 5.38 (d, 5H, J = 2.7 Hz, H4); 5.16–5.02 (m, 10H, H3, H2); 4.61 (d, 5H, J = 7.3 Hz, H1); 4.51 (t, 10H, J = 5.0 Hz, OCH2CH2-triazole); 4.20–4.05 (m, 15H, H5, H6a, H6b); 3.94 (bs, 10H, ArCH2Ar); 3.88–3.77 (m, 15H, OCH2CH2-triazole, β-COCHa); 3.70–3.44 (m, 105H, OCH2, β-COCHb, C(O)NHCH2); 3.29 (s, 15H, ArOCH3); 2.67 (t, 10H, J = 7.6 Hz, triazole-CH2CH2CH2); 2.13 (s, 15H, Ac); 2.02 (s, 15H, Ac); 2.01 (s, 15H, Ac); 1.94 (s, 15H, Ac); 1.71–1.59 (m, 10H, triazole-CH2CH2CH2), 1.59–1.47 (m, 10H, β-COCH2CH2), 1.40–1.23 (m, 50H, CH2 aliphatic chain). 13C NMR (75 MHz, CD3OD) δ ppm: 172.0, 171.5, 171.2 (Ac); 169.6 (C(O)NH); 160.9 (Ar-ipso); 149.0 (C4 triazole); 135.8 (Ar-ortho); 130.7 (Ar-para); 129.7 (Ar-meta); 123.9 (C5 triazole); 102.2 (C1); 72.4 (C3); 71.7 (C5); 71.5, 71.4, 71.3, 70.9 (OCH2); 70.6 (OCH2CH2-triazole); 70.5 (C2); 68.8 (C4); 62.6 (C6); 61.5 (ArOCH3); 51.3 (OCH2CH2-triazole); 41.0 (C(O)NHCH2); 32.0 (ArCH2Ar); 30.6, 30.4, 30.2 (CH2 aliphatic chain); 27.0 (triazole-CH2CH2CH2); 26.3 (CH2 aliphatic chain); 20.8, 20.6, 20.5 (CH3C(O)). HR-ESI-MS(+): m/z 1607.7872 [100% (M + 3Na)3+] calcd: 1607.7813.
Peracetylated-lactosylcalix[5]arene (15)
Calix[5]arene 8 (27.5 mg, 12.0 μmol) and the β-lactoside compound 13 (70.0 mg, 89.0 μmol) were dissolved in 2.5 mL DMF in a microwave tube. CuSO4·5H2O (2.6 mg, 10.4 μmol), sodium ascorbate (4.4 mg, 22.2 μmol) and 0.5 mL H2O were then added. The mixture was heated at 80 °C by microwave irradiation (150 W) for 60 min. When the reaction was completed (checked viaTLC, eluent: CH2Cl2–CH3OH 94:6), it was quenched by addition of water (15 mL) and extracted with AcOEt (5 × 15 mL). The combined organic layers were dried over anhydrous Na2SO4, filtered and the solvent removed in vacuo. The crude was purified by flash chromatography (elution in gradient: CH2Cl2–CH3OH 96:4→95:5) giving product 15 as a yellow oil. Yield: 57%. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.46 (bs, 10H, ArH); 7.40 (s, 5H, H5 triazole); 6.83 (bs, 5H, C(O)NH); 5.33 (d, 5H, J = 3.3 Hz, H4′); 5.17 (t, 5H, J = 9.3 Hz, H3); 5.09 (dd, 5H, J1′–2′ = 7.9 Hz, J2′–3′ = 10.4 Hz, H2′); 4.93 (dd, 5H, J2′–3′ = 10.4 Hz, J3′–4′ = 3.3 Hz, H3′); 4.86 (dd, 5H, J2–3 = 9.3 Hz, J1–2 = 8.1 Hz, H2); 4.50–4.38 (m, 25H, H1′, H1, H6a, OCH2CH2-triazole); 4.15–4.00 (m, 15H, H6b, H6a′, H6b′); 3.90–3.71 (m, 35H, H5′, H4, β-COCHa, ArCH2Ar, OCH2CH2-triazole); 3.65–3.47 (m, 105H, OCH2, H5, C(O)NHCH2); 3.42 (m, 5H, β-COCHb); 3.21 (s, 15H, ArOCH3); 2.65 (t, 10H, J = 7.7 Hz, triazole-CH2CH2CH2); 2.12 (s, 15H, Ac); 2.09 (s, 15H, Ac); 2.04–1.98 (m, 60H, Ac); 1.94 (s, 15H, Ac); 1.65–1.55 (m, 10H, triazole-CH2CH2CH2); 1.55–1.44 (m, 10H, β-COCH2CH2); 1.39–1.15 (m, 50H, CH2 aliphatic chain). 13C NMR (100 MHz, CDCl3): δ ppm 170.4, 170.3, 170.2, 170.1, 169.8, 169.6, 169.1 (Ac); 167.3 (C(O)NH); 159.2 (Ar-ipso); 148.2 (C4 triazole); 134.2 (Ar-ortho); 129.9 (Ar-para); 128.2 (Ar-meta); 121.7 (C5 triazole); 101.1 (C1′); 100.6 (C1); 76.3 (C4); 72.8 (C3); 72.5 (C5); 71.7 (C2); 71.0 (C3′); 70.6 (C5′); 70.5 (OCH2); 70.2 (β-COCH2); 69.8 (OCH2); 69.6 (OCH2CH2-triazole); 69.1 (C2′); 66.6 (C4′); 62.1 (C6); 60.8 (C6′); 60.7 (ArOCH3); 50.0 (OCH2CH2-triazole); 39.7 (C(O)NHCH2); 31.0 (ArCH2Ar); 29.5, 29.3, 25.8, 25.7 (CH2 aliphatic chain); 20.9, 20.8, 20.6, 20.5 (CH3C(O)). HR-ESI-MS(+): m/z 1571.9445 [100% (M + 4Na)4+] calcd: 1571.940.
Galactosylcalix[5]arene (16)
The peracetylated-galactosylcalix[5]arene 14 (45.0 mg, 9.46 μmol) was dissolved in 5 mL of CH3OH, drops of a freshly prepared MeONa in methanol solution were added till pH 8–9. The mixture was stirred at room temperature for 4 h. The progress of the reaction was monitored viaESI-MS analysis. Amberlite resin IR 120/H+ was subsequently added to quench the reaction, and the mixture was gently stirred for 30 min. until neutral pH was reached. The resin was then filtered off and the solvent removed under vacuum to give pure product 16 as a yellow oil. Yield. 90%. 1H NMR (300 MHz, CD3OD): δ (ppm) 8.23 (bs, 5H, C(O)NH); 7.77 (s, 5H, H5 triazole); 7.61 (s, 10H, ArH); 4.51 (t, 10H, J = 5.0 Hz, OCH2CH2-triazole); 4.20 (d, 5H, J = 7.1 Hz, H1); 3.94 (bs, 10H, ArCH2Ar); 3.92–3.80 (m, 20H, H4, β-COCHa, OCH2CH2-triazole); 7.77–7.69 (m, 10H, H6a, H6b); 3.66–3.40 (m, 120H, β-COCHb, H2, H3, H5, OCH2, C(O)NHCH2); 3.29 (s, 15H, OCH3); 2.67 (t, 10H, J = 7.6 Hz, triazole-CH2CH2CH2); 1.72–1.53 (m, 20H, triazole-CH2CH2CH2, β-COCH2CH2); 1.44–1.24 (m, 50H, CH2 aliphatic chain). 13C NMR (75 MHz, CD3OD): δ ppm 169.6 (C(O)NH); 160.9 (Ar-ipso); 149.0 (C4 triazole); 135.8 (Ar-ortho); 130.7 (Ar-para); 129.7 (Ar-meta); 124.0 (C5 triazole); 105.0 (C1); 76.6, 75.1, 72.6 (C2, C3, C5); 71.5, 71.4, 71.3, 70.8, 70.6, 70.4, 70.3 (OCH2, β-COCH2, C4); 62.5 (C6); 61.5 (ArOCH3); 51.3 (OCH2CH2-triazole); 41.0 (C(O)NHCH2); 32.0 (ArCH2Ar); 30.8, 30.6, 30.5, 30.4, 30.3, 27.1 (CH2 aliphatic chain); 26.3 (triazole-CH2CH2CH2). HR-ESI-MS(+): m/z 1305.0569 [100% (M + 3H)3+] calcd: 1305.0601Lactosylcalix[5]arene (17)
The peracetylated-lactosylcalix[5]arene 15 (42.0 mg, 6.8 μmol) was dissolved in 5 mL of CH3OH, and drops of a freshly prepared methanol solution of MeONa were added till pH 8–9. The mixture was stirred at room temperature for 18 h. The progress of the reaction was monitored viaESI-MS analysis. Amberlite resin IR 120/H+ was subsequently added for quenching, and the mixture was gently stirred for 30 min till neutral pH. The resin was then filtered off and the solvent removed under vacuum to give pure product 17 as a yellow oil. Yield: 72%. 1H NMR (300 MHz, CD3OD): δ (ppm) 7.79 (s, 5H, H5 triazole); 7.60 (s, 10H, ArH); 4.51 (t, 10H, J = 5.0 Hz, OCH2CH2-triazole); 4.35 (d, 5H, J = 7.3 Hz, H1′); 4.26 (d, 5H, J = 7.8 Hz, H1); 3.93 (bs, 10H, ArCH2Ar); 3.90–3.65 (m, 40H, H4′, H6ab′, H6ab, Glc β-COCHa, OCH2CH2-triazole); 3.65–3.35 (m, 135H, H3, H4, H5, H2′, H3′, H5′, Glc β-COCHb, OCH2, C(O)NHCH2); 3.28 (s, 15H, OCH3); 3.23 (t, 5H, J = 8.4 Hz, H2); 2.66 (t, 10H, J = 7.6 Hz, triazole-CH2CH2CH2); 1.69–1.53 (m, 20H, CH2 aliphatic chain); 1.42–1.23 (m, 50H, CH2 aliphatic chain). 13C NMR (75 MHz, CD3OD): δ ppm 169.6 (C(O)NH); 160.9 (Ar-ipso); 148.8 (C4 triazole); 135.7 (Ar-ortho); 130.7 (Ar-para); 129.7 (Ar-meta); 124.2 (C5 triazole); 105.1 (C1′); 104.2 (C1); 80.7 (C4); 77.0, 76.5, 76.4, 74.8, 74.7, 72.5 (C2, C3, C5, C2′, C3′, C5′); 71.5, 71.4, 71.3, 70.9, 70.6, 70.4 (OCH2, β-COCH2,); 70.3 (C4′); 62.5, 61.9 (C6, C6′); 61.5 (ArOCH3); 51.4 (OCH2CH2-triazole); 41.0 (C(O)NHCH2); 32.0 (ArCH2Ar); 30.8, 30.6, 30.5, 30.4, 30.2, 27.1 (CH2 aliphatic chain); 26.2 (triazole-CH2CH2CH2). HR-ESI-MS(+) m/z: 1575.1475 [100% (M + 3H)3+] calcd: 1575.1481.17-Azide-3,6,9,12,15-pentaoxaheptadecane-1-aminocarbonyl-p-methoxybenzene (19)
Oxalyl chloride (1.5 mL, 16.0 mmol) was added to a solution of 4-methoxybenzoic acid (0.30 g, 2.0 mmol) in 15 mL of dry CH2Cl2 and the mixture was stirred at room temperature under N2 for 18 h. The solvent was then removed under vacuum and the residue dissolved again in 5 mL of dry CH2Cl2. This solution was added dropwise to a round bottomed flask containing the azidoamine compound 7 (0.91 g, 3.0 mmol) and NEt3 (0.5 mL, 3.0 mmol) in 10 mL of dry CH2Cl2. The mixture was let to react for 20 h at room temperature under an N2 atmosphere. The reaction was monitored viaTLC (eluent: AcOEt). A 1 M HCl solution (20 mL) was then added to quench the reaction, and the product extracted with CH2Cl2 (2 × 20 mL). The combined organic phases were washed with NaHCO3 saturated aqueous solution (15 mL), brine (15 mL), water (15 mL), dried over anhydrous Na2SO4, filtered and the solvent evaporated under reduced pressure. The crude was purified by flash chromatography (eluent: AcOEt–acetone 9:1). Product 19 was obtained pure as a yellow oil. Yield: 20%. 1H NMR (300 MHz, CDCl3): δ (ppm) 7.74 (d, 2H, J = 8.9 Hz, Ar-meta); 6.87 (d, 2H, J = 8.9 Hz, Ar-ortho); 6.78 (bs, 1H, C(O)NH); 3.80 (s, 3H, OCH3); 3.65–3.54 (m, 22H, OCH2, C(O)NHCH2); 3.32 (t, 2H, J = 5.0 Hz, CH2N3). 13C NMR (75 MHz, CDCl3): δ ppm 167.0 (Ac); 162.0 (Ar-ipso); 128.8 (Ar-meta); 126.9 (Ar-para); 113.6 (Ar-ortho); 70.6, 70.5, 70.2, 70.0, 69.9 (OCH2); 55.4 (OCH3); 50.6 (CH2N3); 39.7 (C(O)NHCH2). ESI-MS(+) m/z: 463.0 [100% (M + Na)+]; 435.0 [60% (M − N2 + Na)+].
GM1os-monomer (20)
Starting from compounds 19 and 9, following the same procedure as for compound 1, and using reversed phase column chromatography for the purification (gradient MeOH–H2O–AcOH), monomer 20 was obtained as a white solid in 49% yield. 1H NMR (400 MHz, D2O): δ (ppm) 7.81 (3H, m, triazole, Ar), 6.98 (2H, d, J = 8.4 Hz, Ar), 4.71 (1H, s, H1-GalNAc), 4.39 (1H, d, J = 8.4 Hz, H1-Gal′), 4.42 (1H, m, H1-Gal), 4.38 (1H, m), 4.31, (1H, d, J = 8.2 Hz, H1-Glc), 4.05–4.01 (3H, m), 3.93 (1H, m, H2-GalNAc), 3.75–3.40 (21H, m) , 3.60–3.47 (16H, m), 3.36–3.28 (14H, m), 3.25 (1H, m, H2-Gal), 3.18 (1H, m, H2-Glc), 3.10 (2H, s), 2.54 (1H, m, H3a-Neu5Ac), 2.35 (2H, m, CH2-triazole), 1.92 (3H, s, NHC(O)CH3-Neu5Ac), 1.85 (3H, s, NHC(O)CH3-GalNAc), 1.75 (1H, m, H3b-Neu5Ac), 1.40 (4H, m), 1.11 (2H, m, Glc β-COCH2CH2), 1.03 (8H, m, –CH2CH2CH2–); 13C NMR (75 MHz, D2O): δ (ppm) 174.8, 174.5, 173.9, 168.5, 159.2, 128.4 (2 × CH Ar), 123.1 (C5 triazole), 113.2 (2 × CH Ar) 104.6 (C1-Gal′), 102.7 (C1-Gal), 102.4 (C1-GalNAc), 102.0 (C1-Glc), 101.5 (C2-Neu5Ac), 80.2 (C3-GalNAc), 78.5 (C4-Glc), 77.0 (C4-Gal), 74.7, 74.6, 74.4, 74.2, 74.1, 74.0, 73.9 (C2 Glc), 72.6 (C3 Gal′), 72.3, 72.1 (C2-Gal′), 70.6 (Glc β-COCH2), 70.4 (C2-Gal), 70.4–68.5 (–OCH2–), 68.4 (C4-GalNAc), 67.8, 62.7, 60.9, 60.8, 60.4, 60.0, 51.4, 51.0 (C2 GalNAc), 49.8, 39.5, 36.7, 28.8–28.3 (CH2CH2CH2), 25.0 (Glc β-COCH2CH2), 24.5 (CH2-triazole), 22.5 (NHC(O)CH3-GalNAc), 21.9 (NH(O)CH3-Neu5Ac); HR-ESI-MS(−) m/z: 1587.7009 [100% (M − H)−] calcd: 1587.7034.CTB5 inhibition assays
Each well of a 96-well microtiter plate was coated with a 100 μL native GM1 solution (1.3 μM in ethanol) after which the solvent was evaporated. Unattached GM1 was removed by washing with PBS (3 × 450 μL), the remaining free binding sites were blocked by incubation with 100 μL of a 1% (w/v) BSA solution in PBS for 30 min at 37 °C. Detection limits were determined by placing a CT-horseradish peroxidase conjugate (CT-HRP), without inhibitor, on the plate, which gives the highest response, and the lowest response was determined by the optical density of the blank, i.e. the native GM1-coated well with all components except the inhibitor and the toxin. These two values represent the minimum and the maximum values of optical density, 0% and 100% of binding of the CT to the GM1-coating of the wells. Subsequently, the wells were washed with PBS (3 × 450 μL). In separate vials, a logarithmic serial dilution was performed that started from 2.0 mM of 150 μL saccharide-calixarenes in 0.1% BSA and 0.05% Tween-20 in PBS. Next, each vial was mixed and incubated with 150 μL of a 50 ng mL−1 CTB-HRP solution in the same buffer. This gave an initial inhibitor concentration of 1.0 mM. In the case of potent inhibitors, based on the logarithmic experiments, a more accurate, serial dilution of a factor two was performed around the expected IC50-values. The inhibitor–toxin mixtures were incubated at room temperature for 2 h and then transferred to the coated wells. After 30 min of incubation at room temperature, unbound CTB-HRP-calixarene complexes were removed from the wells by washing with 0.1% BSA, 0.05% Tween-20 in PBS (3 × 500 μL). 100 μL of a freshly prepared OPD solution (25 mg OPD·2HCl, 7.5 mL 0.1 M citric acid, 7.5 mL 0.1 M sodium citrate and 6 μL of a 30% H2O2 solution, pH was adjusted to 6.0 with NaOH) was added to each well and allowed to react with HRP in the absence of light, at room temperature, for 15 minutes. The oxidation reaction was quenched by addition of 50 μL 1 M H2SO4. Within 5 min, the absorbance was measured at 490 nm.Acknowledgements
This work is co-financed by the INTERREG IV A Germany-Netherlands programme through the EU funding from the European Regional Development Fund (ERDF), the Ministry for Economic Affairs, Energy, Building, Housing and Transport of the State of North-Rhine Westphalia, the Dutch Ministry of Economic Affairs, and the Province of Gelderland; it is accompanied by the program management Euregio Rhein-Waal. Additional funding was provided by Italian MIUR projects PRIN 200858SA98 and 2010JMAZML, and the EU COST Action CM1102 ‘MultiGlycoNano’. The CIM (University of Parma) is acknowledged for NMR and mass measurements. Truus Posthuma-Trumpie and Aart van Amerongen (FBR) are acknowledged for their contribution in the ELISA experiments.References
- World Health Organisation, Fact sheet No107, 2011, 2012.
- World Health Organisation, Weekly epidemiological record, 2011, vol. 86, p. 325 Search PubMed.
- N. Lycke, Nat. Immunol., 2012, 12, 592 CrossRef CAS.
- J. Holmgren and A.-M. Svennerholm, Curr. Opin. Immunol., 2012, 24, 343 CrossRef CAS.
- E. A. Merritt, S. Sarfaty, F. v. d. Akker, C. l'Hoir, J. A. Martial and W. G. J. Hol, Protein Sci., 1994, 3, 166 CrossRef CAS.
- W. E. Minke, C. Roach, W. G. J. Hol and C. L. M. J. Verlinde, Biochemistry, 1999, 38, 5684 CrossRef CAS.
- W. B. Turnbull, B. L. Precious and S. W. Homans, J. Am. Chem. Soc., 2004, 126, 1047 CrossRef CAS.
- N. Sahyoun and P. Cuatrecasas, Proc. Natl. Acad. Sci. U. S. A., 1975, 72, 3438 CrossRef CAS; M. D. Hollenberg, P. H. Fishman, V. Bennett and P. Cuatrecasas, Proc. Natl. Acad. Sci. U. S. A., 1974, 71, 4224 CrossRef.
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. D. Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K.-E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. Murphy, C. Nativi, S. Oscarson, S. Penadés, F. Peri, R. J. Pieters, O. Renaudet, J.-L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schäffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709–4727 RSC; T. R. Branson and W. B. Turnbull, Chem. Soc. Rev., 2013, 42, 4613–4622 RSC.
- E. Fan, Z. Zhang, W. E. Minke, Z. Hou, C. L. M. J. Verlinde and W. G. J. Hol, J. Am. Chem. Soc., 2000, 122, 2663 CrossRef CAS.
- Z. Zhang, J. C. Pickens, W. G. J. Hol and E. Fan, Org. Lett., 2004, 6, 1377 CrossRef CAS.
- Z. Zhang, J. Liu, C. L. M. J. Verlinde, W. G. J. Hol and E. Fan, J. Org. Chem., 2004, 69, 7737 CrossRef CAS.
- J. P. Thompson and C. L. Schengrund, Glycoconjugate J., 1997, 14, 837 CrossRef CAS.
- A. Bernardi, L. Carrettoni, A. Grosso Ciponte, D. Monti and S. Sonnino, Bioorg. Med. Chem. Lett., 2000, 10, 2197 CrossRef CAS.
- A. Bernardi, D. Arosio, D. Potenza, I. Sànchez-Medina, S. Mari, F. J. Cañada and J. Jiménez-Barbero, Chem.–Eur. J., 2004, 10, 4395 CrossRef CAS.
- D. Arosio, I. Vrasidas, P. Valentini, R. M. J. Liskamp, R. J. Pieters and A. Bernardi, Org. Biomol. Chem., 2004, 2, 2113 CAS.
- D. Arosio, M. Fontanella, L. Baldini, L. Mauri, A. Bernardi, A. Casnati, F. Sansone and R. Ungaro, J. Am. Chem. Soc., 2005, 127, 3660 CrossRef CAS.
- F. Sansone and A. Casnati, Chem. Soc. Rev., 2013, 42, 4623–4639 RSC.
- F. Sansone, L. Baldini, A. Casnati and R. Ungaro, New J. Chem., 2010, 34, 2715 RSC.
- A. Dondoni and A. Marra, Chem. Rev., 2010, 110, 4949 CrossRef CAS.
- A. V. Pukin, H. M. Branderhorst, C. Sisu, C. A. G. M. Weijers, M. Gilbert, R. M. J. Liskamp, G. M. Visser, H. Zuilhof and R. J. Pieters, ChemBioChem, 2007, 8, 1500 CrossRef CAS.
- C. Sisu, A. J. Baron, H. M. Branderhorst, S. D. Connell, C. A. G. M. Weijers, R. de Vries, E. D. Hayes, A. V. Pukin, M. Gilbert, R. J. Pieters, H. Zuilhof, G. M. Visser and W. B. Turnbull, ChemBioChem, 2009, 10, 329 CrossRef CAS.
- D. R. Stewart, M. Krawiec, R. P. Kashyap, W. H. Watson and C. D. Gutsche, J. Am. Chem. Soc., 1995, 117, 586 CrossRef CAS.
- D. R. Stewart and C. D. Gutsche, Org. Prep. Proced. Int., 1993, 25, 137 CrossRef CAS.
- S. E. J. Bell, J. K. Browne, V. McKee, M. A. McKervey, J. F. Malone, M. O'Leary and A. Walker, J. Org. Chem., 1998, 63, 489 CrossRef CAS.
- J. C. Duff, J. Chem. Soc., 1941, 547 RSC.
- W. E. Smith, J. Org. Chem., 1972, 37, 3972 CrossRef CAS.
- A. W. Schwabacher, J. W. Lane, M. W. Scheisher, K. M. Leigh and C. W. Johnson, J. Org. Chem., 1998, 63, 1727 CrossRef CAS.
- S. S. Iyer, A. S. Anderson, S. Reed, B. Swanson and J. G. Schmidt, Tetrahedron Lett., 2004, 45, 4285 CrossRef CAS.
- A. V. Pukin, C. A. G. M. Weijers, B. van Lagen, R. Wechselberger, B. Sun, M. Gilbert, M.-F. Karwaski, D. E. A. Florack, B. C. Jacobs, A. P. Tio-Gillen, A. van Belkum, H. P. Endtz, G. M. Visser and H. Zuilhof, Carbohydr. Res., 2008, 343, 636 CrossRef CAS.
- G. Zemplén and E. Pascu, Ber. Dtsch. Chem. Ges., 1929, 62, 1613 CrossRef.
- M. Matarella, J. Garcia-Hartjes, T. Wennekes, H. Zuilhof and J. S. Siegel, Org. Biomol. Chem., 2013 10.1039/c3ob40438b , in press.
- C. D. Gutsche, Calixarenes Revisited, Royal Society of Chemistry, Cambridge, 1998 Search PubMed.
- C. A. G. M. Weijers, M. C. R. Franssen and G. M. Visser, Biotechnol. Adv., 2008, 26, 436 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Copies of NMR spectra for all reported new compounds. See DOI: 10.1039/c3ob40515j |
| ‡ Both authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2013 |