 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Nanomolar cholera toxininhibitors based on symmetrical pentavalent ganglioside GM1os-sym-corannulenes†
Martin
Mattarella‡
a,
Jaime
Garcia-Hartjes‡
b,
Tom
Wennekes
b,
Han
Zuilhof
*bc and
Jay S.
Siegel
*a
aInstitute of Organic Chemistry, University of Zurich, Winterthurerstrasse 190, 8057 Zurich, Switzerland. E-mail: jss@oci.uzh.ch; Tel: +41 446354281
bLaboratory of Organic Chemistry, Wageningen University, Dreijenplein 8, 6703 HB, Wageningen, The Netherlands
cDepartment of Chemical and Materials Engineering, King Abdulaziz University, Jeddah, Saudi Arabia. E-mail: han.zuilhof@wur.nl; Tel: +31 317482361
First published on 4th June 2013
Abstract
Eight symmetric and pentavalent corannulene derivatives were functionalized with galactose and the ganglioside GM1-oligosaccharide (GM1os) via copper-catalyzed alkyne-azidecycloaddition (CuAAC) reactions. The compounds were evaluated for their ability to inhibit the binding of the pentavalent cholera toxin to its natural ligand, ganglioside GM1. In this assay, all ganglioside GM1os-sym-corannulenes proved to be highly potent nanomolar inhibitors of cholera toxin.
1. Introduction
The World Health Organization (WHO) estimates that annually 3–5 million people worldwide are infected with cholera, resulting in over a hundred thousand fatalities.1 The responsible pathogen, the Vibrio cholerae bacterium, produces the cholera toxin (CT) protein that is responsible for the severe clinical symptoms. CT belongs to the protein family of AB5 bacterial toxins.2 The structure and activity of CT and other AB5toxins have been investigated in detail.3 These proteins consist of two distinct domains with different roles. The A-subunit is an enzyme that – when delivered inside a host cell – is toxic and responsible for the subsequent disease symptoms. The cholera toxin B-subunit (CTB) is a lectin and plays a crucial role in the recognition and interaction with its natural ligand, ganglioside GM1 (Fig. 1a), on the periphery of intestinal cells. The crystal structure of CT4 shows that the protein complex consists of five identical monomeric CTB subunits, arranged in a pentagonal symmetry, and each of these subunits can bind the ganglioside GM1 in a one-to-one stoichiometry. Detailed calorimetric studies revealed that CT exhibits allosteric cooperativity,5 which contributes to increasingly higher binding affinities to CT when more ligands are bound.6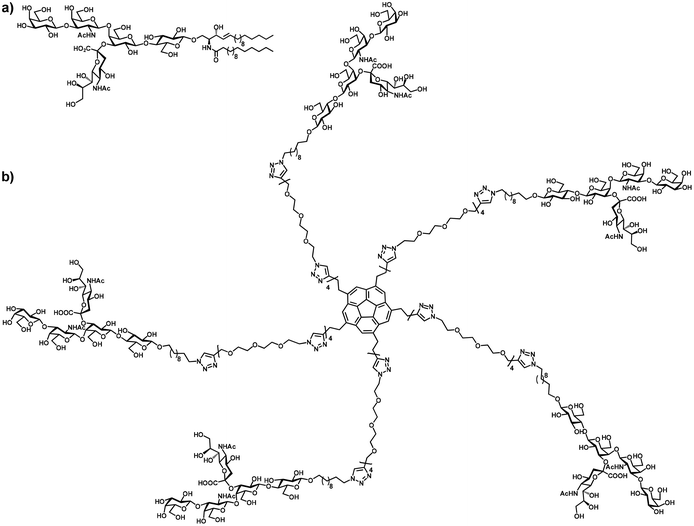 | ||
| Fig. 1 (a) Structure of CTB's natural ligand, ganglioside GM1; (b) structure of the pentavalent GM1os-functionalized sym-corannulene, a nanomolar inhibitor of cholera toxin. | ||
One of the possible approaches for the design of CT inhibitors aims to prevent the receptor-recognition process.7 Two major routes can be discerned in the literature to achieve this goal. The first strategy, monovalent receptor-binding approach, focuses on strong binding interactions, and it is based on the design and synthesis of ligands that closely mimic the natural ligand on the cell surface8 in order to obtain a strong interaction with the CTB receptor. The second approach, multivalent receptor-binding,9 exploiting chelate cooperativity, takes advantage of the pentavalent character of the ligand-binding sites of CT. This approach is based on the synthesis of a functionalized branched system, in which each branch carries a single-site inhibitor, like galactose10,11 or lactose,12 leading to a compound that has an overall stronger interaction with the toxin than the sum of the independent inhibitors. The synthesis of dendritic multivalent inhibitors, functionalized with the GM1os, has been reported;13 these displayed unprecedented high inhibitory potencies for CTB, in the picomolar range.
An important improvement in the design of multivalent binders was achieved with symmetrical pentameric molecules based on the concept of “finger-linker-core” systems:12 the pentavalent “core” is connected by flexible “linkers” to “fingers” that include the monovalent receptor-binding ligand. Pentavalent CT inhibitors were synthesized using various “cores”: acylated pentacyclen,12a a large cyclic peptide12e and calix[5]arene.14
This paper details a study that combines the two strategies to obtain an optimal binding by the design and synthesis of pentavalent GM1os-presenting compounds based on a 5-fold symmetrical sym-substituted15 corannulene scaffold (Fig. 1b). Binding assays allowed the determination of the interaction with CT-B5 (the protein complex without the toxic A part) for these and analogous pentavalent galactose corannulenes. This revealed a high inhibitory potency towards CTB by this new class of inhibitors.
2. Results and discussion
Apart from its obvious 5-fold symmetry, the corannulene scaffold is a good candidate to function as the “core” of pentavalent receptor-binding inhibitors for AB5toxins on the basis of the following observations: the recent development of a kilogram-scale synthesis of corannulene (1),16 its further functionalization to sym-pentachloro-corannulene (2),17 and the growing number of robust and flexible procedures for the preparation of molecular pentapods displaying functional groups and bioconjugated moieties, by iron-catalyzed18 cross-coupling and copper-catalyzed azide-alkynecycloaddition (CuAAC) reaction.The synthetic pathway towards PEG-corannulene systems starts from the conjugation of the terminal acetylene318 by an efficient copper nanoparticle-catalyzed CuAAC reaction with n-mers of α-azidoethyl-ω-propargyl diglyme (n = 1, 2, 4) (Fig. S1 in the ESI†).19 This route gave satisfactory results for the preparation of other sym-bioconjugated corannulenes, and was used here to prepare the five-fold symmetric compounds 5, 6 and 7 in good yield and purity. The PEG arms function as linkers of different lengths and improve water solubility and flexibility of the inhibitor. The terminal TIPS-protected acetylenes were deprotected by reaction with TBAF19 yielding the alkyne-terminated PEG-corannulenes 8, 9 and 10 (Scheme 1).
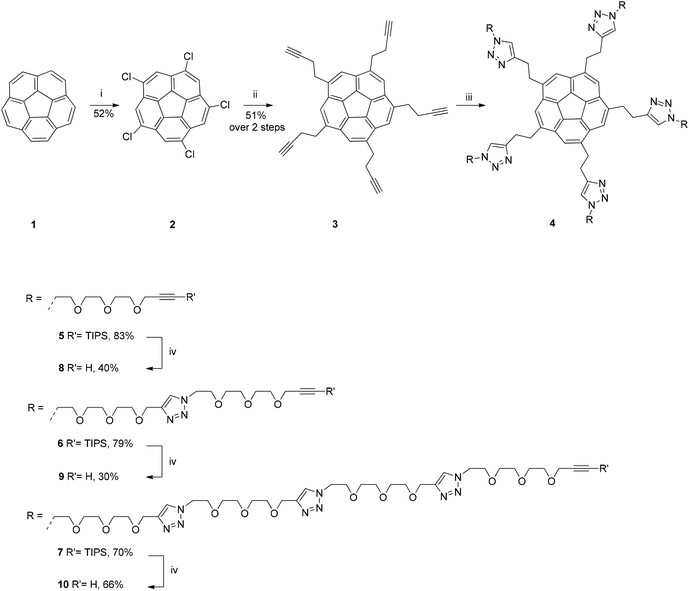 | ||
| Scheme 1 Synthesis route of the corannulene core and ethylene glycol spacers. (i) ICl, DCM, r.t., 72 h; (ii) 1. Fe(acac)3, 1-bromo-4-trimethylsilyl-3-butyne, THF–NMP, r.t., 2.5 h; 2. NaOH, MeOH–H2O, r.t., 24 h; (iii) Cu nanoparticles, DMF, microwave, 60 °C, 2 h; (iv) TBAF, THF, r.t., 2 h. | ||
After preparing the three PEG-corannulenes the focus shifted to attaching the (oligo)saccharides. In this work, two different CTB binders were used: galactose, an easily obtainable but quite poor binder of CTB, and the oligosaccharide of ganglioside GM1 (GM1os), the natural ligand of CTB.
In order to introduce the galactose-based “finger” to the terminal acetylenes3, 8, 9 and 10, galactoside11 was functionalized at the anomeric position with a short PEG chain that bears an azido group (compound 12, Scheme 2). An analogous compound without ethylene oxide moieties belonging to this inhibitor family was previously synthesized18 by CuAAC reaction on the terminal acetylene3 and 2-azidoethyl-β-D-galactopyranoside.20,21 The synthetic pathway for the synthesis of azido-PEG-galactoside 14 starts with a Koenigs–Knorr-type glycosylation of azido-PEG hydroxide 1222,23 with peracetylated galactosyl bromide 11 to afford β-galactoside 13. The deprotected compound 14 was then obtained by treatment of 13 with sodium methoxide. The pentavalent Gal-functionalized sym-corannulene inhibitor was prepared in good yield by employing the microwave-assisted CuAAC reaction on the terminal acetylenes3, 8, 9 and 10 with the azido galactose derivative 14 affording the four galactose-based CTB inhibitors15–18 (Scheme 3).
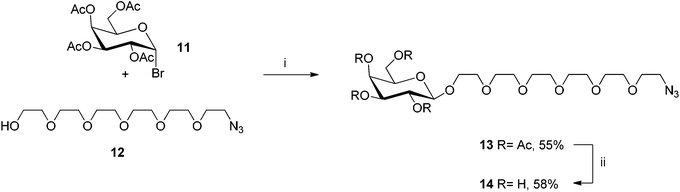 | ||
| Scheme 2 Synthesis of azido-linked galactoside14. (i) HgBr2, DCM, r.t., 3 d; (ii) (1) MeONa, MeOH, r.t., 48 h; (2) DOWEX-H+. | ||
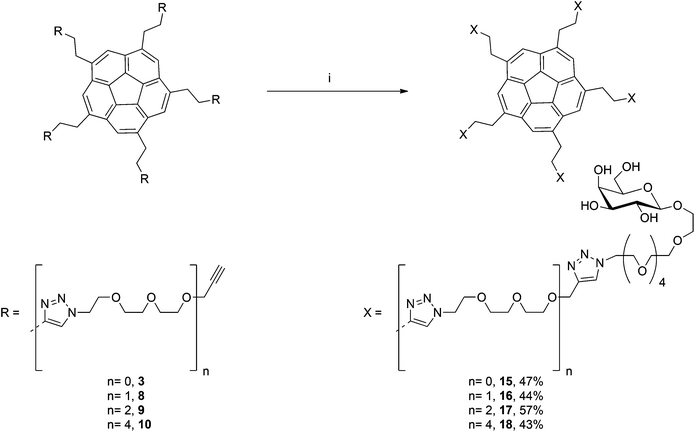 | ||
| Scheme 3 Microwave-assisted CuAAC-based synthesis of penta-galactose corannulenes. (i) 14, Cu nanoparticles, DMF, microwave, 80 °C, 2 h. | ||
The second family of pentavalent inhibitors displays five GM1os moieties (Scheme 4). The chemo-enzymatic synthesis from lactose of the azide-terminated GM1os oligosaccharide was previously reported.24 The pentavalent GM1os inhibitors20, 21 and 22 were synthesized, in the presence of copper nanoparticles, by microwave-assisted CuAAC reaction of azido-pentasaccharide 19 with the terminal acetylenes8, 9 and 10. Because of the low solubility of the GM1 analog in DMF, the reactions were performed in water, allowing a complete dissolution of 19 in the reaction media.
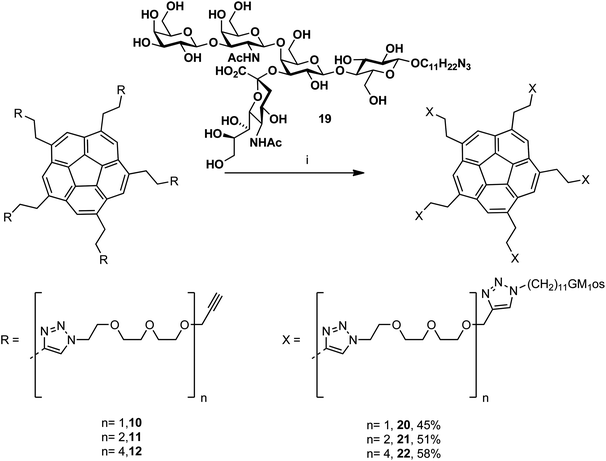 | ||
| Scheme 4 Synthesis of the GM1os-functionalized PEG-corannulenes. (i) Cu nanoparticles, H2O, microwave, 80 °C, 2 h. | ||
The inhibitory efficiency of the synthesized five-fold symmetric pentavalent CT ligands (15–18 and 20–22) was evaluated by an ELISA-type assay on 96-well plates.25,26 Solubility issues at concentrations >1 mM limited the experiments intended to find an IC50-value for galactose-based compounds 15–18. When compared to previously reported IC50-values for multivalent ligands functionalized with galactose,10,11 these concentrations should not have been limiting. One hypothesis to account for this observation is that the supramolecular aggregation competes against binding to the pentad-complex.
Further investigations by 1H-NMR, UV-Vis and fluorescence spectroscopy on the properties of C5-symmetric galactose conjugated corannulene in aqueous solution suggest the formation of supramolecular aggregates of this amphiphilic molecule in water. If the formation of the supramolecular assemblies is thermodynamically and kinetically favored over the interaction with the CTB, the competition of these two processes might be a plausible explanation for the unexpected results obtained from the ELISA assays; however, the GM1os-PEG-corannulenes showed high, nanomolar inhibitory potencies (Fig. 2; Table 1).
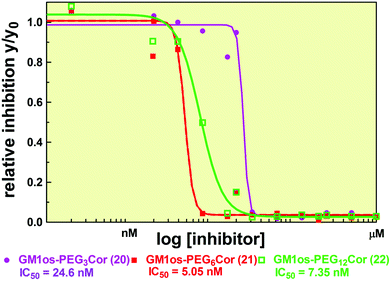 | ||
| Fig. 2 Fitted inhibition curves of compounds GM1os-PEG3 corannulene 20 (purple line), GM1os-PEG6 corannulene 21 (red line), and GM1os-PEG12 corannulene 22 (green line). | ||
The pentavalent GM1os-functionalized inhibitors, 20, 21, and 22, bind CTB at lower IC50 values than monomeric GM1os (cf. Table 1). The lowest IC50-value measured is for 21 (Table 1, entry 2) with an inhibition potency that is nearly 4000 times stronger than that of monomeric GM1os.12f The lower binding affinity for CTB by compounds 20 and 22 (Table 1, entries 1 and 3) is most probably due to the effects related to the linker length: if the linker is not long enough (compound 20), not all of the five ligands can simultaneously bind the five binding sites of CTB. In the opposite case (i.e. compound 22), binding would lead to a substantial ordering in the chain, making it entropically less favorable, while enthalpically the necessary folding of the longer linkers to enable binding of CTB by the GM1os fingers might create sufficient steric hindrance to cause an increased IC50-value. A further complicating factor is the possibility that galactose and GM1os derivatives undergo supramolecular self-aggregation that competes with binding to CTB and results in apparent higher inhibitory concentrations.
Caveats notwithstanding, 21 is as of yet one of the strongest multivalent CT inhibitors,22,27 and displays the power of combining the natural valency with the natural ganglioside to reach optimal blocking of multivalent lectins. Design efforts to create systems that optimize spacer geometry and avoid self complexation will likely lead to new derivatives with inhibitory concentrations more favorably comparable to other previously reported GM1os-based inhibitors.
3. Conclusions
This manuscript reports a powerful method for the synthesis of a new class of cholera toxininhibitors with a design based on a pentavalent sym-substituted corannulene as the core unit and equipped with the galactose and the GM1os as CTB binders. Microwave-assisted CuAAC reactions, catalyzed by copper nanoparticles, were employed for the conjugation of the monovalent CTB ligands (galactose and GM1os) to the corannulene core viaazide-presenting PEG-linkers of various lengths. The potent CTB inhibition of 25, 5.0, and 7.3 nM observed for the penta-GM1os corannulenes, 20, 21, and 22, respectively, prove that multivalent systems functionalized with strong CTB binders represent a solid strategic approach for the synthesis of CT inhibitors with high potency in comparison with previously reported monovalent inhibitors.The developed method allows for the use of sym-substituted corannulenes as a possible core unit for the development of new multivalent binders of cholera toxin or other possible biological targets that rely on multivalent binding of their target ligand.
4. Representative experimental procedures
Compound 6
A mixture of 3 (9.4 mg, 18 μmol), the proper azide-functionalized PEG (83.0 mg, 0.14 mmol) and copper nanoparticles (11.7 mg, 0.18 mmol) in DMF (1.0 mL) was loaded in a microwave vessel and was heated at 80 °C in a microwave reactor for 2 h. The reaction mixture was filtrated over celite and the solvent evaporated. The crude mixture was purified by column chromatography on silica gel (from DCM–MeOH 94![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :6 to DCM–MeOH 9:1) to yield a reddish oil (49 mg, 79%). 1H-NMR (500 MHz, CDCl3): δ 7.69 (s, 5H), 7.64 (s, 5H), 7.53 (s, 5H), 4.62 (s, 10H), 4.48 (q, J = 5.5 Hz, 20 H), 4.21 (s, 10H), 3.82 (t, J = 5.0 Hz, 20H), 3.69–3.47 (m, 90H), 3.26 (t, J = 8.0 Hz, 10H), 1.04 (s, 105H). 13C-NMR (125 MHz, CDCl3): δ 147.07, 144.81, 140.42, 134.88, 129.74, 123.91, 123.06, 122.47, 103.21, 87.81, 70.55, 70.54 70.51, 70.47, 69.68, 69.64, 69.52, 68.72, 64.58, 59.28, 50.32, 50.27, 33.38, 28.49, 18.66, 11.20. HRMS (ESI) m/z: found 1163.6590 (M + 3Na); calc. (C175H280N30Na3O30Si5) 1163.6609.
:6 to DCM–MeOH 9:1) to yield a reddish oil (49 mg, 79%). 1H-NMR (500 MHz, CDCl3): δ 7.69 (s, 5H), 7.64 (s, 5H), 7.53 (s, 5H), 4.62 (s, 10H), 4.48 (q, J = 5.5 Hz, 20 H), 4.21 (s, 10H), 3.82 (t, J = 5.0 Hz, 20H), 3.69–3.47 (m, 90H), 3.26 (t, J = 8.0 Hz, 10H), 1.04 (s, 105H). 13C-NMR (125 MHz, CDCl3): δ 147.07, 144.81, 140.42, 134.88, 129.74, 123.91, 123.06, 122.47, 103.21, 87.81, 70.55, 70.54 70.51, 70.47, 69.68, 69.64, 69.52, 68.72, 64.58, 59.28, 50.32, 50.27, 33.38, 28.49, 18.66, 11.20. HRMS (ESI) m/z: found 1163.6590 (M + 3Na); calc. (C175H280N30Na3O30Si5) 1163.6609.
Compound 9
A solution of TBAF in THF (1 M, 1.0 mL, 1.0 mmol) was added to a solution of 6 (103 mg, 30.1 μmol) in THF (1.0 mL) and the reaction mixture was stirred at r.t. for 2 h. The solution was then diluted with a saturated aqueous solution of NH4Cl and extracted with ethyl acetate. The collected organic phases were dried over NasSO4 and evaporated to yield the crude product. The crude was then heated at 75 °C under high vacuum for 18 h. The product was then purified by column chromatography on silica gel (DCM–MeOH 9:1) to yield a reddish oil (24 mg, 30%). 1H-NMR (500 MHz, CDCl3): 1H-NMR (500 MHz, CDCl3): δ 7.70 (s, 5H), 7.64 (s, 5H), 7.53 (s, 5H), 4.62 (s, 10H), 4.50–4.46 (m, 20 H), 4.16 (d, J = 2.5 Hz, 10H), 3.82 (t, J = 5.0 Hz, 20H), 3.64–3.51 (m, 90H), 3.23 (t, J = 8.0 Hz, 10H), 2.44 (d, J = 2.5 Hz, 5H). 13C-NMR (125 MHz, CDCl3): δ 147.11, 144.83, 140.47, 134.89, 129.76, 123.97, 123.07, 122.47, 79.70, 74.86, 70.57, 70.55, 70.47, 69.65, 69.52, 69.14, 64.72, 58.48, 50.26, 50.20, 33.39, 28.52. HRMS (ESI) m/z: found 529.2767 (M + 5H); calc. (C130H185N30O30) 529.2769.
Compound 17
A mixture of 9 (8.5 mg, 3.2 μmol), 14 (11.3 mg, 24.1 μmol) and copper nanoparticles (1.5 mg, 23.6 μmol) in DMF (300 μL) was loaded into a microwave vessel and was heated at 80 °C in a microwave reactor for 2 h. The mixture was filtrated over celite and the solid was washed with water. The crude was lyophilized and purified by size exclusion chromatography (Sephadex® G-25, water) to yield a reddish solid (9.1 mg, 57%). 1H-NMR (500 MHz, D2O/d4-MeOD): δ 8.01 (s, 5H), 7.88 (s, 5H), 7.68 (s, 5H), 7.60 (s, 5H), 4.58–4.39 (m, 50H), 4.08–4.06 (m, 5H), 3.98–3.12 (m, 255H). 13C-NMR (125 MHz, D2O/d4-MeOD): δ 147.90, 144.84, 141.31, 134.78, 130.36, 126.25, 126.05, 124.76 124.23, 103.86, 76.12, 73.69, 71.73, 70.69, 70.57, 70.52, 70.42, 70.41, 70.35, 70.27, 70.24, 69.86, 69.79, 69.73, 69.65, 69.61, 69.54, 63.99, 61.93, 50.90, 50.77, 49.52, 33.97, 28.11. HRMS (ESI) m/z: found 854.2378 (M + 6Na); calc. (C220H355N45Na6O85) 854.2366.Compound 21
A mixture of 9 (2.88 mg, 1.09 μmol), 19 (11.1 mg, 9.3 μmol) and copper nanoparticles (0.76 mg, 12.0 μmol) in water (300 μL) was loaded into a microwave vessel and was heated at 80 °C in a microwave reactor for 2 h. The mixture was filtrated over celite and the solid was washed with water. The crude was lyophilized and purified by size exclusion chromatography (Sephadex® G-25, water) to yield a colorless solid (4.8 mg, 51%). 1H-NMR (500 MHz, D2O/d4-MeOD): δ 7.96–7.75 (m, 20H), 4.57–4.54 (m, 70H), 4.18–4.05 (m, 45H), 3.94–3.34 (m, 286H), 2.70 (d, J = 10.0 Hz, 5H), 2.06 (d, J = 15.0 Hz, 35H), 1.98–1.94 (t, J = 10.0 Hz, 5H), 1.62–1.45 (m br, 25H), 1.28–1.25 (m br, 20H), 1.11–0.90 (m br, 75H). 13C-NMR (125 MHz, D2O/d4-MeOD): δ 175.96, 175.06, 105.70, 103.61, 103.48, 103.18, 102.70, 75.83, 75.32, 73.47, 71.66, 70.66, 70.64, 70.62, 70.52, 70.50, 70.48, 70.47, 70.45, 69.59, 69.57, 61.90, 52.61, 52.12, 51.13, 51.11, 29.73, 26.58, 26.23, 23.58, 22.99. HRMS (ESI) m/z: found 1721.1778 (M − 5H); calc. (C370H590N55O175) 1720.7797.ELISA assays
Each well of a 96-well microtiter plate was coated with a 100 μL native GM1 solution (1.3 μM in ethanol) after which the solvent was evaporated. Unattached GM1 was removed by washing with PBS (3 × 450 μL), and the remaining free binding sites were blocked by incubation with 100 μL of a 1% (w/v) BSA solution in PBS for 30 min at 37 °C. Subsequently, the wells were washed with PBS (3 × 450 μL). In separate vials, a logarithmic serial dilution, starting from 2.0 mM, of 150 μL saccharide-corannulenes in 0.1% BSA, 0.05% Tween-20 in PBS, mixed with 150 μL of a 50 ng mL−1 CTB–HRP solution in the same buffer, were incubated. This gave an initial inhibitor concentration of 1.0 mM. In the case of potent inhibitors, based on the logarithmic experiments, a more accurate, serial dilution of a factor of two was performed around the expected IC50 values. The inhibitor-toxin mixtures were incubated at room temperature for 2 h and then transferred to the coated wells. After 30 min of incubation at room temperature, unbound CTB–HRP–corannulene complexes were removed from the wells by washing with 0.1% BSA, 0.05% Tween-20 in PBS (3 × 500 μL). 100 μL of a freshly prepared OPD solution (25 mg OPD·2HCl, 7.5 mL 0.1 M citric acid, 7.5 mL 0.1 M sodium citrate and 6 μL of a 30% H2O2 solution, pH was adjusted to 6.0 with NaOH) was added to each well and allowed to react with HRP in the absence of light, at room temperature, for 15 min. The oxidation reaction was quenched by the addition of 50 μL 1 M H2SO4. Within 5 min, the adsorbance was measured at 490 nm.Acknowledgements
Funding from the Swiss National Foundation (SNF) and INTERREG IV a Germany–Netherlands programme of a) the European Regional Development Fund (ERDF); b) the Ministry for Economic Affairs, Energy, Building, Housing and Transport of the State of North-Rhine Westphalia; and, c) the Dutch Ministry of Economic Affairs and the Province of Gelderland.Notes and references
- World Health Organization, W. H. O., 2011; vol. 2012, p Fact sheet No107.
- (a) B. D. Spangler, Microbiol. Rev., 1992, 56, 622 CAS; (b) E. A. Merritt and W. G. J. Hol, Struct. Biol., 1995, 5, 165 CAS.
- For review, see: E. Fan, C. J. O'Neal, D. D. Mitchell, M. A. Robien, Z. Zhang, J. C. Pickens, X. J. Tan, K. Korotkov, C. Roach, B. Krumm, C. L. M. J. Verlinde, E. A. Merritt and W. G. J. Hol, Int. J. Med. Microbiol., 2004, 294, 217 CrossRef CAS.
- I. Lonnroth and J. Holmgren, J. Gen. Microbiol., 1973, 76, 417 CrossRef CAS.
- The following reviews and references therein: (a) C. A. Hunter and H. L. Anderson, Angew. Chem., Int. Ed., 2009, 48, 7488 CrossRef CAS; (b) G. Ercolani and L. Schiaffino, Angew. Chem., Int. Ed., 2011, 50, 1762 CrossRef CAS.
- D. E. Schafer and A. K. Thakur, Cell Biophys., 1982, 4, 25 CAS.
- E. Fan, E. A. Merritt, C. L. M. J. Verlinde and W. G. J. Hol, Curr. Opin. Struct. Biol., 2000, 10, 680 CrossRef CAS.
- A. Bernardi, A. Checchia, P. Brocca, S. Sonnino and F. Zuccotto, J. Am. Chem. Soc., 1999, 121, 2032 CrossRef CAS.
- A. Bernardi, J. Jiménez-Barbero, A. Casnati, C. D. Castro, T. Darbre, F. Fieschi, J. Finne, H. Funken, K.-E. Jaeger, M. Lahmann, T. K. Lindhorst, M. Marradi, P. Messner, A. Molinaro, P. Murphy, C. Nativi, S. Oscarson, S. Penadés, F. Peri, R. J. Pieters, O. Renaudet, J.-L. Reymond, B. Richichi, J. Rojo, F. Sansone, C. Schäffer, W. B. Turnbull, T. Velasco-Torrijos, S. Vidal, S. Vincent, T. Wennekes, H. Zuilhof and A. Imberty, Chem. Soc. Rev., 2013, 42, 4709 RSC; Y. M. Chambre and R. Roy, Chem. Soc. Rev., 2013, 42, 4657 RSC; T. R. Branson and W. B. Turnbull, Chem. Soc. Rev., 2013, 42, 4613 RSC.
- (a) I. Vrasidas, N. J. de Mol, R. M. J. Liskamp and R. J. Pieters, Eur. J. Org. Chem., 2001, 4685 CrossRef CAS; (b) D. Arosio, I. Vrasidas, P. Valentini, R. M. J. Liskamp, R. J. Pieters and A. Bernardi, Org. Biomol. Chem., 2004, 2, 2113 RSC; (c) T. Hayama, Ph.D. Thesis, University of Zurich, 2008.
- H. M. Branderhorst, R. M. J. Liskamp, G. Visser and R. J. Pieters, Chem. Commun., 2007, 47, 5043 RSC.
- (a) E. Fan, Z. S. Zhang, W. E. Minke, Z. Hou, C. L. M. J. Verlinde and W. G. J. Hol, J. Am. Chem. Soc., 2000, 122, 2663 CrossRef CAS; (b) E. A. Merritt, Z. S. Zhang, J. C. Pickens, M. Ahn, W. G. J. Hol and E. Fan, J. Am. Chem. Soc., 2002, 124, 8818 CrossRef CAS; (c) Z. S. Zhang, E. A. Merritt, M. Ahn, C. Roach, Z. Hou, C. L. M. J. Verlinde, W. G. J. Hol and E. Fan, J. Am. Chem. Soc., 2002, 124, 12991 CrossRef CAS; (d) J. C. Pickens, D. D. Mitchell, J. Liu, X. Tan, Z. Zhang, C. L. M. J. Verlinde, W. G. J. Hol and E. Fan, Chem. Biol., 2004, 11, 1205 CrossRef CAS; (e) Z. Zhang, J. Liu, C. L. M. J. Verlinde, W. G. J. Hol and E. Fan, J. Org. Chem., 2004, 69, 7737 CrossRef CAS; (f) A. V. Pukin, Ph.D. Thesis, Wageningen University, February 2010.
- A. V. Pukin, H. M. Branderhorst, C. Sisu, C. A. G. M. Weijers, M. Gilbert, R. M. J. Liskamp, G. M. Visser, H. Zuilhof and R. J. Pieters, ChemBioChem, 2007, 8, 1500 CrossRef CAS.
- (a) J. Garcia-Hartjes, S. Bernardi, C. A. G. M. Weijers, T. Wennekes, F. Sansone, A. Casnati and H. Zuilhof, Org. Biomol. Chem., 2013 10.1039/C3OB40515J; (b) F. Sansone, L. Baldini, A. Casnati and R. Ungaro, New J. Chem., 2010, 34, 2715 RSC.
- Prefix sym is used here to indicate the C5-symmetric-substituted corannulene corresponding to 1,3,5,7,9-pentasubstitution.
- A. M. Butterfield, B. Gilomen and J. S. Siegel, Org. Process Res. Dev., 2012, 16, 664 CrossRef CAS.
- G. H. Grube, E. J. Elliott, R. J. Steffens, C. S. Jones, K. K. Baldridge and J. S. Siegel, Org. Lett., 2003, 5, 713 CrossRef CAS.
- M. Mattarella and J. S. Siegel, Org. Biomol. Chem., 2012, 10, 5799 CAS.
- S. Binauld, C. J. Hawker, E. Fleury and E. Drockenmuller, Angew. Chem., Int. Ed., 2009, 48, 6654 CrossRef CAS.
- M. E. Jung, E. C. Yang, B. T. Vu, M. Kiankarimi, E. Spyrou and J. Kaunitz, J. Med. Chem., 1992, 42, 3899 CrossRef.
- F. Fazio, M. C. Bryan, O. Blixt, J. C. Paulson and C.-H. Wong, J. Am. Chem. Soc., 2002, 124, 14397 CrossRef CAS.
- A. Bouzide and G. Sauve, Org. Lett., 2002, 4, 2329 CrossRef CAS.
- M. K. Mueller and L. Brunsveld, Angew. Chem., Int. Ed., 2009, 48, 2921 CrossRef CAS.
- A. V. Pukin, C. A. G. M. Weijers, B. van Lagen, R. Wechselberger, B. Sun, M. Gilbert, M.-F. Karwaski, D. E. A. Florack, B. C. Jacobs, A. P. Tio-Gillen, A. van Belkum, H. P. Endtz, G. M. Visser and H. Zuilhof, Carbohydr. Res., 2008, 343, 636 CrossRef CAS.
- R. M. Dawson, J. Appl. Toxicol., 2005, 25, 30 CrossRef CAS.
- A. M. Svennerholm and J. Holmgren, Curr. Microbiol., 1978, 1, 19 CrossRef CAS.
- C. Sisu, A. J. Baron, H. M. Branderhorst, S. D. Connell, C. A. G. M. Weijers, R. de Vries, E. D. Hayes, A. V. Pukin, M. Gilbert, R. J. Pieters, H. Zuilhof, G. M. Visser and W. B. Turnbull, ChemBioChem, 2009, 10, 329 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c3ob40438b |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2013 |