Continuous hydrothermal synthesis of surface-functionalised nanophosphors for biological imaging†
Robert I.
Gruar
a,
Christopher J.
Tighe
a,
James
Muir
b,
Joseph T.
Kittler
b,
Maciej
Wodjak
c,
Anthony. J
Kenyon
c and
Jawwad A.
Darr
*a
aChristopher-Ingold Laboratories, Department of Chemistry, University College London, 20 Gordon Street, London, WC1H 0AJ, UK. Fax: +44 (0)20 45 7679 7463; Tel: +44 (0)20 7679 4345E-mail: j.a.darr@ucl.ac.uk
bDepartment of Neuroscience, Physiology and Pharmacology, University College London, London, UK, WC1E 6BT
cDepartment of Electronic & Electrical Engineering, University College London, Torrington Place, London, WC1E 7JE
First published on 16th August 2012
Abstract
A crystalline and highly luminescent nanoparticle red phosphor with average particle size of 35 nm (nominal 4 mol% Eu in Y2O3) was prepared using flash heat-treatment of a nanoparticle precursor (crystals of the corresponding doped oxyhydroxide). The nanoparticle precursors (which also show strong red emission over the 600–630 nm region under broad UV excitation from transitions of 5D0 → 7F2 in europium) were prepared in a single step using a continuous hydrothermal process (utilising supercritical water) operated at ca. 380 °C and 24.1 MPa. Photoluminescence (PL) and time-resolved PL measurements were performed on selected heat-treated nanomaterials and revealed a significantly extended lifetime of >2.25 ms (bulk material typically ca. 1.7 ms). Increases in the emission lifetime as a function of increased heat-treatment time were attributed to inter-particle effects. Surface-functionalized nanoparticles were prepared and further evaluated as probes for biological imaging with the initial precursor phosphor and the highly luminescent oxide variant both being clearly resolved in cell imaging studies under an excitation of 470 nm, using a wide pass band filter centered at 640 nm. Thus, the method employed herein holds promise for readily formulated stable colloids for luminescent security inks and as biological imaging probes.
Introduction
Nanosized phosphor materials have a number of advantages over their bulk material counterparts, such as often having a more uniform size distribution, ease of formulation into stable dispersions (e.g. for use in biological imaging) and higher packing densities for applications in phosphor inks, such as those used for security applications.1 Typically, nanosized phosphor materials show reduced quantum efficiency when compared to their bulk material counterparts due to their structural defects and lower crystallinity, limiting their application in many fields.2 However, the luminescent characteristics and chemical stability of rare-earth-activated phosphors have attracted significant interest as both bulk and nanosized materials for various applications, especially those related to biological imaging and as security taggants (luminescent inks).3Phosphor nanoparticles have been synthesized by a number of synthetic methods based on both bottom-up and top-down strategies; for example, solid state, sol–gel, gas phase/deposition methods, co-precipitation, alkali reduction and combustion/flame syntheses are all well represented in the literature.3 Despite the prospect of lanthanide-doped nanoparticles in labelling and other applications where a nano-sized material is desirable, their application in many fields is often limited by the lack of suitable materials. Lanthanide-doped nanoparticles reported in the literature are typically produced via high temperature (energy intensive) processes and subsequently require lengthy post processing to suitably formulate a nanosized material.4 Particles from many synthetic processes are formed without any ligands on the surface, thus they have limited or no dispersibility in solvents (a basic requirement for application of many phosphors as nanomaterials).3 Therefore, further refinement in the synthetic process is desirable to eliminate the need for high-temperature heat-treatment of as-prepared materials and, hence, improve energy efficiency and reduce the number of synthesis steps to make the process simpler and with more consistent product quality. The products obtained from low temperature solution-based synthesis methods are often used as precursor materials to produce the material of interest.4 Some phosphor systems have been prepared as colloidal dispersions and several research groups have reported the synthesis of well dispersed colloids of yttrium vanadate,5,6 lanthanum fluoride7,8 and lanthanum phosphate4 nanoparticles doped with lanthanide(III) ions. In addition, most lanthanide-doped nanoparticles used for biological studies are typically made in an aqueous solution. However, many known phosphor compositions are not accessible using these synthesis methodologies as the reaction temperatures involved are too low, giving amorphous products.9
A common dopant for the synthesis of nanoparticle phosphors is trivalent europium (Eu3+) due to its broad UV excitation and strong red emission spectrum originating from 5DJ (J = 0,1,2) → 7FJ (J = 0,1,2,3) transitions. Europium (Eu3+)-doped rare-earth oxides, oxysulfides, hydroxides, phosphates and fluorides have been investigated as possible alternatives to conventional fluorescent probes and many have been produced and evaluated as nanomaterials.3 Briefly, the literature highlights lanthanide-doped phosphor materials as having several advantages over both conventional organic and functionalised toxic heavy metal-containing quantum dots, including high photochemical stability (non-blinking), narrow emission, low cytotoxicity and tunable emission. Significant research effort has focused on producing these materials as nanoparticles in a crystalline form.4
In the manufacture of crystalline inorganic nanomaterials, hydrothermal methods (using supercritical or subcritical water) often require relatively lower synthesis temperatures compared to more conventional methods. The process of continuous hydrothermal flow synthesis (CHFS) has been used to make crystalline nanoparticles of many different materials e.g. components of solid oxide fuel cells, photocatalysts and materials with biomedical applications.10–17 Particles form within CHFS (and related hydrothermal processes) via mixing a stream of pressurised heated water (300–450 °C, 24.1 MPa) with a stream of metal ions in solution, with the rapid change in the environment of the metal ions causing homogenous precipitation. Metal oxides are rapidly formed through hydrolysis and dehydration reactions, which occur as a function of temperature, resulting in the formation of crystalline nanoparticles. In situ synchrotron-based measurements have shown that particle nucleation occurs immediately upon mixing and data related to modelling and detailed experimental evaluations of temperature have also been presented.18,19–23 The method by which the superheated water is contacted with the solution of metal salt (precursors) can influence the properties of the nanoparticles (e.g. size distribution) and the reproducibility of their synthesis. Indeed, the authors recently published work related to the complete automation of CHFS allowing the rapid synthesis of Y1 − xEux(OH)3 (where, x = 0.05–0.30) nanoparticles as precursor solids for the low temperature production of (Y1 − xEux)2O3. This allowed rapid optimisation of luminescence efficiency.24 The same study also suggested that as heat-treatment temperatures of the nanoprecursors continue, the process of sintering can lead to defects (and loss of luminescence intensity) until sintering is complete and larger or bulk highly crystalline material is formed again.24
CHFS-type methods have also been used to produce crystalline phosphor materials based on a YAG (Yttrium Aluminium Garnet), europium-doped YAG (YAG:Eu3+) and terbium-doped YAG25 (YAG:Tb). For example, Hakuta et al. synthesised YAG:Tb directly using a reaction temperature of 400 °C in a continuous tubular reactor, where aqueous solutions of metal salts containing Al3+, Y3+ and Tb3+ ions were used as precursors.26 Single phase YAG was produced using a stoichiometric (Y + Tb)/Al ratio ((Y + Tb)/Al = 0.6). The particles appeared to be single crystals and showed the characteristic emission of a YAG:Tb phosphor with a comparable maximum intensity to phosphors produced through a solid state reaction.27
Herein, the synthesis of a crystalline and highly luminescent nanoparticle red phosphor (nominal 4 mol% Eu in Y2O3) from flash heat-treatment of precursor (Y1–xEux)OOH nanoparticles is presented. The precursor is also a known phosphor material and has, to the best of the authors knowledge, not been prepared as a phase pure nano-sized material via. CHFS. The materials have been characterized by powder X-ray diffraction, thermogravimetric analysis, transmission electron microscopy (TEM), high resolution TEM, photoluminescence measurements (PL) and time-resolved PL. Moreover, the method provides precursor materials that can be readily formulated into stable doped Y2O3-based colloids for luminescent security inks and probes for biological imaging as discussed later within the text.
Materials and methods
Materials
Yttrium nitrate, Y(NO3)3·6H2O (99.99%), citric acid, C6H8O7 (99%), HCl (99%), NaCl (99%), NaOH (99%) and H2O2 (30% v/v) were supplied by Sigma-Aldrich Chemical Co. (Dorset, U.K.). [Eu(NO3)3·6H2O] (99.99%) was supplied by Neo Europe (Abingdon, U.K.). KOH (96%) was supplied by VWR International (Leicestershire, U.K.). All experiments were conducted using deionized water (>10 MΩ). Metal salt solutions of 0.05 M with a desired Y:Eu salt solution stoichiometry were used throughout. Table 1 provides a summary of the syntheses presented.| Run | Nominal composition | Reaction additives (M) | XRD | Reaction yield (%) | Cs (nm) | BET (m2 g−1) | TEM | |
|---|---|---|---|---|---|---|---|---|
| Avg (nm) | SD | |||||||
| a Key: flow rates used in all syntheses were fixed at 25 mL min−1 (P1), 5 mL min−1 (P2) and 5 mL min−1 (P3) generating a reaction point temperature of 382° C and residence time of 1.69 s. Nominal compositions are based on the use of 0.05 M metal salt feeds within the process. b Use of KOH as an auxiliary reagent. c Use of H2O2 as a precursor at the noted concentration. d Use of 0.1 M KOH as an auxiliary reagent added to the H2O2 solution. | ||||||||
| 1 | Y0.96Eu0.04 | 0.5b | Y(OH)3/YOOH | 84 | — | — | — | — |
| 2 | Y0.96Eu0.04 | 1.0b | YOOH | 82 | 35.7 | — | — | — |
| 3 | Y0.96Eu0.04 | 2.0b | YOOH | 87 | 41.1 | — | — | — |
| 4 | Y0.96Eu0.04 | 0.2cd | YOOH | 81 | 43.8 | — | — | — |
| 5 | Y0.96Eu0.04 | 0.4cd | YOOH | 79 | 46.5 | — | — | — |
| 6 | Y0.96Eu0.04 | 0.6cd | YOOH | 83 | 34.3 | — | — | — |
| 7 | Y0.96Eu0.04 | 0.8cd | YOOH | 80 | 33.0 | — | — | — |
| 8 | Y1.0Eu0.00 | 0.8cd | YOOH | 83 | 37.6 | 33.0 | 35.6 | 7.8 |
| 9 | Y0.98Eu0.02 | 0.8cd | YOOH | 75 | 40.8 | 34.3 | 37.4 | 6.7 |
| 10 | Y0.96Eu0.04 | 0.8cd | YOOH | 73 | 40.8 | 31.2 | 35.8 | 9.4 |
| 11 | Y0.94Eu0.06 | 0.8cd | YOOH | 79 | 45.4 | 31.9 | 40.1 | 10.3 |
| 12 | Y0.92Eu0.08 | 0.8cd | YOOH | 73 | 37.4 | 34.8 | 39.6 | 6.9 |
| 13 | Y0.90Eu0.10 | 0.8cd | Y(OH)3/YOOH | 74 | — | 27.8 | 56.7 | 7.4 |
| 14 | Y0.88Eu0.12 | 0.8cd | Y(OH)3/YOOH | 76 | — | 29.3 | 54.7 | 15.6 |
| 15 | Y0.86Eu0.14 | 0.8cd | Y(OH)3/YOOH | 52 | — | 22.7 | 67.9 | 17.2 |
Materials synthesis
Fig. 1 shows a schematic representation of the CHFS process equipment used in this work. Briefly, a three pump CHFS process was used for materials synthesis, broadly analogous to the one used by Boldrin et al. and constructed entirely of Swagelok™ components (with certain modifications as identified).10 Pumps 2 and 3 were dedicated to pumping premixed yttrium/europium nitrate solutions (0.05 M) and KOH (0.05–0.2 M), the combined output of this feed being referred to hereafter as “precursor”. Both reagent solutions were pumped at flow rates of 5 mL min−1 and water was pumped through the heater using P1 at flow rate of 25 mL min−1 and pre-heated to 450 °C in a custom made heater.10,19 A T-piece (1/8′′) was used to mix the feed issuing from P2 and P3 before meeting the superheated water feed (T in Fig. 1) to ensure complete precipitation of the component metal ions in situ at pH 11. This precipitate was then brought into contact with a supercritical water feed in a co-current mixing (reaction) geometry made using off-the-shelf Swagelok™ fittings, R in Fig. 1, resulting in rapid crystallisation of the respective products (patent application; GB1008721). The products of each reaction were collected at the exit of the back pressure regulator, after the mixture was cooled in flow. In a typical experiment, a sample volume of ca. 0.5 litres of nanoparticle-laden slurry was collected and recovered by centrifugation and washed with deionised water until the pH was neutral using repeating cycles of dispersion and centrifugation. The concentrated slurry was subsequently frozen at −40 °C and then dried in a freeze drier by slowly heating a sample from −40 °C to 25 °C over 24 h under a vacuum of 100 mTorr to recover the product for characterisation.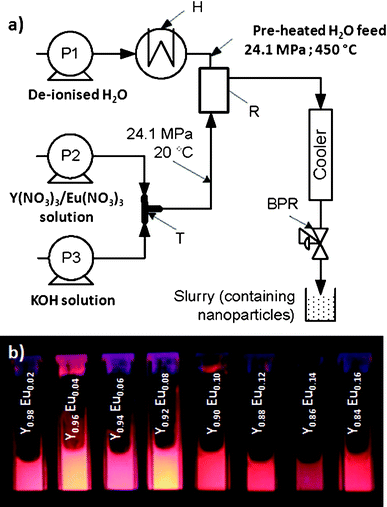 | ||
| Fig. 1 (a) Schematic of the continuous hydrothermal flow synthesis process used to make as produced phosphor nanomaterials (P = pump, H = heater, BPR = back-pressure regulator, R = Reactor, T = T-piece mixer). (b) Particle slurries of (Y1–xEux)OOH nanoparticles for values of x = 0.02–0.14 (runs 9–15) as obtained from the CHFS process under UV excitation (λ = 254 nm). | ||
The maximum theoretical temperature was calculated for the outlet of the mixer and has been quoted as the reaction point temperature throughout this publication.19 This was determined from the overall enthalpy balance, as described in our previous publication.19,22 The temperature within the reactor was also verified experimentally. The specific enthalpies and the temperature at 24.1 MPa were determined from the IAPWS Formulation 1995 for the Thermodynamic Properties of Ordinary Water Substance for General and Scientific using the associated FLUIDCAL software.27 This software was also used to calculate the density and viscosity of water for any given temperature at 24.1 MPa and used to derive the reactor residence times quoted in Table 1.
A flash heat-treatment protocol was used for controlled conversion of as prepared nanoprecursor (Y1–xEux)OOH to give the corresponding oxide. Samples (0.5 g of freeze-dried powder in a Pt crucible) were fed into the middle of a pre-heated tube furnace set to 550 °C and held at the center of it for the desired time, before being removed from the hot zone and allowed to cool. All materials characterisation was undertaken on materials obtained directly from the flash heat-treatment without further processing.
Selected nanomaterials were then prepared as dispersions. Both (Y0.96Eu0.04)OOH or Y0.96Eu0.04O3 were accurately weighed and dispersed ultrasonically using a tip sonicator (Branson USP 4000) at 80 W for 5 min in DI water. In a typical preparation, 0.5 g of nanoparticles were dispersed in DI water (10 MΏ) the dispersion forming solution 1. The dispersion was adjusted to pH 4 using HCl after the addition of 2.0 g of citric acid forming solution 2. Solution 2 was vigorously stirred using a magnetic stirrer and heated to a temperature of 60 °C. The reaction of solution 2 was allowed to proceed overnight. After completion, solution 2 was dialysed against DI water to remove un-bound citric acid and neutralise the solution against a buffer solution of DI water. After dialysis, the samples were retained as both dispersions and as freeze-dried powders for further characterisation.
Dispersions of nanoparticles were prepared for fluorescence microscopy by first filtering the dispersions through a 0.22 μm filter (Millipore, UK) and dispersing an appropriate volume in 1.5 × PBS so the final concentration of nanoparticles in 1 × PBS was 166 μg mL−1 . The concentration of nanoparticles in each aqueous preparation was determined by drying the dispersion in an oven at 300 °C and weighing the dry mass. Fluorescence microscopy was performed on cover slips seeded with COS7 cells (derived from African green monkey kidney cells). Cells with different nanoparticle loadings were incubated for 4 h prior to assay in PBS.
Characterisation
All materials characterisation was performed on materials that had been freeze-dried using a Genesis 35 XL freeze dryer. A JEOL 4000F Transmission Electron Microscope (400 kV accelerating voltage) was used for generating HRTEM micrographs of the particles. Alternatively, TEM images were generated on a JEOL 100 keV (JEOL1000 CX) system and data coming from the two microscopes is clearly identified within the text. Samples were prepared for TEM by dispersing the particles ultrasonically in ethanol (99.9%, Sigma) and dropping onto holey carbon film grids (400 mesh) (Agar Scientific, UK). Elemental analysis was performed using an Oxford Instruments Inca 300 energy dispersive X-ray (EDX) detector connected to a JEOL JSM-6300 scanning electron microscope. Brunauer–Emmett–Teller (BET) surface area measurements were carried out using N2 in a Micrometrics ASAP 2420 instrument. The samples were degassed at 120 °C (12 h) in nitrogen before the BET measurements. The equivalent sphere diameter from BET was calculated using the equation Cs = 6000/(ρ × S. A.) where Cs is the equivalent sphere diameter, ρ is the material density (calculated from the diffraction data) and S. A. is the BET surface area. XRD patterns were collected on a Bruker D4 diffractometer using Cu–Ka radiation (0.15418 nm) over the 2θ range 10–80 with a step size of 0.02° and a count time of 2 s. Yttria (Y2O3) was used as standard for the estimation of instrumental peak broadening. Refinement of the XRD data using the Le-Bail method was performed using GSAS.Thermogravimetric analysis of the samples was performed on a Netzsh STA 449C instrument under a constant flow of air, at a heating rate of 5 K min−1 from room temperature to 1500 °C (20 mg of each sample was used in each analysis). A Perkin Elmer LS55 spectrometer was used for fluorescence measurements on the as-synthesised powder samples and dispersions of particles at known concentrations in quartz cuvettes. Heat-treated samples were prepared for PL measurements by mounting the powder between two glass slides. PL measurements were performed using a 473 nm DPSS laser as an excitation source, the emitted luminescence was dispersed with a Bentham M300 monochromator and detected using a Hamamatsu photomultiplier. To obtain time-resolved PL, the laser beam was modulated with Pockels cell and PL transients were recorded with a digital oscilloscope. DLS and electrophoretic mobility measurements were taken using a Malvern Instruments Zetasizer nano series (Malvern, UK). Electrophoretic mobility measurements were taken at constant electrolyte concentrations (5 mM NaCl) using NaOH and HCL as titrants. An Olympus BX51 microscope (BX51WI) using standard continuous excitation from a 150 W Mercury Xenon lamp (Hamamatsu, L7047) was used to capture fluorescence in fluorescence assays. Digital images of phosphor particles or cellular autofluorescence were taken with a EM-CCD camera (Ixon, Andor) and the exposure was optimized for each experiment and they were analysed using freely available software Image J.
Results and discussion
Effect of CHFS reaction conditions on nanoprecursors
Phosphor materials were synthesised using a CHFS system operated under the reaction conditions summarised in Table 1. All materials were obtained in high yield (ca. 78–92%). A reaction point temperature of ca. 383 °C with 0.5 M KOH added to the auxiliary feed (run 1) produced a mixed phase product of Y(OH)3/YOOH-type phases, as determined by XRD (similar to ICDS card numbers 20098 and 28442, respectively). In contrast, the addition of KOH at concentration of 1.0–2.0 M was sufficient to produce a phase pure monoclinic YOOH (run 2 and 3; good agreement with ICDS pattern 28442). This suggests that high KOH concentrations further facilitated partial dehydration of Y(OH)3 to YOOH. Similarly, the decomposition of nitrate species in supercritical water has also been shown to form oxidizing species, including hydroxyl radicals, O2 and NO2, although alone these appeared to be insufficient to produce the YOOH phase.28 The addition of H2O2 up to 0.8 M (previously used by the authors as an oxidant aiding dehydration) to the metal feed did not alter the phase of the material produced (runs 5–8).10 This suggests a thermodynamic limitation to the direct synthesis of Y2O3 under the conditions used here. These observations differ from the author’s previous reports due to the different temperature profile (as measured between a counter current mixing geometry and the reaction point used in this work).22To investigate the effect of increasing dopant concentration on the phase behavior of (Y1 − xEux)OOH, a series of compositions were produced in the nominal range x = 0–0.14. Samples made in the range x = 0–0.08 were identified by XRD as being phase pure (ICDS pattern 28442 for monoclinic (P121/M1) YOOH) and showed bright red emission (λexitation = 254 nm) as both aqueous particle slurries (Fig. 1b) or freeze-dried powders (Fig. S4†). Le-Bail fitting of the diffraction data is presented in supplementary Table ST1† and shows a slight increase in unit cell volume as a function of dopant concentration. The metal ratios in each sample were confirmed by EDX (Fig. S2†). In contrast, materials produced in the range x = 0.10–0.14 were a mixture of Y(OH)3/YOOH phases (similar to ICDS patterns 20098 and 28442, respectively).
A comparison of the nominal and measured Eu content (measured by EDX) in the samples produced in experimental runs 9–15 (x = 0.02–0.14) is presented in Fig. S2†. The measured composition values were in good agreement with nominal values in the sample series identified as (Y1-xEux)OOH for values of x = 0.02–0.08. The largest divergence between nominal and measured composition was found in the 14 mol% Eu sample (run 15) with a measured 11.6 mol% Eu content. This discrepancy has been attributed to the incomplete precipitation of the Eu3+ ion during the reaction.
TGA of the products from runs 10 and 14, respectively, showed an overall weight loss in the region 250–700 °C of ca. 18.45%, which is in good agreement with the theoretical weight loss (19.57%) for complete dehydration of Y(OH)3 to Y2O3 (equation S1†). The TGA for the sample produced in run 10 gave a single weight loss of 6.02% in the range 400–700 °C, as expected for dehydration of YO(OH) to Y2O3 (equation S2†) (data not shown).
Photoluminescence excitation and emission spectra were measured for the samples synthesised in experimental runs 9–15 are presented in Fig. S4 and S5†. The excitation spectra of (Y1 − xEux)OOH (where x = 0.02–0.14) showed regions of strong absorbance in the 200–500 nm region attributed to transitions from charge transfer states (europium–oxygen bonds).29 Absorbance in the region 350–500 nm can be attributed to transitions from 7F0 to excited states; each transition is denoted in Fig. S4†). The emission spectra measured for samples 13–19 showed strong emission in the 600–630 nm regions corresponding to transitions of 5D0 → 7F2. A weak emission region (580–600 nm) was observed, which can be assigned to the transitions of 5D0 → 7F1.30 The intensity of the emission spectra for materials synthesised in runs 4–7 was almost identical, suggesting significant similarity in the reaction products (data not shown). In Fig. S5† it can be seen that concentration quenching appears to occur at >4 mol% europium for phase pure (Y1 − xEux)OOH where x = 0.02–0.10. The (Y1 − xEux)OOH emission spectra showed characteristic emission in the 600–630 nm regions corresponding to transitions of 5D0 → 7F2(1–4). A weak emission region (580–600 nm) was observed, which can be assigned to the transitions of 5D0 → 7F1(1–2).29 The intensity of the emission spectra was similar for materials synthesised in runs x = 0.02–0.08, however, in runs x = 0.10–0.14 the intensity was lower, which is not surprising in light of the mixed phase product identified using XRD.
Time-resolved photoluminescence was also used to investigate the dynamics of luminescence for nanoprecursors Y1 − xEuxOOH (range x = 0.02–0.08) (runs 9–12). This technique is sensitive to local variations in electron density (caused by differences in the site of europium within a host lattice) and can therefore detect defect structures and be used to infer the mechanisms responsible for the emission lifetimes. Fig. 2 displays the time-resolved luminescence data at an emission wavelength of 617 nm using pulsed laser excitation at a wavelength of 473 nm. The photoluminescence lifetimes were obtained from a single-exponential fit to the decay curves as follows (eqn (1)):
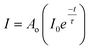 | (1) |
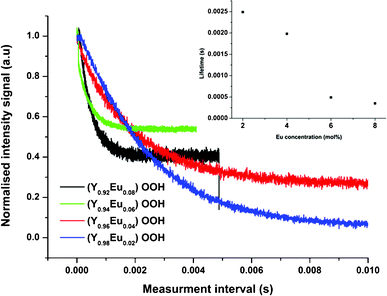 | ||
| Fig. 2 Photoluminescence lifetime measurements recorded for the 5D0 → 7F2 transition measured at 617 nm in (Y1 − xEux)OOH (where x = 0.00–0.08) (runs 9–12) obtained directly from the CHFS process. Inset shows the variation in the lifetime determined by single exponential fitting as a function of nominal Eu concentration. | ||
Where I is the signal intensity, I0 is the maximum intensity, t is the time, τ is the decay constant and Ao is a term describing the background. An increase in dopant concentration resulted in a decrease in the emission lifetime (λem 617 nm) of the materials. The longer lifetime observed for the 5D0 → 7F1 transition (3.04 ms) and those observed for 5D0 → 7F2 and 5D0 → 7F4 transitions (0.50 and 0.42 ms, respectively), are indicative of electronic dipole transitions and magnetic dipole transitions, as reported by Holsa.32 The observed decrease in photoluminescence lifetime as a function of Eu3+ substitution is consistent with an increase in non-radiative emission, which is supported by PL data showing an apparent decrease in emission intensity at 617 nm (Fig. S5†). Typically, non-radiative decay increases with dopant substitution, leading to either a reduction in local symmetry surrounding the activator ion or activator segregation with more atoms located at or near the particle surface and an increase in the proportion of defects as the host lattice is strained to accommodate the larger dopant ion.27 These defects may increase the degree of disorder and lower the local symmetry of Eu3+ ions located in/near the surface of the particles. As a consequence, the emission of 5D1–7F2 decreases.
A comparison of the crystallite sizes obtained for the diffraction data (Scherrer equation), BET data (the equivalent sphere diameter calculated measurements) and the crystallite size determined from TEM shows significant consistency (summarised in Table 1). TEM was also used to assess the crystallite morphology and particle size distribution of phosphor nanoparticles (Y1 − xEux)OOH (where x = 0.00–0.14). Fig. 3 shows the morphology and crystallite size of the (Y1 − xEux)OOH nanoprecursors, which appear to transition from spherical crystallites at lower values of x (range = 0.02–0.10) to a rod-like morphology at higher values of x (range = 0.12–0.14). Particle size data obtained for each composition are presented in Table 1. The morphology of samples produced for x in the range 10–14 mol% were identified as “Y(OH)3”, which is a similar shape to Y(OH)3 nanoparticles synthesised using other batch hydrothermal methods.32
 | ||
| Fig. 3 TEM images of the nanoparticles produced in the composition series (Y1−xEux)OOH: (a) x = 0.02, (b) x = 0.04, (c) x = 0.06, (d) x = 0.08, (e) x = 0.10 and (f) x = 0.12. | ||
HREM was used to further assess the crystalline structure of the materials produced with the nominal composition (Y0.96Eu0.04)OOH and (Y0.90Eu0.14)(OH)3 as identified by XRD (experimental runs 10 and 15) and Fig. 4a and b, respectively. HREM images confirmed that the materials were crystalline and sample (Y0.90Eu0.14)(OH)3 showed lattice fringes extending towards the surface of the crystal allowing visualisation of the (210) and (201) lattice planes with measured d-spacings of 3.12 and 2.13 Å, respectively. These were in good agreement with theoretical values of 3.13 and 2.14 Å, respectively (ICDD 23811). Selected area electron diffraction (SAED) patterns were used to confirm the identity of this phase (data not shown). Lattice fringes corresponding to the (101) plane with a measured d-spacing of 4.01 Å (theoretical, 4.03 Å) were observed at the edge of selected crystals. The material produced with the nominal composition (Y0.96Eu0.04)OOH was identified as a candidate for further heat-treatment to investigate the influence of a flash heating strategy on the formation of a nanosized (Y0.96Eu0.04)2O3 analogue of the phosphor produced directly by CHFS.
![(a) A HREM image of (Y0.90Eu0.14)(OH)3 produced using CHFS (run 14) (inset; indexed SAED pattern confirming the phase). (b) A HREM image of (Y0.96Eu0.04)OOH (run 10) showing visualization of the [101] lattice fringe with a measured d-spacing of 4.01 Å (run 10) (inset; indexed SAED pattern). (c) A HREM image of (Y0.96Eu0.04)2 O3 produced at 550 °C (inset; indexed SAED pattern).](/image/article/2012/RA/c2ra21798h/c2ra21798h-f4.gif) | ||
| Fig. 4 (a) A HREM image of (Y0.90Eu0.14)(OH)3 produced using CHFS (run 14) (inset; indexed SAED pattern confirming the phase). (b) A HREM image of (Y0.96Eu0.04)OOH (run 10) showing visualization of the [101] lattice fringe with a measured d-spacing of 4.01 Å (run 10) (inset; indexed SAED pattern). (c) A HREM image of (Y0.96Eu0.04)2 O3 produced at 550 °C (inset; indexed SAED pattern). | ||
Flash heat-treatments
In addition to size-related variables, the influence of post-synthetic processes that are often necessary to produce optical materials must be investigated, as many of the properties depend on many factors, including synthesis method, size of the particles, post-treatment temperature and the existence of segregated phases. Nanoparticle heat-treatment is a common necessity to optimise the optical properties of many phosphor systems, herein, a rapid heat-treatment protocol was tested during the current study, and XRD (Fig. 5) and fluorescence were used to analyse the products (Fig. 6). Heat-treatment is often used to process nanophase phosphor material to convert the material to a single phase or improve crystallinity. It is widely reported that the presence of OH species within nanoparticles or on the surface of nanoparticles results in reduced fluorescence yields. However, heat-treatment may induce a degree of grain growth, and can also result in the sintering of particles. These processes need to be minimized for some applications that require disperse particles.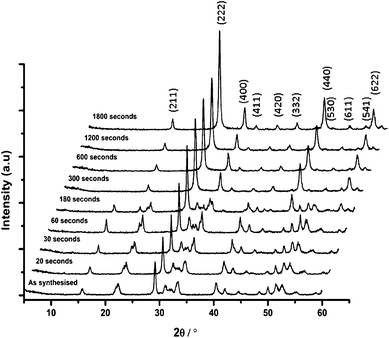 | ||
| Fig. 5 XRD patterns of (Y0.96Eu0.04)OOH (run 10) heat-treated at 550 °C for the indicated time (Miller indices for the cubic oxide system are indicated on the figure). | ||
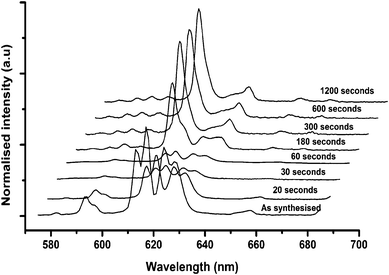 | ||
| Fig. 6 Photoluminescence spectra excited at 473 nm of the nanomaterials precursor obtained from run 11 forming (Y0.96Eu0.04)2O3 after heat-treatment at 550 °C for the indicated time. | ||
A batch of samples from run 13 (nominal composition (Y0.96Eu0.04)OOH) were heat-treated for one of the following time periods at 550 °C; 20, 30, 60, 120, 180, 300, 600, 1200 or 1800 s. Fig. 5 shows the stacked XRD patterns for these heat-treated samples and shows that at less than 60 s the dominant product was monoclinic (Y0.96Eu0.04)OOH, and between 60 and 300 s the precursor did not completely dehydrate to the corresponding oxide composition. At >300 s, the oxide was exclusively formed as a cubic phase in good agreement with ICDS pattern 23811. The Scherrer equation was applied to the XRD peak breadths to assess crystallite size to infer the degree of grain growth through coarsening vs. heat-treatment time and the data is summarized in Fig. 7.
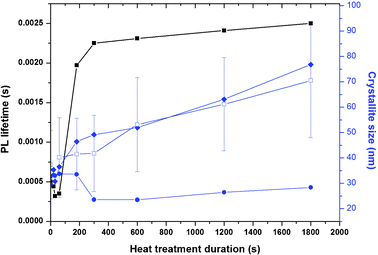 | ||
| Fig. 7 ■ Measured emission lifetime; □ crystallite size by TEM (error bars = Cv); ♦ BET equivalent sphere diameter; • crystallite size from diffraction (N. B lifetime quoted for < 180 s measured at 617 nm (5D0 → 7F2) in (Y0.96Eu0.04)OOH and >300 s at 612 nm (5D0 → 7F2) in (Y0.96Eu0.04)2O3). Dashed line represents the PL lifetime for bulk Eu-doped Y2O3. | ||
TEM images of the material heat-treated for 300 s (Y0.96Eu0.04)2O3 suggested that significant sintering of the nanoprecursor had occurred with an evident increase in primary crystallite size. Fig. S6† shows a compilation of TEM images taken for the samples produced from heat-treatment for different treatment durations. The particle size distributions measured for all heat-treated samples compared to the as-synthesised material are presented in Fig. 7. HREM was also used to assess the crystalline structure of the (Y0.96Eu0.04)2O3 after heat-treatment at 550 °C for 300 s (Fig. 4c). Fig. 4c (inset) shows the indexed selected area electron diffraction (SAED) pattern of cubic Y0.96Eu0.04O3, confirming the phase identification made using XRD. Fig. 4c shows lattice fringes allowing visualisation of the (211) and (222) planes with measured d-spacings of 4.33 and 3.08 Å (in good agreement with the expect values of 4.36 and 3.10 Å from ICDD pattern 23811, allowing for imperfect alignment of the crystal). Direct observation of the crystallites in this manner allowed confirmation of the complete recrystallization of the precursor nanomaterial to the corresponding oxide product.
A comparison of the crystallite sizes obtained from XRD and TEM is summarised in Table 1. A discrepancy between the crystallite size calculated from the Scherrer equation and TEM images suggests that coarsening occurs through a necking mechanism. The crystallite size of the precursor nanomaterials is suggested to decrease until a phase change occurred (of course this could occur from the initial reduction in size from dehydration of the precursor nanomaterial). Surface areas of the samples measured using the BET method (Fig. 7) showed a reduction in surface area as a function of heat-treatment time. Calculation of the equivalent sphere diameter for this data provided values that were consistent with TEM data, providing further evidence for a necking mechanism of particle growth and size distribution coarsening.
Photoluminescence spectra (excitation wavelength of 473 nm) for the heat-treated samples are shown in Fig. 6. The spectra clearly show a reduction in luminescence intensity from the 5D0 → 7F2 transitions in (Y0.96Eu0.04)OOH until a partial phase transition to the cubic oxide at ca. 180 s. For 473 nm excitation of samples heat-treated for 300 s at 550 °C, the emission spectrum for (Y0.96Eu0.04)2O3 was observed as rather broad red emission peaks at 581 (corresponding to 5D0–7F0), 587, 593, 600 (corresponding to 5D0–7F1(1–3)), 612 nm (highest intensity and asymmetric peak corresponding to 5D0–7F2 transition with Eu3+ ions) and dominant 630 nm (corresponding to 5D0–7F3 transition) peak.27 The luminescence intensity of the latter peak increased with heat-treatment time used, which is consistent with the literature.4 This suggests a decrease in the surface-to-volume ratio that also coincides with an increase in radiative intensity for (Y0.96Eu0.04)2O3.
The emission lifetime is a dependent parameter that can be influenced by many intrinsic structural properties of the material, particularly the presence of defects and the crystal symmetry surrounding the europium ion and the crystal field. The effect of heat-treatment time on emission lifetime was investigated for the nanoprecursor (Y0.96Eu0.04)OOH. The measured emission lifetime (λemis 617 nm [5D0 → 7F2]) for samples prepared with short heat-treatment times was < 5 ms (60 s or less heat-treatment). The emission spectrum of the material heat-treated for ca. 180 s showed an emission profile including peaks for the expected (Y0.96Eu0.04)OOH and (Y0.96Eu0.04)2O3 phase contributions. Deconvolution of peaks was difficult and was treated as an empirical comparison.33 For the samples shown to be (Y0.96Eu0.04)2O3 the lifetime measured (5D0 → 7F2 at 612 nm) for samples heat-treated for 300 s or higher gradually increased from 2.25 ms to a value of 2.45 ms with increasing heat-treatment time (data summarized in Fig. 7). The observed lifetimes are considerably greater than those reported for bulk (Y1 − xEux)2O3, which is typically ca. 1.7 ms.27 Other investigators have found that the lifetime of the 5D0–7F2 transition is actually longer by as much as a factor of three for nanocrystalline Y2O3:Eu (4.2 ms), rather than a shorter as one might expect if surface recombination relaxation is enhanced.34,35 The existence or density of defects (e.g. grain boundaries) will affect the measured lifetime of the material. A possible explanation for the increased lifetimes may be due to breakdown of the crystal field symmetry of europium at or near the surface of the nanoparticle.4 The measured lifetime arises from combined effects of 5D0 → 7F2 transition and surface states that are difficult to distinguish. With the increased heat-treatment duration, the inter-granular growth and coarsening of particles may have increased, giving longer emission lifetimes through reducing non-radiative de-excitation through the rearrangement of the crystal at growth interfaces.4 Evidence for grain growth is presented in Fig. 7, where the BET surface area of heat-treated samples reduced sequentially with increasing heat-treatment time. Schmechel and co-workers suggested that the increased lattice strain they observed in nanocrystalline Y2O3 (10.641 vs. 10.604 Å in bulk) was responsible for the increased lifetime of the 5D0 state, because “forced” electric dipole transitions are hypersensitive to small changes in local structure and are broadly consistent with our findings.33,34 Our results suggest only minimal change in crystallisation of (Y0.96Eu0.04)2O3 as a function of heat-treatment time, thus, the increase in lifetime probably arises as a result of increasing crystallite size and the dominance of the surface states over inter-particle interaction.33
Investigation of nanophosphor dispersions for biomedical use
As a high proportion of atoms are at or near the surface for nanoparticles, the adsorption of small molecules on the surface of phosphor nanoparticles can greatly alter the luminescence efficiency of the material. For aqueous dispersions of nanoparticles stabilised with citric acid, most of the surface will be covered by O–H and C–H groups that are known to actively quench the luminescence of certain lanthanide ions.3 As such, nanoparticles of both (Y0.96Eu0.04)2O3 and (Y0.96Eu0.04)OOH coated with citric acid were produced by the method detailed in the synthesis section. The obtained particle dispersions were assessed by DLS (dynamic light scattering) and the Z-average hydrodynamic diameter of each sample was measured as 88.2 and 160.3 nm with polydispersity values of 0.213 and 0.290 for (Y0.96:Eu0.04)OOH and (Y0.96Eu0.04)2O3, respectively (Fig. S6b†). All of the samples showed a distribution of particle sizes ranging from ca. 40 nm to 450 nm when the intensity weighted data was used for size evaluation if the distributions were corrected for particle number the distribution width and hydrodynamic diameter averaged between 35–80 nm as shown in Fig. S6b†. Fig. S6b† inset shows a TEM image of citric acid-coated (Y0.96Eu0.04)2O3 nanoparticles, revealing them to be well dispersed by direct observation.The stability of the dispersions was assessed using laser Doppler electrophoresis to determine the electrophoretic mobility of the material. The zeta-potential was calculated from the electrophoretic mobility data by applying the Smoluchowski approximation, assuming a material refractive index close to that of bulk YOOH (2.10). Fig.S6a† shows the zeta potential of citric acid-coated (Y0.96Eu0.04)OOH and (Y0.96Eu0.04)2O3 nanoparticles suspended in aqueous solution titrated from pH 3 to 10. The dispersions were characterised as stable above pH 7 with a measured zeta-potential >−30 mV, consistent with that expected for the de-protonation of carboxylic acid groups on the surface of the nanoparticles. The surface charge reduced at around pH 5, which is consistent with the suppression of dissociation of the weakest acid group reported for citric acid (pKa 6.40) in agreement with literature data, suggesting co-ordination to the particle surface through the two strongest acid groups of citric acid.35
Photoluminescence emission spectra (λex 254 nm) recorded for phosphors dispersions and comparative measurements between the precursor nanoparticles and coated derivatives are presented in Fig. S7a†. In both samples, a significant reduction in fluorescence intensity is observed when the coated samples are compared to the precursor particles. Concerning Eu-doped nanophosphors, the partial OH quenching of the Eu red emission appears to be significant, since the energy of the 5D0–7F2 transition in both (Y0.96: Eu0.04)OOH and (Y0.96Eu0.04)2O3 nearly corresponds to the third harmonic of the vibration mode of the OH oscillator and significant adsorption of energy by the carboxylate groups is likely the cause of reduced PL intensity. Similarly, co-ordination of acids to phosphor particle surfaces is also known to induce non-radiative de-excitation processes, which would also contribute to the reduced PL intensity.5
In biological imaging applications where UV radiation can damage organic structures, blue excitation has been employed with Eu3+ complexes and other lanthanide-doped nanophosphors as a suitable excitation wavelength.36 To demonstrate the use of both citric acid coated (Y1 − xEux)OOH and Y1 − xEuxO3 nanoparticles as potential probes for conventional bioimaging, optical phantoms containing various concentrations of coated nanoparticles (0.01, 0.05, 0.1, 0.5, 1.0 mg mL−1) were analyzed in an photoluminescence spectrometer in a quartz cuvette using an excitation wavelength of λex = 473 nm. The fluorescence intensity (expressed as relative intensity) was measured and plotted as a function of particle concentration (Fig. S7b†). The nanoparticles displayed a near linear relationship of luminescence with concentration, suggesting potential application in quantitative assays. To demonstrate the potential application of citric acid-coated (Y0.96Eu0.04)OOH nanoparticles as fluorescent probes, they were visualised under conditions analogous to a typical in vitro fluorescence imaging experiment. Particles were sparsely dispersed on cover-glass and visualised using 470 nm excitation/620 nm emission and resolvable fluorescence from both dispersions of particles was observed (Fig. S8†). Histograms of observed emission from over 50 down-converted luminescent spots are presented as insets in Fig. S8†. The diameter of fluorescence signals was typically in the order of 1–2 pixels (ca. 1 pixel = 270 nm), suggesting that most of the luminescence observed is from single citric acid-coated nanoparticles when the approximated wave spread function of the microscope is considered (λem/2 = 320 nm). The size data determined from optical methods was consistent with the data presented from light scattering techniques (Fig. S6b†). To further assess the visualization of phosphors in the dispersed phosphor systems (Y0.96Eu0.04)2O3 (squares) and (Y0.96Eu0.04)OOH (circles), an assay was designed to evaluate if fluorescence from phosphor nanoparticles could be resolved from cellular autofluorescence. In the assay, phosphor nanoparticles (filtered through a 0.22 μm syringe filter) were dispersed in PBS to a concentration of 166 μg mL−1 and COS7 cells were incubated in the solution for 4 h. We predicted that constitutive uptake of the particles would be consistent with previous publications.35,36Fig. 8a shows a bright-field image of a COS7 cell following incubation with nanoparticles of coated (Y0.96Eu0.04)OOH and (Y0.96Eu0.04)2O3, respectively, showing some sub-micron particle agglomerates formation surrounding the cell membrane. Agglomerate formation was expected as the electrostatically stabilized dispersion suffers compression of the double layer at physiological salt concentrations resulting in agglomeration. Fig. 8b shows a COS7 cell imaged using 470 nm excitation/620 nm emission showing resolvable fluorescence from the particles. A degree of cellular autofluorescence was observed using 470 nm excitation/620 nm (likely to arise from flavoproteins). Fig. 8B shows the extent of cellular autofluorescence observed using instead a 470 nm excitation/540 nm emission optical configuration. However, no fluorescence from the phosphor nanoparticles was observed at this measurement wavelength, consistent with our fluorescence measurements.
 | ||
| Fig. 8 Visualisation of COS7 cells using citric acid-coated (Y0.96Eu0.04)OOH and (Y0.96Eu0.04)2O3 nanoparticles. Data generated from (Y0.96Eu0.04)OOH nanoparticles: (a) A bright-field image of a cell incubated with (Y0.96Eu0.04)OOH NPs, (b) shows cellular autofluorescence measured at 470 nm excitation/540 nm emission. (c) Down converted luminescence following 470 nm excitation/620 nm emission shows uptake. Data generated for the (Y0.96Eu0.04)2O3 nanoparticles is presented in (d–f): (d) A bright-field image of a cell incubated with NPs, (e) cellular autofluorescence measured at 470 nm excitation/540 nm emission, (f) luminescence following 470 nm excitation/620 nm emission shows uptake (scale bar, 30 μm). | ||
Conclusions
We have used a rapid, direct and continuous hydrothermal process for the bottom-up synthesis of highly crystalline nanoprecursors that showed visibly bright red emission based on the composition (Y1− xEux)OOH. The rapid and efficient conversion of a nanoprecursor to the corresponding oxide phosphor (Y0.96Eu0.04)2O3 was achieved using a flash heat-treatment strategy, showing that complete dehydration of the corresponding (Y0.96Eu0.04)OOH precursor nanoparticles can be achieved with limited agglomeration and particle growth. The nanoprecursor and its flash heat-treated product were suitable as probes in biological imaging (after suitable formulation) and it was observed that both the as-synthesised composition (Y0.96Eu0.04)OOH and its heat-treated analogue (Y0.96Eu0.04)2O3 were both visualised in a cell imaging experiment. Both (Y0.96Eu0.04)OOH and (Y0.96Eu0.04)2O3 could be readily dispersed to form stable nanoparticle dispersions and could be readily visualised in a typical fluorescence assay using 470 nm excitation (exciting the 7F0 → 5D0 transitions in both materials) and detection at 620 nm. Due to the excitation and emission characteristics of the nanoparticles, both compositions could be readily resolved from cellular autofluorescence. The work has also demonstrated that solid state conversion of a precursor nanomaterial to a more widely recognised phosphor whilst retaining its small size yielded little overall benefit over the direct solution-based processing of precursor material due to the physicochemical characteristics of the nanosized oxide phosphor. In closing, the results presented herein highlight a desirable methodology for the synthesis of nanosized phosphors as the strategy proposed here eliminates the need for high-temperature and prolonged heat-treatment of as-prepared materials when nanosized precursors are used. The continuous nature of the initial synthesis process is amenable to scale-up to multiple kg per day scale, the process for which the authors have developed and the results of which will be reported in due course.Acknowledgements
EPSRC are thanked for funding a project entitled “Continuous hydrothermal synthesis of nanomaterials: from laboratory to pilot plant.” EPSRC Reference: EP/E040551/1 (RG, JAD, CT). EPSRC are also thanked for funding the access to the TEM instruments in Oxford Materials under the Materials Equipment Access scheme, grant reference: EP/F01919X/1 and Dr Teck Lim is thanked for assistance in taking the HRTEM images.References
- M. Bruchez, M. Moronne, P. Gin, S. Weiss and A. P. Alivisatos, Science, 1998, 281, 2013 CrossRef CAS PubMed.
- B. M. Tissue, Chem. Mater., 1998, 10, 2837 CrossRef CAS.
- V. Buissette, D. Giaume, T. Gacoin and J. P. Boilot, J. Mater. Chem., 2006, 16, 529 RSC.
- C. R. Ronda, T. Justel and H. Nikol, J. Alloy. Compd., 1998, 275, 669 CrossRef.
- K. Riwotzki and M. Haase, J. Phys. Chem. B., 2001, 105, 12709 CrossRef CAS.
- K. Riwotzki, H. Meyssamy, H. Schnablegger, A. Kornowski and M. Haase, Angew. Chemie. Int. Ed., 2001, 40, 573 CrossRef CAS.
- J. W. Stouwdam, M. Raudsepp and F. C. J. M. van Veggel, Langmuir, 2005, 21, 7003 CrossRef CAS PubMed.
- J. W. Stouwdam and F. C. J. M. van Veggel, Langmuir, 2004, 20, 11763 CrossRef CAS PubMed.
- T. Adschiri, Y. W. Lee, M. Goto and S. Takami, Green Chem., 2011, 13, 1380 RSC.
- P. Boldrin, A. K. Hebb, A. A. Chaudhry, L. Otley, B. Thiebaut, P. Bishop and J. A. Darr, Ind. Eng. Chem. Res., 2007, 46, 4830 CrossRef CAS.
- A. A. Chaudhry, J. Goodall, M. Vickers, J. K. Cockcroft, I. Rehman, J. C. Knowles and J. A. Darr, J. Mater. Chem., 2008, 18, 5900 RSC.
- J. A. Darr and M. Poliakoff, Chem. Rev., 1999, 99, 495 CrossRef CAS PubMed.
- R. Gruar, C. J. Tighe, L. M. Reilly, G. Sankar and J. A. Darr, Solid State Sci., 2010, 12, 1683 CrossRef CAS.
- X. Weng, B. Perston, X. Z. Wang, I. Abrahams, T. Lin, S. Yang, J. R. Evans, D. J. Morgan, A. F. Carley, M. Bowker, J. C. Knowles, I. Rehman and J. A. Darr, Appl. Cat. B. Environ., 2009, 90, 405 CrossRef CAS.
- Z. Zhang, J. B. Goodall, S. Brown, L. Karlsson, R. J. Clark, J. L. Hutchison, I. Rehman and J. A. Darr, Dalton Trans., 2010, 39, 711 RSC.
- Z. Zhang, J. B. Goodall, D. J. Morgan, S. Brown, R. J. Clark, J. C. Knowles, N. J. Mordan, J. R. Evans, A. F. Carley, M. Bowker and J. A. Darr, J. Eur. Ceram. Soc., 2009, 29, 2343 CrossRef CAS.
- M. Taguchi, S. Takami, T. Adschiri, T. Nakane, K. Sato and T. Naka, Cryst. Eng. Comm., 2011, 13, 2841 RSC.
- C. Y. Ma, C. J. Tighe, R. I. Gruar, T. Mahmud, J. A. Darr and X. Z. Wang, J. Sup. Fluids., 2011, 57, 236 CrossRef CAS.
- C. J. Tighe, R. I. Gruar, C. Y. Ma, T. Mahmud, X. Z. Wang and J. A. Darr, J. Sup. Fluids, 2012, 62, 165 CrossRef CAS.
- M. Chen, C. Y. Ma, T. Mahmud, J. A. Darr and X. Z. Wang, J. Sup. Fluids., 2011, 59, 131 CrossRef CAS.
- C. Y. Ma, X. Z. Wang, C. J. Tighe, X. Z. Wang, R. I. Gruar and J. A. Darr, AIChE. Submitted manuscript, 2012 Search PubMed.
- C. J. Tighe, R. I. Gruar and J. A. Darr, Scale-up of confined jet reactors for the continuous synthesis of nanoparticles, Ind. Eng. Chem. Res. Submitted., 2012 Search PubMed.
- C. Y. Ma, T. Mahmud, X. Z. Wang, C. J. Tighe, R. I. Gruar and J. A. Darr, Chem. Prod. Proc., 2011, 2, 123 Search PubMed.
- T. Lin, S. Kellici, K. Gong, K. Thompson, J. R. Evans, X. Wang and J. A. Darr, J. Combi. Chem., 2010, 12, 383 CrossRef CAS PubMed.
- V. Middelkoop, P. Boldrin, M. Peel, T. Buslaps, P. Barnes, J. A. Darr and S. D. Jacques, Chem. Mater., 2009, 21, 2430 CrossRef CAS.
- Y. Hakuta, K. Seino, H. Ura, T. Adschiri, H. Takizawa and K. Arai, J. Mater. Chem., 1999, 9, 2671 RSC.
- R. S. Meltzer, S. P. Feofilov, B. Tissue and H. B. Yuan, Phys. Rev. B., 1999, 60, 14012 CrossRef.
- H. G. Brittain and J. V. Posluszny, Thermochimica Acta, 1987, 118, 25 CrossRef CAS.
- J. Holsa, J. Phys. Sci., 1990, 45, 173 CAS.
- J. Holsa, T. Leskela and M. Leskela, J. Inorg. Chem., 1985, 24, 1539 CrossRef CAS.
- J. Holsa, J. Phys. Chem., 1990, 94, 4835 CrossRef CAS.
- N. Li, K. Yanagisawa and N. Kumada, Crys. Growth. Des., 2009, 9, 978 CrossRef CAS.
- R. Schmechel, M. Kennedy, H. von Seggern, H. Winkler, M. Kolbe, R. A. Fischer, X. M. Li, A. Benker, M. Winterer and H. Hahn, J. of Appl. Phys., 2001, 89, 1679 CrossRef CAS.
- R. Schmechel, H. Winkler, X. M. Li, M. Kennedy, M. Kolbe, A. Benker, M. Winterer, R. A. Fischer, H. Hahn and H. von Seggern, Scripta Materialia, 2001, 44, 1213 CrossRef CAS.
- N. T. Thanh and L. A. Green, Nano Today, 2010, 5, 213 CrossRef CAS.
- C. Sun, C. Carpenter, G. Pratx and L. Xing, Nanoscale Res. Lett., 2011, 6, 1432 Search PubMed.
Footnote |
| † Electronic Supplementary Information (ESI) available: Fig. S1–S9 and supplementary equations 1 and 2. See DOI: 10.1039/c2ra21798h |
| This journal is © The Royal Society of Chemistry 2012 |