Real time plasmonic spectroscopy of the interaction of Hg2+ with single noble metal nanoparticles†
M. S.
Bootharaju
a,
Kamalesh
Chaudhari
ab and
T.
Pradeep
*a
aDST Unit of Nanoscience and Thematic Unit of Excellence, Department of Chemistry, Indian Institute of Technology, Madras, Chennai - 600 036, India. E-mail: pradeep@iitm.ac.in
bDepartment of Biotechnology, Indian Institute of Technology, Madras, Chennai - 600 036, India
First published on 28th August 2012
Abstract
In this work, we use dark-field optical microscopy (DFM) and hyper spectral imaging (HSI) to study the interactions of single Ag and Au nanoparticles (NPs) with Hg2+ in real time, at room temperature (25 °C). NPs were immobilized on glass substrates using 3-aminopropyltrimethoxysilane (APTMS) as the anchoring agent. Red, green and blue colors were assigned to the particles in hyper spectral images on the basis of their relative scattering intensities at 640, 550 and 460 nm, where the particles showed maxima in their scattering spectra. While Ag NPs showed all the colors, Au NPs were mostly red and rarely green in DFM images. The scattering spectra of Ag NPs were more blue shifted (with an average shift of 46 nm in the case of red particles) in a given time compared to Au NPs, after passing over Hg2+ solution and these shifts increased with time. Depending on the extent of blue shift, the colors of the particles got modified. Red particles appeared more reactive than green and blue, as revealed from the larger extent of shifts and their time dependence. The greater reactivity of red particles is attributed to their anisotropic nature possessing reactive tips, edges and more surface area due to their large size. The effect of quality of water on the scattering spectrum was checked by passing over deionized (DI) and tap waters separately, which showed that the effect is minimal compared to the presence of Hg2+, when data at a given time, flow rate and temperature were compared. Solution phase interactions of NPs with Hg2+ were also performed for comparison. These were characterized by UV-vis absorption spectroscopy, transmission electron microscopy (TEM) and energy dispersive analysis of X-rays (EDAX). Solution phase experiments showed citrate-induced aggregation of Ag NPs and partial reduction of Hg2+ to Hg0 upon exposure to Hg2+. Immobilized particles cannot aggregate and they show only reduction.
Introduction
Noble metal nanoparticles (NPs) are well known to exhibit unique optical, catalytic and electronic properties which offer numerous opportunities for fundamental studies.1 These characteristics are due to the coherent oscillations of free electrons in the conduction band of the NPs, when they are illuminated with electromagnetic radiation, a phenomenon called localized surface plasmon resonance (LSPR).2,3 The growing field of research of such light–metal interactions is known as ‘plasmonics’.4 LSPR is measured as an extinction spectrum (scattering and absorption) of these NPs which depends on various parameters such as size,5 shape,6 inter-particle spacing,7 composition,8 refractive index,9 local dielectric environment of the medium9 and temperature.10 NPs possess extremely high absorption and scattering cross sections11 compared to molecular dyes due to LSPR (for e.g., fluorescein dye and 60 nm silver NPs have absorption and extinction cross sections of 3.5 × 10−16 and 2.5 × 10−10 cm2, respectively)12 which help in sensing applications. Excitation of LSPR results in wavelength-selective absorption with extremely high molar extinction coefficients (∼3 × 1011 M−1 cm−1).13 SPR can be tuned from the visible to the infrared regions11 for various applications in miniaturized optical devices, chemical/biological sensing, photonic circuits as well as in medical diagnostics and therapeutics.3 In the recent past, noble metal NPs have been found to have applications in water purification by removing pesticides,14,15 mineralizing halogenated hydrocarbons,14,16 decontaminating toxic metal ions such as mercury, cadmium and lead,17etc. Typically, nanoparticles in solution undergo aggregation leading to a red shift in their characteristic surface plasmon resonance upon exposure to the analyte.The scattering properties of NPs have been used for sensing applications as extinction-based methods require relatively smaller quantities of NPs in comparison to other colorimetric reagents. Scattering intensity is highly advantageous as a single 80 nm Au particle exhibits a light scattering power equivalent to the emission of ∼106 fluorescein molecules.12 Recently, a technique called single particle plasmon spectroscopy18 has evolved which measures the scattering spectrum of isolated plasmonic NPs. This spectroscopy has several advantages including greater sensitivity and smaller sample volumes than traditional methods.19 The NPs can be imaged by collecting scattered light from the particles and the image is called a hyper spectral image (HSI). This technique has been utilized for the real time study of chemical reactions,20 bio-molecule binding,18 detection of conformational changes of protein,21 sensing of small organic molecules22 and receptors23 as well as for DNA hybridization24. The application of such a technique is necessary to understand the real time interactions of NPs with inorganic contaminants such as heavy metal ions (Hg2+, Cd2+, Pb2+, etc.) and organic contaminants such as phenols, nitro aromatics, etc. present in water. Mercury is known to cause the minamita25 disease and its permissible limit in drinking water is 2 ppb set by the US environmental protection agency (EPA).26 Contamination of mercury in the environment occurs due to natural and anthropogenic processes.26 Various sensors such as monolayer and protein protected noble metal quantum clusters,26,27 NPs,28etc. have been reported for mercury sensing where the interactions are ensemble averaged. Recently, functionalized single plasmonic gold NPs have been utilized for Hg2+ sensing.29 In this paper, the mechanistic aspects of sensing are not discussed. There is no study so far on the utilization of scattering spectra of Ag NPs for understanding the real time interactions with Hg2+.
In this work, we studied the interaction of immobilized Ag and Au@citrate NPs with Hg2+ in real time using DFM and X-ray photoelectron spectroscopy (XPS). Interactions of Hg2+ with mobile (solution phase) NPs were also done for comparison, which were characterized with UV-vis spectroscopy, TEM and EDAX. After interaction of immobilized NPs with Hg2+, representative colors of NPs were changed with a blue shift and decrease in the intensity of the scattering. The color change and blue shifts were attributed to the redox reaction of Ag NPs and reduction of Hg2+ leading to the formation of Hg0. It may be pointed out that the particles are immobile which prevents consequent aggregation and red shift. The effect of water quality on the scattering spectrum of NPs was also studied. The decrease in silver content due to the reaction was confirmed by XPS quantification data. A visual detection limit of Hg2+ on the basis of color change of NPs in HSI is 1 ppm in our experimental conditions. This can be brought down further to ppb level by optimizing the experimental conditions such as decreasing the flow rate of Hg2+ and increasing the incubation time. Here, our objective was to understand the real time interactions of Ag NPs with Hg2+ spectroscopically and microscopically. Enhancing the detection limit was not an aspect of the investigation.
Experimental section
Materials
Silver nitrate, tetrachloroauric acid trihydrate (CDH, India), trisodium citrate (TSC, Qualigens), mercuric acetate (Ranbaxy), 3-aminopropyltrimethoxysilane (APTMS, Sigma-Aldrich), chloroform, hydrogen peroxide, and sulphuric acid were purchased and used as such without further purification. For flow cell experiments, 1.1 mm thick 40 mm round glass substrates and 0.145 mm thick 30 mm round glass cover slips from Warner instruments were used. Before immobilization thorough cleaning of the substrates was done as mentioned in the section on immobilization of NPs (below). For dark-field microscopy, a type A immersion oil (Cargille) was used as received without purification.Synthesis of Ag and Au@citrate NPs
The Ag@citrate NPs were prepared according to the literature.17 In this method, to a boiling 50 mL silver nitrate (1 mM) solution, 2 mL of 1 wt% TSC was added, and heating was continued further for a few minutes. The solution turned light yellow in color, indicating the formation of NPs. The suspension was cooled in an ice bath to allow the growth of NPs. The Au@citrate NPs (red color solution) were synthesized similar to Ag@citrate NPs according to the literature.30Immobilization of NPs on glass substrates
Immobilization of Ag and Au@citrate NPs (after twenty times dilution of the as-prepared particles) was done on a glass substrate in a plastic glove box in which N2 atmosphere was maintained. Before that, it was thoroughly cleaned using aqua regia (3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 volume mixture of conc. HCl and HNO3) several times. Later, it was immersed in piranha solution (2:1 volume mixture of H2SO4 and H2O2) for 30 min to create Si–OH bonds. Then, the substrate was rinsed with DI water followed by drying in N2 flow. About 100 μL of 0.3 M APTMS in chloroform was dropped on the substrate with a pipette and dried for 5 min. Then it was rinsed with excess chloroform followed by DI water to remove excess APTMS. After that, the glass substrate was dipped in the dilute NP solution for 2 min in such a way that the solution contacted only one surface. Negatively charged Ag and Au@citrate NPs bind with positively charged amino groups of APTMS through electrostatic interaction. Then it was rinsed with DI water and kept in a beaker containing DI water till it was used. Binding of NPs to APTMS is shown schematically as given below (inset A of Scheme 1).
:1 volume mixture of conc. HCl and HNO3) several times. Later, it was immersed in piranha solution (2:1 volume mixture of H2SO4 and H2O2) for 30 min to create Si–OH bonds. Then, the substrate was rinsed with DI water followed by drying in N2 flow. About 100 μL of 0.3 M APTMS in chloroform was dropped on the substrate with a pipette and dried for 5 min. Then it was rinsed with excess chloroform followed by DI water to remove excess APTMS. After that, the glass substrate was dipped in the dilute NP solution for 2 min in such a way that the solution contacted only one surface. Negatively charged Ag and Au@citrate NPs bind with positively charged amino groups of APTMS through electrostatic interaction. Then it was rinsed with DI water and kept in a beaker containing DI water till it was used. Binding of NPs to APTMS is shown schematically as given below (inset A of Scheme 1).
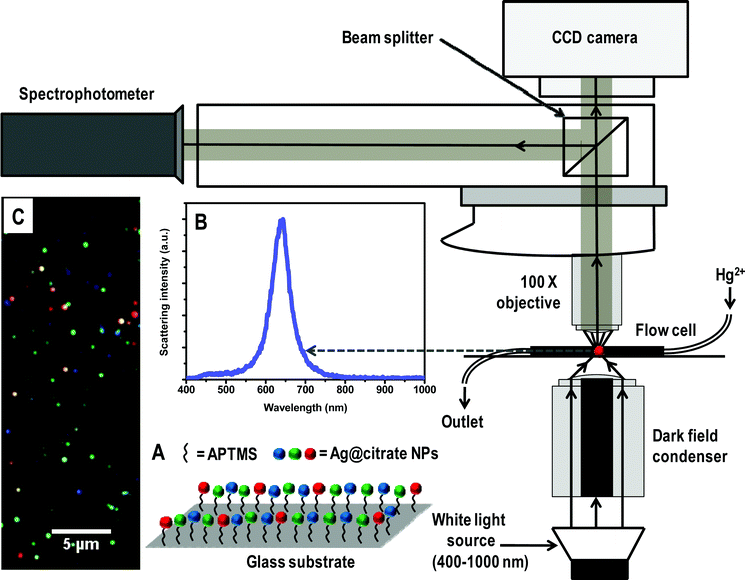 | ||
| Scheme 1 Schematic representation of HSI set-up. A is a representation of immobilized Ag@citrate NPs using APTMS on glass substrate. B and C are the scattering spectrum of a silver particle and HSI of Ag@citrate NPs, respectively. | ||
Real time monitoring of interaction between NPs and Hg2+
For this, a flow cell set-up which is shown in Scheme 1 was used. Initially, DI water was allowed to flow over the immobilized NPs for 5 min. Then, 5 ppm Hg2+ solution was continuously passed at a flow rate of 3.5 mL h−1 and HSI images from a particular area were collected at different times. In control experiments, DI and tap water were also passed to check their effect on the scattering spectrum of immobilized NPs.Instrumentation
Hyper spectral measurements were done with the Cytoviva HSI system containing an Olympus BX-41 microscope equipped with a Dage high resolution camera and a Specim V10E spectrometer which scans in the range of 400–1000 nm, divided into 462 bands which gives a spectral resolution of ±1.5 nm. The color of the particle was fixed on the basis of its relative scattering intensity at 640, 550 and 460 nm, assigned to red, green and blue, respectively. UV-vis spectra were measured with a PerkinElmer Lambda 25 instrument in the range of 200 to 1100 nm. High resolution transmission electron microscopy (HRTEM) of the samples was carried out using a JEOL 3010 instrument with a UHR polepiece. TEM specimens were prepared by drop-casting one or two drops of the aqueous solution on carbon-coated copper grids and allowed to dry at room temperature overnight. All measurements were done at 200 kV to minimize the damage of the sample. X-ray photoelectron spectroscopy (XPS) measurements were conducted using an Omicron ESCA Probe spectrometer with unmonochromatized Al Kα X-rays (energy = 1486.6 eV). The X-ray power applied was 300 W. The pass energy was 50 eV for survey scans and 20 eV for specific regions. The base pressure of the instrument was 2.0 × 10−10 mbar. The binding energy was calibrated with respect to the adventitious C 1s feature at 285.0 eV. Most of the spectra were deconvoluted to their component peaks using the software, CASA-XPS. Inductively coupled plasma-optical emission spectroscopic (ICP-OES) measurements were done using a PerkinElmer Optima 5300 DV instrument. Field-emission scanning electron microscopic (FESEM) measurements were carried out using an FEI Nova NanoSEM 600 instrument with a chamber pressure of 1 × 10−4 Torr, at a working distance of 3–4 mm. Instrumental conditions were optimized to avoid surface charging.Results and discussion
Characterization of NPs
The Ag and Au@citrate NPs were characterized extensively previously30,31 and also in this study. We are giving here only the essential data. The Au particles show an absorption maximum at 522 nm and TEM analysis shows the size of particles as 10 ± 5 nm suggesting the narrower size distribution as shown in Fig. S1 of ESI†. As-synthesized Ag@citrate NPs show surface plasmon resonance around 417 nm as shown in Fig. S2A. The TEM image shows that they are polydispersed (Fig. S2B). Particles are mostly spherical along with some triangles, prisms, rods, pentagons, etc. (Fig. S3) which are expected in the case of Ag@citrate NPs.22 The sizes are in the range of 15–60 nm. XPS data suggest the presence of silver in the zero-valent state. This data will be discussed later in the text.Immobilized particles were subjected to HSI analysis and a large area hyper spectral image is shown in Fig. 1A. It shows different representative particles in red, green and blue. The scattering spectra from single red, green and blue (traces a, b and c, respectively) particles are shown in Fig. 1B. The corresponding particles are shown in the inset of B in which particles are marked with dotted circles. In our specific case, the red, green and blue particles show maxima at 659, 540 and 470 nm, respectively. It is reported that when Au NP monomer is converted to its dimer, a color change from green to yellow is observed with red shift in the scattering spectrum.32 Intensities of scattering spectra of blue particles are less than green and green is lesser than red. It is known that particles of high aspect ratio and larger size scatter more light than the smaller objects.20 To verify the existence of isolated nanoparticles on the glass substrates, FESEM measurements were carried out on the nanoparticle-immobilized surfaces (inset of Fig. 1A). The large area FESEM image of the same sample is shown in Fig. S4A. The EDAX spectrum clearly shows the presence of silver in a single NP (Fig. S4B). FESEM images reveal that each colored particle in HSI is a single particle with polydispersity in size. The scattering position is therefore related to the size of the particle. To validate the nature of a single particle in HSI, we have compared the number of particles in HSI and FESEM in a given area. For an area of 7.2 × 6.2 μm2, the number of particles in HSI and FESEM images are 33 ± 3 and 35 ± 3, respectively (Fig. S5). However, as the particles appear larger in HSI images, they seem to present a larger number density.
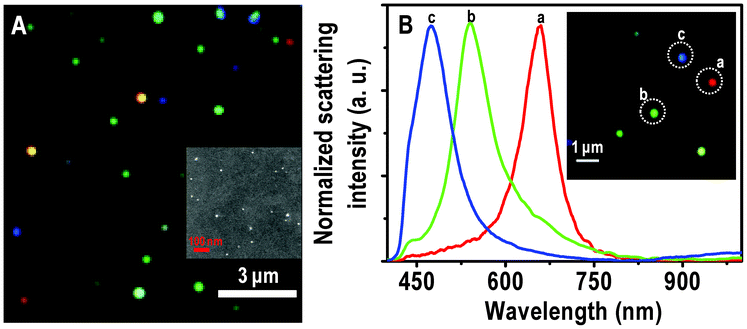 | ||
| Fig. 1 (A) Large area HSI of immobilized Ag@citrate NPs. (B) Scattering spectra of Ag@citrate particles which are shown in the inset. Particles from which spectra are collected are labelled in the inset. The particles and the spectral traces have the same colors. Scale bars: 3 μm in (A) and 1 μm in the inset of (B), showing the typical distances between the particles. The sizes of the particles are exaggerated as they are imaged under the optical diffraction limit. Inset of A is an FESEM image of immobilized Ag@citrate NPs on a glass substrate. | ||
Interaction of Ag@citrate NPs with Hg2+
A Hg2+ solution of 5 ppm was passed over the immobilized Ag@citrate NPs in a flow cell at a flow rate of 3.5 mL h−1. At different times of interaction, HSI analyses were done. The scattering spectra of all the colored particles were seen to be blue shifted simultaneously with a change in color as a function of time. The large area HSI of Ag particles before and after treating Hg2+ are shown in Fig. S6. The scattering spectra of almost all particles are blue shifted. One such single red particle is completely converted to green and is shown in Fig. 2A. The intermediate colors, red–yellow and yellow–green are seen after 1.5 and 2.5 h (d and f, respectively in Fig. 2A). Normalized scattering spectra of the same particle at different times are shown in Fig. 2B. The λmax of the red particle at 0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0 and 6.0 h of interaction with Hg2+ are 633, 621, 612, 607, 602, 599, 592, 588 and 585 nm, respectively (traces a–i, respectively). The peak maxima have an uncertainty of 1.5 nm. A blue shift of 12 nm within 30 min indicates a fast interaction of Ag@citrate NP with Hg2+. From the HSI images, it is noted that at 3.0 h, a particular red particle is completely transformed to a green one with a λmax of 592 nm (trace g). Note that the extent of blue shift varies from one red particle to another red particle. Even after an exposure for 3.0 h, no further color change is seen. But only a blue shift of 7 nm is noticed. This indicates that there could be adsorption of Hg2+ on the surface of NP which diminishes further interaction Hg2+ in solution (discussed later). The color change may be due to oxidation of Ag0 in the process of reducing Hg2+ to Hg0 and subsequent changes. At least part of the Ag+ formed may dissolve in water. Simultaneously, the Hg0 formed diffuses into the nanoparticles changing their composition (may lead to the formation of amalgam). Gradual conversion of another red particle is shown in Fig. S7. The λmax at 0.0 and 6.0 h exposure are 632 and 605 nm, respectively (Fig. S7B). This change is reflected in a color change of deep red to red–yellow.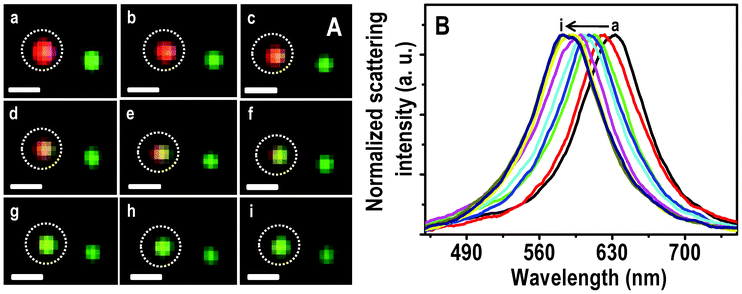 | ||
| Fig. 2 HSI (A) of immobilized Ag@citrate NPs after passing 5 ppm Hg2+ for 0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0 and 6.0 h (a–i, respectively). Scattering spectra (B) of a red particle with time (a–i). Scale bars of images in A are 500 nm. The particle from which spectra are collected is marked. | ||
Similarly, conversion of a green particle to blue is shown in Fig. 3. The λmax of the particle at 0.0, 0.5 and 6.0 h of passing Hg2+ are 528, 518 and 501 nm, respectively. The particle is almost completely converted to blue at 3.0 h (Fig. 3Ag) itself and a further blue shift of the scattering position is less than 5 nm. This indicates that similar to the red particle, there could be adsorption of Hg2+ followed by reduction on NPs which prevents subsequent interaction with Hg2+. The HSI of the blue particle with time is shown in Fig. S8. The maxima were observed at 495, 491, 484 and 478 nm at 0.0, 0.5, 1.0 and 6.0 h of treatment of Hg2+. Seemingly interaction of the blue particle with Hg2+ is less compared to red and green particles, under the given experimental conditions.
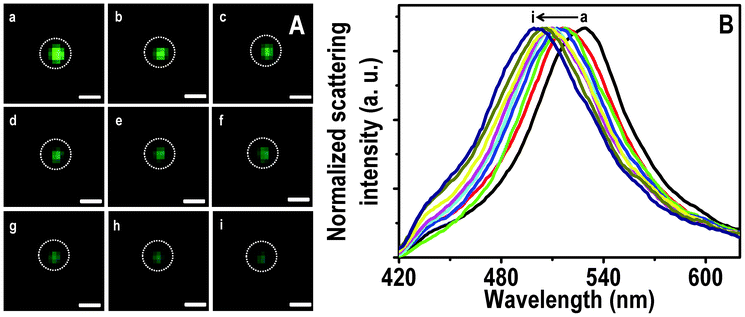 | ||
| Fig. 3 HSI (A) of immobilized Ag@citrate NPs after passing 5 ppm Hg2+ for 0.0, 0.5, 1.0, 1.5, 2.0, 2.5, 3.0, 4.0 and 6.0 h (a–i, respectively). Scattering spectra (B) of a green particle (marked) with time (a–i). Scale bars of images in A are 400 nm. | ||
Interestingly, some particles (red and green) show a blue shift with decrease in scattering intensity after reacting with Hg2+ for 1.5 h. These effects were attributed to a redox reaction.33,34 Thereafter, they show a small red shift and increase in scattering intensity up to 4.0 h (Fig. S9). These were due to the deposition of reduced mercury on partially reacted silver particles leading to a small increase in size. After 4.0 h, a small blue shift with diminishing scattering intensity was noticed which may be due to the continuous reactivity of Ag particle. The dominant blue shift (due to oxidation of Ag0) compared to red shift (due to deposition of reduced mercury) of scattering wavelength may be due to the requirement of oxidation of two Ag0 for reduction of one Hg2+. Uptake of mercury by the particle was manifested in the EDAX spectrum of Ag@citrate particles immobilized on the functionalized glass substrate. The FESEM image of the particle from which the EDAX was taken is shown in Fig. S10 as an inset. The presence of Hg and Ag was confirmed by the EDAX spectrum collected from one of the particles (Fig. S10).
To understand the interactions better, HSI and scattering spectra of 5 particles of each color (red, green and blue) were collected. The average of shifts (Δλmax) of each colored particle with time of treatment with Hg2+ are plotted in Fig. 4. Note that the bars (I) represented in the graph are not error bars. They show the range of Δλmax in which all the five particles are present. Shifts are larger for red particles and smaller for green and very small for blue particles, at a given time of exposure. From this, it appears that red particles are most reactive towards Hg2+ in comparison to blue particles. There are two issues being discussed here. One is the larger shift of the red particles and the second is the nature of the shift itself. While the plasmon resonances are normally found to red shift upon interaction with analytes, a blue shift is observed here.
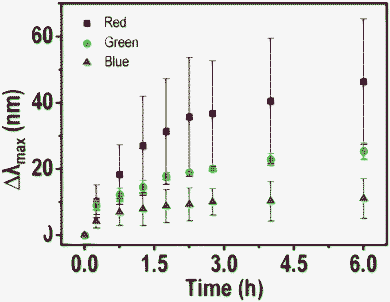 | ||
| Fig. 4 A plot of average blue shift (Δλmax) of 5 particles of each color (red, blue and green) with time. A large shift is observed in the case of red particles and it decreases for green and blue analogues. | ||
The apparent larger reactivity of red particles may be due to the following reasons. 1. As in the Ag@citrate particle system, there is a possibility of non-spherical particles to exist which possess reactive sharp edges and tips.35,36 Anisotropic particles are expected to scatter light of higher wavelengths due to large aspect ratios (red colored particles). Maybe due to the greater reactivity of these anisotropic particles, more of a blue shift was seen in the case of red particles. Although we looked for anisotropic particles and their shape dependent Hg2+ reaction leading to inhomogeneous mercury distributions on them in TEM, these effects were not detected. It could be that mercury diffusion in such nanoscale particles was fast and imaging time scale was slow to detect inhomogeneity in the distribution. 2. From the FESEM and TEM images, we know that the particles are polydispersed in size and shape. The red particles in HSI are bigger in size. Due to the greater surface area of bigger particles, a greater extent of reactivity may be expected in such red particles.
Various research groups reported the red shift of the scattering spectrum of NPs when molecules/ions interact/bind with NPs.18,20,37 They attributed the red shift to an increase in the local refractive index.38 In our experiment, we see a blue shift which may be due to Hg2+ interaction with the NP core. There is the feasibility of a redox reaction which may be dominating here as particles were immobilized whereas in the mobilized case, aggregation is dominant (discussed later). The possibility of reduction of Hg2+ by citrate was avoided as there was a continuous flow which flushes excess citrate. The reduced Hg2+ may interact with the remaining silver core and change its composition leading to the formation of an amalgam, shifting the plasmon resonance. This leads to a blue shift.39 The deposited Ag+, likely to be in the form of poorly soluble salts reduces further interaction of the core with Hg2+ and as a result, the shift reduces with time. The Ag+ ions formed are partly detectable in the solution. Another reason for the blue shift is the reduction in the size of the silver particles.33 Further support for this suggestion comes from XPS (see below). The results presented suggest that all of these events occur and the observed process is predominantly due to redox chemistry in the case of silver. However, such reduction slows down with time as Ag+ is deposited on the surface of the particle.
Similarly immobilized Au@citrate NPs were also treated with 5 ppm Hg2+. HSI images with time are shown in Fig. S11. Initially, Au particles were in green and red colors (with maxima ∼540 and ∼640 nm, respectively). Some of the red particles turned red–yellow after treating with Hg2+. Here again the scattering spectra were blue shifted due to reduction of Hg2+ leading formation of an amalgam.34 The magnitude of the shifts was less compared to Ag particles for a given time which indicates more reactivity of silver compared to gold particles in our experimental conditions. This suggests that nano silver reduces Hg2+ more efficiently than nano gold. Ag particles of red color show an average blue shift of 12 nm within 30 min whereas Au particles of red color show a shift of 15 nm after 2.0 h (Fig. S12). It could be due to the strong metallophilic interactions of Au1+–Hg2+.27,40 Since large shifts were seen in the case of silver, more analyses were done in this case compared to gold NPs.
XPS analysis was performed to deduce the elemental composition of immobilized Ag NPs before and after passing Hg2+ for 6.0 h. The XPS survey spectra and Ag 3d regions of immobilized Ag@citrate NPs are shown in Fig. 5A and B, respectively. Fig. 5A shows the presence of all possible elements C, O (from APTMS or TSC), N (from APTMS), Si (from glass substrate and APTMS) and Ag (from Ag@citrate NPs) before as well as after treatment with Hg2+ (traces a and b, respectively). The presence of N confirms the immobilization of NPs on APTMS. The presence of Ag 3d5/2 before and after treatment of Hg2+ at 368.2 eV suggests silver in the zero-valent state17 (Fig. 5B). But we were unable to detect the presence of mercury which may be due to its ultra low concentrations. The full width at half maximum (FWHM) of Ag 3d5/2 before and after passing Hg2+ over are 1.3 and 1.7 eV, respectively under the same conditions of XPS measurements. The increase of FWHM may be due to presence of mixed silver oxidation states (Ag+ and Ag0). The elemental quantification data suggest that atomic Ag% decreases with respect to Si% after treating with Hg2+. The atomic ratios of Ag to Si before and after Hg2+ treatment were 0.09 and 0.05 as shown in Table 1. This may support a decrease in the particle size and blue shift of λmax after passing Hg2+ over. This could be due to the dissolution of Ag NPs as Ag0 gives electrons for the reduction of Hg2+ and the ions may dissolve partially in water. Si was taken as an internal reference element because it is largely due to the glass substrate. To check the presence of Ag in the outlet of the flow cell, ICP-OES analysis was performed. An output solution of 20 mL was concentrated using a rotavapor to 2 mL in which 50 ± 7 ppb silver was detected.
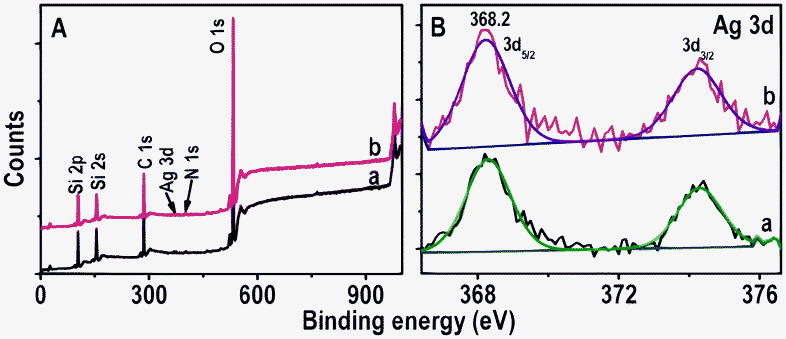 | ||
| Fig. 5 XPS survey spectra and Ag 3d regions (A and B, respectively) of immobilized Ag@citrate NPs before and after passing Hg2+ (traces a and b, respectively). The spectra have been normalized with respect to the Si 2p feature in A and Ag 3d5/2 in B, but shifted vertically for clarity. | ||
| Si(At.%) | Ag(At.%) | Atomic ratio of Ag to Si | |
|---|---|---|---|
| Before | 91.55 | 8.44 | 0.09 |
| After | 95.24 | 4.76 | 0.05 |
Effect of water quality on the scattering spectrum
It is important that these spectral changes are compared with changes observed in real water samples. DI (specific resistance = 18.2 MΩ cm) and tap water (in which concentrations of Ca2+, Mg2+, Cl−, NO3−, SO42− were 52 ± 5, 10 ± 2, 80 ± 6, 4 ± 1 and 31 ± 3 ppm, respectively) were separately passed over immobilized Ag@citrate NPs under the same experimental conditions as that for Hg2+. HSI and scattering spectra of all colored particles were collected. The average shifts of five particles of each color were plotted at different times. The data shown in Fig. S13 suggest that DI and tap waters also cause spectral shifts. In both the cases, i.e. DI and tap waters (Fig. S13A and B, respectively), a maximum shift of 15 nm is observed at 6.0 h with a flow rate of 3.5 mL h−1 at 25 °C. In the case of 5 ppm Hg2+, a maximum shift of 46 nm was observed after 6.0 h. This indicates that under the above experimental conditions, the effect of typical ions in water on the position of the scattering spectrum is minimal.Solution phase experiments
To understand the interactions of NPs with Hg2+, solution phase reactions were performed. For this, 5 mL of as-synthesized Ag, Au@citrate NPs were centrifuged to remove excess TSC if any, and to the centrifugate, 5 mL of 5 ppm Hg2+ solution was added. This solution was stirred for 6.0 h at room temperature and it was analyzed with UV-vis absorption spectroscopy, XPS, TEM and EDAX. UV-vis absorption of Ag@citrate NPs (after centrifuging followed by redispersion) shows a peak at 418 nm (Fig. 6A, trace a). After 6.0 h, Hg2+ treated NPs show a broad peak around 464 nm with a decrease in intensity along with a hump around 384 nm (Fig. 6A, trace b). This red shift of SPR may be due to aggregation of NPs. Added Hg2+ interacts with carboxylate groups of citrate which leads to aggregation.17 The blue shift of SPR (similar to scattering spectra of single particles) may be due to reduction of Hg2+ to Hg0 leading to the formation of an amalgam.39 Formation of Hg0 was further confirmed by XPS analysis (see below). The absorption spectrum showed dominant aggregation than reduction whereas in HSI we see only blue shift due to reduction of Hg2+ in a given experimental condition. It clearly indicates that in solution phase as particles were mobile, there was more chance to bring NPs closer by interlocking Hg2+ with the carboxylate groups present on the surface of nanoparticles. In the case of immobilized particles, interaction with the core occurred leading to the reduction of Hg2+ resulting in a large blue shift of the scattering wavelength. Aggregation of NPs in the sample was confirmed by TEM analysis (Fig. S14A). The presence of mercury and silver in aggregate of NPs is supported by elemental mapping and EDAX spectrum (Fig. S14). The reaction mixture was centrifuged at 5000 rpm and the centrifugate was analyzed using XPS to deduce the oxidation states of Hg and Ag. XPS data are shown in Fig. 6B–D. Fig. 6B is the survey spectrum which shows the presence of all possible elements; C, O, Ag, Hg and Na, the last is from TSC. Peaks of Hg 4f7/2 are seen at 99.7 and 101.5 eV (Fig. 6C). The peak at 101.5 eV suggests that most of the mercury is present as Hg2+ and the peak at 99.7 eV indicates that a small percentage of Hg0 was also formed26 which supports the blue shift of UV-vis (trace b, Fig. 6A). The presence of Ag 3d5/2 at 368.0 eV confirms that silver is in the zero-valent state (Fig. 6D).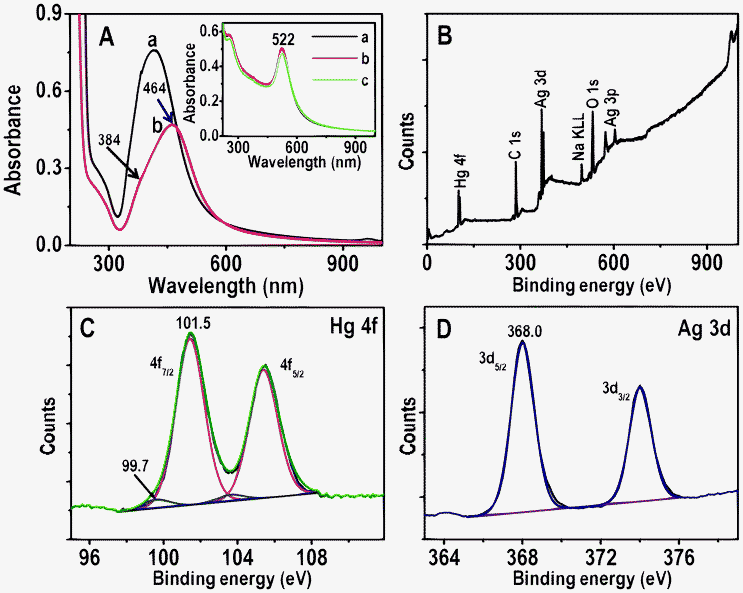 | ||
| Fig. 6 A) UV-vis absorption spectra of Ag@citrate NPs before and after treatment with 5 ppm Hg2+ for 6.0 h (traces a and b, respectively). B–D are XPS survey spectrum, Hg 4f and Ag 3d regions, respectively of Ag@citrate NPs treated with 5 ppm Hg2+ for 6.0 h. Inset: UV-vis absorption spectra of Au@citrate NPs and 5 ppm Hg2+ after interaction for 5 min, 3.0 and 6.0 h (a, b and c, respectively). | ||
Time-dependent UV-vis absorption spectra of Au@citrate particles and 5 ppm Hg2+ solution are shown in the inset of Fig. 6A. Unlike in the case of Ag particles, no shift of plasmon peak is seen but a small decrease in the absorbance value is noticed after 6.0 h. This supports the poor interaction of Hg2+ with gold particles compared to silver in a given time (6.0 h) and Hg2+ concentration (5 ppm). The reaction mixture of Au NPs and 5 ppm Hg2+ (after 6.0 h) was centrifuged at 5000 rpm. The residue obtained was analyzed with XPS and data are compared with parent Au NPs (Fig. 7). Survey spectra of NPs before and after treatment of Hg2+ (traces a and b, respectively in Fig. 7A) show the presence of expected elements Au, C, Na and O. Gold is present in the zero-valent state (Au 4f7/2 at 84.1 eV) before and after treatment of Hg2+ (traces a and b, respectively in Fig. 7B). The Hg 4f region from Hg2+ treated NPs is shown as an inset in Fig. 7A. Its absence indicates that no or poor interaction of Hg2+ occurs with Au NPs. However, Au@citrate NPs were found to interact with mercuric ion41 at higher concentrations (∼60 ppm, data not shown).
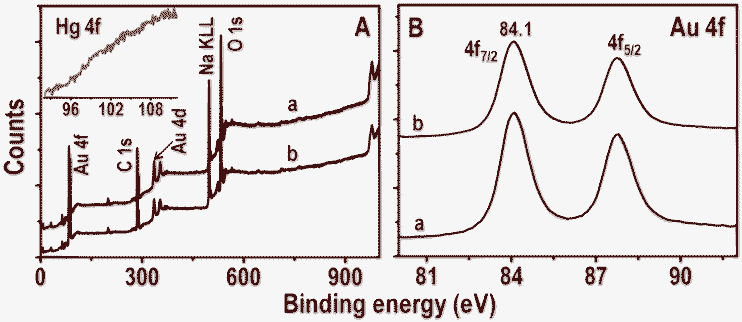 | ||
| Fig. 7 XPS survey spectra and Au 4f regions (A and B, respectively) of Au@citrate NPs before and after treatment with 5 ppm Hg2+ for 6.0 h (traces a and b, respectively). Inset of A is the Hg 4f region of trace b. | ||
The reduction of Hg2+ to Hg0 is also supported by redox potentials of Ag+/Ag, Hg2+/Hg+ and Hg+/Hg0 systems. The standard reduction potentials of Ag+/Ag, Hg2+/Hg+ and Hg+/Hg0 are 0.8, 0.91 and 0.82 V, respectively. The cell emf (electromotive force) for the reactions, Ag0 + Hg2+ → Ag+ + Hg+ and Ag0 + Hg+ → Ag+ + Hg0 are therefore +0.11 and +0.02 V, respectively. But at the nanoscale, the reaction may be more feasible. The lower reactivity of gold particles with Hg2+ may be due to the negative emf (−0.5 V) for the reduction (2Au0 + 3Hg2+ → 2Au3+ + 3Hg0) of Hg2+. Although Au+ could be formed, this reaction is also not feasible (Au0 + Hg2+ → Au+ + Hg+, emf: −0.77 V).
Summary and conclusions
A real time study of interactions of Ag and Au@citrate NPs with Hg2+ was performed using hyperspectral imaging. The feasibility of the redox reaction of Ag@citrate with Hg2+ leads to the oxidation of silver and its dissolution. Consequently the reduced Hg interacts with the nanoparticle left behind and the amalgam formed blue shifts the scattering spectrum. It is found that red particles are more reactive compared to green and blue as they show a larger shift in the scattering spectrum upon reaction with Hg2+ which may be due to their larger size and consequent surface area. As NPs are immobilized, aggregation is avoided. Whereas in the solution phase, aggregation of NPs along with partial reduction of Hg2+ is seen, which was confirmed by XPS. The effect of ions present in tap water on the scattering spectrum of NP is minimal for a given time. Greater reactivity of silver (in the present study) compared to gold particles with Hg2+ in a given time is observed due to the feasibility of the redox reaction for the former. To the best of our knowledge, this is the first attempt to study real time interactions of single silver particles with mercuric ions. In terms of sensitivity, although our study reveals a visual detection limit (in HSI) of 1 ppm Hg2+, the sensing capability of these NPs may be increased to detect lower mercury levels by functionalizing with thiols, increasing in incubation time, decreasing the flow rate, etc.Acknowledgements
We thank the Department of Science and Technology (DST), Government of India, for constantly supporting our research program on nanomaterials. Thanks are also due to Prof. G. U. Kulkarni of Jawaharlal Nehru Centre for Advanced Scientific Research, Bangalore for FESEM measurements. M.S.B. thanks the CSIR for a senior research fellowship.References
- I. Pastoriza-Santos and L. M. Liz-Marzan, J. Mater. Chem., 2008, 18, 1724–1737 RSC.
- J. Homola, Chem. Rev., 2008, 108, 462–493 CrossRef CAS.
- M. E. Stewart, C. R. Anderton, L. B. Thompson, J. Maria, S. K. Gray, J. A. Rogers and R. G. Nuzzo, Chem. Rev., 2008, 108, 494–521 CrossRef CAS.
- E. Ozbay, Science, 2006, 311, 189–193 CrossRef CAS.
- M. A. El-Sayed, Acc. Chem. Res., 2001, 34, 257–264 CrossRef CAS.
- R. Jin, Y. Cao, C. A. Mirkin, K. L. Kelly, G. C. Schatz and J. G. Zheng, Science, 2001, 294, 1901–1903 CrossRef CAS.
- J. Yu, H. Tao and B. Cheng, ChemPhysChem, 2010, 11, 1617–1618 CrossRef CAS.
- E. Ringe, J. M. McMahon, K. Sohn, C. Cobley, Y. Xia, J. Huang, G. C. Schatz, L. D. Marks and R. P. Van Duyne, J. Phys. Chem. C, 2010, 114, 12511–12516 CAS.
- P. Mulvaney, Langmuir, 1996, 12, 788–800 CrossRef CAS.
- S. Link and M. A. El-Sayed, J. Phys. Chem. B, 1999, 103, 4212–4217 CrossRef CAS.
- L. Slaughter, W.-S. Chang and S. Link, J. Phys. Chem. Lett., 2011, 2, 2015–2023 CrossRef CAS.
- J. Yguerabide and E. E. Yguerabide, Anal. Biochem., 1998, 262, 157–176 CrossRef CAS.
- T. R. Jensen, M. D. Malinsky, C. L. Haynes and R. P. Van Duyne, J. Phys. Chem. B, 2000, 104, 10549–10556 CrossRef CAS.
- T. Pradeep and S. Anshup, Thin Solid Films, 2009, 517, 6441–6478 CrossRef CAS.
- M. S. Bootharaju and T. Pradeep, Langmuir, 2012, 28, 2671–2679 CrossRef CAS.
- A. S. Nair and T. Pradeep, Curr. Sci., 2003, 84, 1560–1564 CAS.
- M. S. Bootharaju and T. Pradeep, J. Phys. Chem. C, 2010, 114, 8328–8336 CAS.
- G. Raschke, S. Kowarik, T. Franzl, C. Sönnichsen, T. A. Klar, J. Feldmann, A. Nichtl and K. Kürzinger, Nano Lett., 2003, 3, 935–938 CrossRef CAS.
- Y. Liu, J. Ling and C. Z. Huang, Chem. Commun., 2011, 47, 8121–8123 RSC.
- C. Novo, A. M. Funston and P. Mulvaney, Nat. Nanotechnol., 2008, 3, 598–602 CrossRef CAS.
- W. P. Hall, J. Modica, J. Anker, Y. Lin, M. Mrksich and R. P. Van Duyne, Nano Lett., 2011, 11, 1098–1105 CrossRef CAS.
- A. D. McFarland and R. P. Van Duyne, Nano Lett., 2003, 3, 1057–1062 CrossRef CAS.
- T. Huang, P. D. Nallathamby, D. Gillet and X.-H. N. Xu, Anal. Chem., 2007, 79, 7708–7718 CrossRef CAS.
- G. L. Liu, Y. Yin, S. Kunchakarra, B. Mukherjee, D. Gerion, S. D. Jett, D. G. Bear, J. W. Gray, A. P. Alivisatos, L. P. Lee and F. F. Chen, Nat. Nanotechnol., 2006, 1, 47–52 CrossRef CAS.
- E. Sumesh, M. S. Bootharaju, S. Anshup and T. Pradeep, J. Hazard. Mater., 2011, 189, 450–457 CrossRef CAS.
- M. S. Bootharaju and T. Pradeep, Langmuir, 2011, 27, 8134–8143 CrossRef CAS.
- Y.-H. Lin and W.-L. Tseng, Anal. Chem., 2010, 82, 9194–9200 CrossRef CAS.
- C.-J. Yu and W.-L. Tseng, Langmuir, 2008, 24, 12717–12722 CrossRef CAS.
- H. D. Song, I. Choi, Y. I. Yang, S. Hong, S. Lee, T. Kang and J. Yi, Nanotechnology, 2010, 21, 145501 CrossRef.
- A. S. Nair and T. Pradeep, J. Nanosci. Nanotechnol., 2007, 7, 1871–1877 CrossRef CAS.
- M. V. Cañamares, J. V. Garcia-Ramos, J. D. Gómez-Varga, C. Domingo and S. Sanchez-Cortes, Langmuir, 2005, 21, 8546–8553 CrossRef.
- B. M. Reinhard, M. Siu, H. Agarwal, A. P. Alivisatos and J. Liphardt, Nano Lett., 2005, 5, 2246–2252 CrossRef CAS.
- L.-X. Qin, C. Jing, Y. Li, D.-W. Li and Y.-T. Long, Chem. Commun., 2012, 48, 1511–1513 RSC.
- W. Qi, Y. Wang, J. Wang and C. Huang, Sci. China: Chem., 2012, 55, 1445–1450 CrossRef CAS.
- R. Narayanan and M. A. El-Sayed, J. Phys. Chem. B, 2005, 109, 12663–12676 CrossRef CAS.
- T. Mokari, E. Rothenberg, I. Popov, R. Costi and U. Banin, Science, 2004, 304, 1787–1790 CrossRef CAS.
- T. Huang, P. D. Nallathamby and X.-H. N. Xu, J. Am. Chem. Soc., 2008, 130, 17095–17105 CrossRef CAS.
- J. N. Anker, W. P. Hall, M. P. Lambert, P. T. Velasco, M. Mrksich, W. L. Klein and R. P. Van Duyne, J. Phys. Chem. C, 2009, 113, 5891–5894 CAS.
- A. Henglein and C. Brancewicz, Chem. Mater., 1997, 9, 2164–2167 CrossRef CAS.
- J. Xie, Y. Zheng and J. Y. Ying, Chem. Commun., 2010, 46, 961–963 RSC.
- I. Ojea-Jiménez, X. López, J. Arbiol and V. Puntes, ACS Nano, 2012, 6, 2253–2260 CrossRef.
Footnote |
| † Electronic Supplementary Information (ESI) available: UV-vis absorption spectra and TEM images of Ag and Au@citrate NPs. EDAX spectrum, scattering spectra, FESEM and HSI images of Hg2+ treated NPs. See DOI: 10.1039/c2ra21384b |
| This journal is © The Royal Society of Chemistry 2012 |