Highly efficient and stable deep-blue emitting anthracene-derived molecular glass for versatile types of non-doped OLED applications†
Illhun
Cho‡
,
Se Hun
Kim‡
,
Jong H.
Kim
,
Sanghyuk
Park
and
Soo Young
Park
*
Creative Research Initiative Center for Supramolecular Optoelectronic Materials and WCU Hybrid Materials Program, Department of Materials Science and Engineering, Seoul National University, 1 Gwanak-ro, Gwanak-gu, Seoul, 151-744, Korea. E-mail: parksy@snu.ac.kr; Fax: +82-2-886-8331; Tel: +82-2-880-8327
First published on 20th October 2011
Abstract
New anthracene-based deep-blue emitting molecular glass, 9-(9-phenylcarbazole-3-yl)-10-(naphthalene-1-yl)anthracene (PCAN), which is asymmetrically functionalized with N-phenylcarbazole and naphthalene, has been designed, synthesized, and characterized. The deep-blue emitting PCAN is efficiently secured for color purity, has high glass transition temperature (Tg = 151 °C), and has excellent solubility (>100 mg mL−1 in toluene), due to its highly tilted asymmetric molecular conformation. Using processing versatility of PCAN, vacuum-deposited and solution-processed non-doped deep-blue fluorescent organic light-emitting diodes (OLEDs) were prepared, which employ PCAN as an emitter. The vacuum deposited, non-doped EL device exhibited not only the excellent luminance efficiency and external quantum efficiency (as high as 3.64 cd A−1 and 4.61%, respectively) for the saturated deep-blue CIE chromaticity coordinates of (0.151, 0.086), but also stable performance and a good device lifetime. Furthermore, the solution processed EL device also exhibited an encouraging level of performance (1.24%, 1.15 cd A−1) and deep-blue emission (0.159, 0.105).
Introduction
Organic light-emitting diodes (OLEDs) have emerged as the most promising next-generation flat-panel display technology in the decades since the pioneering work of Tang et al.1 Although OLEDs have recently been developed that can be employed in small display panels, there remain several obstacles to be overcome before they can be applied in the mainstream display panel market. A critical drawback is the poor performance of saturated deep-blue OLEDs compared with green and red OLEDs. Because of the intrinsic wide-band-gap nature of deep-blue emitters, balancing the various requirements is a significant challenge in the development of new OLEDs.2 For proper non-doped (ND) blue electroluminescence (EL) devices, the requirements of high efficiency, thermal stability, sufficiently long device lifetime, and saturated deep-blue emission with a Commission Internationale de l'Eclairage (CIE) coordinate of y < 0.10 that can be matched to the National Television System Committee (NTSC) (0.14, 0.08) must be concurrently satisfied.3 In addition, amenability to wet processing, which is an inevitable prerequisite for large area, low cost mass production, must be taken into consideration.4Since the first reports on the efficient emission properties and excellent stability of 9,10-di-(2-naphthyl)anthracene (ADN),5 there has been intense interest in the development of anthracene derivatives as promising blue emitters for EL devices.6 Recent reports by Shu et al. and Park et al. showed luminance efficiencies of 5.5 cd A−1 and 3.1 cd A−1 for anthracene derivatives applied as non-doped blue EL devices, but insufficient deep-blue emission was shown, with CIE coordinates of (0.15, 0.12) and (0.16, 0.13), respectively.7,8 Lee et al. reported a new blue emitter with saturated color coordinates of (0.149, 0.086), but a maximum luminance efficiency of only 2.63 cd A−1.9 Lai et al. and Park et al. reported efficient saturated blue-emitting materials ((0.15, 0.07), 3.2 cd A−1, (0.156, 0.088), 3.64 cd A−1).10,11 However, these data cannot be fully interpreted, because no mention was made of device lifetime. Despite the fact that device lifetime is one of the most important requirements for EL devices, reports of highly efficient deep-blue emitting materials possessing sufficiently long device lifetime are still rare. In addition, small molecular deep-blue anthracene OLEDs that can be wet-processed have not yet been reported. To realize highly efficient and stable deep-blue emitters with wet processibility, these materials must possess highly efficient emitting centers and amorphous glass characteristics, with not only thermal and morphological stability and a high glass transition temperature (Tg), but also high solubility through a bulky backbone structure.
Here we describe our discovery, in the course of investigating anthracene-derived deep-blue emitting materials, of 9-(9-phenylcarbazole-3-yl)-10-(naphthalene-1-yl)anthracene (PCAN), which is asymmetrically functionalized with N-phenylcarbazole and naphthalene, and which showed saturated deep-blue emission (0.151, 0.086) and highly efficient device performance (3.64 cd A−1, 4.61%), with long device lifetime. Furthermore, using the high solubility and amorphous-film-formation properties of PCAN, we demonstrated a wet-processed EL device with saturated blue emission (0.159, 0.105) and high efficiency (1.15 cd A−1, 1.24%). Herein, we report the synthesis, characterization and electroluminescent device results of PCAN.
Result and discussion
Synthesis
The molecular structure and synthesis route for PCAN are depicted in Scheme 1. Briefly, compound 1 was synthesized by reaction of 3-iodo-9-phenylcarbazole with n-BuLi at −78 °C, followed by lithium exchange with 2-isopropoxy-4,4,5,5-tetramethyl-1,3,2-dioxaborolane. Compound 2 was synthesized by the Suzuki cross-coupling reaction of 9-bromoanthracene with 1-naphthyl boronic acid, in the presence of a catalytic amount of Pd(PPh3)4, 2 N NaOH aqueous solution, and a toluene/ethanol 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 mixed solution. Compound 3 was synthesized by the NBS bromination of compound 2 in CHCl3. The final product, PCAN, was synthesized by the Suzuki cross-coupling reaction of compound 3 with compound 1. PCAN was obtained by using the same reaction conditions with compound 2. The molecular structure was characterized by 1H NMR, 13C NMR, elemental analysis (EA), and mass spectrometry (MS).
:1 mixed solution. Compound 3 was synthesized by the NBS bromination of compound 2 in CHCl3. The final product, PCAN, was synthesized by the Suzuki cross-coupling reaction of compound 3 with compound 1. PCAN was obtained by using the same reaction conditions with compound 2. The molecular structure was characterized by 1H NMR, 13C NMR, elemental analysis (EA), and mass spectrometry (MS).
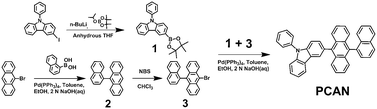 | ||
| Scheme 1 Synthesis of PCAN. | ||
Theoretical calculations
To understand the optimized geometry and the frontier molecular orbital energy levels, we also carried out density functional theory (DFT) calculations at the B3LYP/6-31G* using Gaussian 03.12 As shown in Fig. 1, the calculated highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) were found to be mainly localized on the anthracene moiety. Because of large torsional stresses, the 9,10-substituents are tilted almost 90° to the anthracene emitting center. Consequently, PCAN has a bulky, non-coplanar structure that can efficiently prevent molecular recrystallization through intermolecular interactions, resulting in an emitting center that is totally isolated from neighboring molecules that can induce excimer or exciplex formation.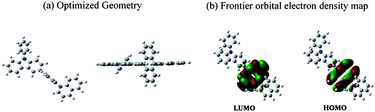 | ||
| Fig. 1 Density functional theory (DFT) calculation. (a) Three-dimensional optimized geometry of PCAN, and (b) the calculated HOMO and LUMO electron density map of PCAN. | ||
Thermal and morphological properties
The thermal properties of PCAN were characterized with differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) in an N2 atmosphere. Although PCAN is a low molecular weight material, it showed a high Tg of 151 °C, and a melting temperature (Tm) was not observed from 50 °C to 300 °C. The onset-of-decomposition temperature (Td), defined as the temperature at which 5% mass loss occurs, as measured by TGA, was determined to be 374 °C (see ESI†).Because of its highly bulky backbone structure and the included carbazole moiety, PCAN exhibits a molecular glass character (i.e., it is a thermally stable, glass-like solid that does not undergo a first-order melting transition).13 We probed these molecular glass features, and the birefringence, using a polarized optical microscope (POM). In the glass state, PCAN showed a fine surface, transparency, and emitted in the deep blue under irradiation with UV light. Birefringence was not shown at all directions (see Fig. 2).
 | ||
| Fig. 2 Polarized optical microscope (POM) images of PCAN molecular glass ((a) PCAN molecular glass images and (b) polarized angular position images for the birefringence test). | ||
To confirm the thermal stability and morphological durability against thermal stress, we also carried out an atomic force microscopy (AFM) study of a vacuum-deposited (VD) PCAN film (see Fig. 3). Samples were prepared using the thermal vapor deposition technique on a silicon wafer substrate of about 40 nm thickness to match that used in the actual electroluminescent device. For a direct comparison, we used ADN as a control material. We prepared thermally treated sample films at 100 °C with times of 0 h, 12 h, and 24 h under an N2 atmosphere. The untreated ADN VD film had a root mean square roughness (Rrms) of 15.4 nm, and the PCAN VD film had an Rrms of 5.5 nm. In the ADN case, the Rrms significantly increased (48.2 nm at 12 h, 55.6 nm at 24 h) during the thermal treatment, due to recrystallization. In contrast, the PCAN VD film showed a negligible change in its Rrms (±1 nm from 0 h to 24 h). Due to the Joule heat generated during device operation, high thermal and morphological stability against heat in the organic material is critically important. Consequently, we expect that PCAN materials having high thermal and morphological stability will maintain a uniform and stable emitting layer interface during device operation, which is a basic factor contributing to long device operation lifetime.
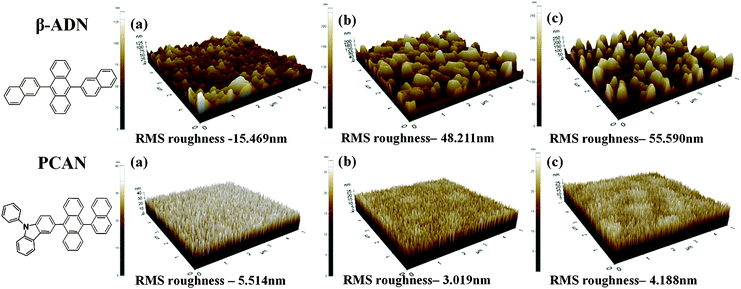 | ||
| Fig. 3 Atomic force microscope (AFM) topographic images of PCAN and ADN with thermal treated samples ((a) no treatment, (b) 100 °C 12 h, and (c) 100 °C 24 h). RMS roughness = Rrms | ||
Photophysical and electrochemical properties
The photophysical properties of PCAN were explored using diluted THF solutions, and in solid thin films VD on quartz plates. The absorption spectra in the diluted solution expressed the archetypal vibronic structure of isolated anthracene (358, 375 and 396 nm). In the case of PL, although the emission λmax of the VD film was slightly red-shifted compared with the diluted solution (430 nm for the solution and 455 nm for the VD film), the total emission range was mainly located in the deep-blue region, and the two emission spectra showed almost the same emission band edge (see Fig. 4(a)). The resulting emission spectra revealed that, through the isolation of the anthracene emitting core from the neighboring molecules, excimer formation, significantly red-shifted emission and concentration quenching occurring due to π–π interaction were effectively inhibited, as predicted by the DFT calculations.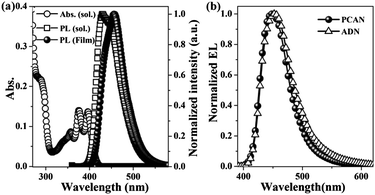 | ||
| Fig. 4 (a) The absorption and PL spectra of PCAN in dilute THF solution, and in vacuum-deposited film. (b) EL spectra of PCAN and ADN. | ||
The energy levels of PCAN were also measured by cyclic voltammetry (CV) and optical band-gap measurements. The HOMO energy level measured using CV was −5.49 eV, and the LUMO energy level calculated from the optical band gap (Eg) was −2.51 eV (Table 1). The Eg calculated from the threshold of optical absorption was 2.98 eV.
| Compound | λ abs a/nm | λ em b/nm | λ em c/nm | T g/Tm/Td/°C | HOMO/LUMOd/eV |
|---|---|---|---|---|---|
| a Maximum absorption wavelength, measured in THF (10−5). b Maximum emission wavelength, measured in THF (10−5). c Maximum emission wavelength, measured in film state (films prepared on the quartz plate by the thermal evaporation method). d The HOMO energy level was measured by CV, and the LUMO energy level was determined from the HOMO level and optical band-gap (N.D.: not detected). | |||||
| PCAN | 358, 375, 396 | 430 | 455 | 151/N.D./374 | −5.49/−2.51 |
Electroluminescent properties
To explore the OLED characteristics, we fabricated ND EL devices with a device structure of ITO (150 nm)/2-TNATA (60 nm)/NPB (20 nm)/blue emitting layer (PCAN-40 nm and ADN-40 nm)/Alq3 (20 nm)/LiF (1 nm)/Al (100 nm), where 2-TNATA = 4,4′,4′′-tris(N-(2-naphthyl)-N-phenyl-amino)triphenylamine, NPB = N,N′-bis(naphthalene-1-yl)-N,N′-bis(phenyl)-benzidine, Alq3 = tris(8-hydroxy-quinolinato)aluminium. To evaluate the efficiencies and device lifetime of PCAN objectively, we used ADN that we synthesized (under exactly the same environmental conditions as were used for the synthesis of PCAN) as a control material. Fig. 4(b) shows the EL spectra of PCAN and ADN at 10 mA cm−2. The emission λmax features of PCAN and ADN were located at 447 nm and 452 nm, and the full width at half maximum (FWHM) values were 54 nm and 60 nm, respectively.As a direct consequence of the emission properties, the CIE chromaticity coordinates (x,y) of PCAN and ADN EL emissions were (0.151, 0.086) and (0.157, 0.111) at 10 mA cm−2. As expected, PCAN showed much more pure deep-blue emission than ADN, due to the former molecule's bulky and asymmetric molecular structure. As shown in Fig. 5, the luminance efficiency, power efficiency, and external quantum efficiency values for PCAN were 3.64 cd A−1, 1.41 lm W−1, and 4.61% at 10 mA cm−2, respectively. The values for ADN were 3.09 cd A−1, 1.24 lm W−1, and 3.22%, respectively, at 10 mA cm−2 (Table 2). Notably, PCAN not only had deeper blue emission than ADN, it also showed higher performance for all measures of efficiency. Furthermore, the CIE value deviation of the PCAN EL spectra in response to the current change was negligible (x = 0.000, y = 0.001) from 10 mA cm−2 to 100 mA cm−2 (see Fig. 6(a)). Through the excellent color stability of the PCAN EL device, we confirmed that the electronic energy level matching with neighboring layers was finely tailored, and charge carrier injection and recombination occurred efficiently at the PCAN emitting layer at all current values. To investigate the device operation stability, the lifetime of PCAN and ADN EL devices was measured as the operational half lifetime (t1/2) under an initial luminance of 400 cd m−2 at a constant current (see Fig. 6(b)). The PCAN EL device showed a lifetime (610 h) that was approximately 5 times longer than that of the ADN device (133 h), as expected by the AFM thermal properties study.
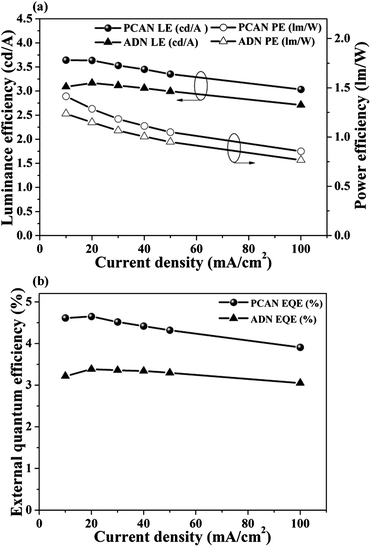 | ||
| Fig. 5 (a) Luminance efficiency (LE) and power efficiency (PE) of the blue non-doped device with PCAN and ADN. (b) External quantum efficiency (EQE) of the blue non-doped device with PCAN and ADN. | ||
| EML | V on a/V | V b , c/V | η c c | η p d | η ex e | CIExy | T 1/2 f |
|---|---|---|---|---|---|---|---|
| a Turn-on voltage, at 1 cd m−2 (V). b At 10 mA cm−2 (V). c Luminance efficiency (cd A−1). d Power efficiency (lm W−1). e External quantum efficiency (%). f Device half-lifetime at 400 cd m−2 (h). | |||||||
| PCAN | 4.6 | 8.10 | 3.64 | 1.41 | 4.61 | 0.15, 0.08 | 610 |
| (11.15) | (3.04) | (0.86) | (3.90) | (0.15, 0.08) | |||
| ADN | 4.6 | 7.85 | 3.09 | 1.24 | 3.22 | 0.15, 0.11 | 133 |
| (11.12) | (2.71) | (0.77) | (3.05) | (0.15, 0.10) |
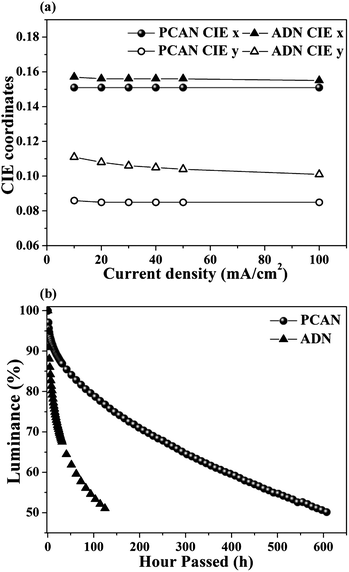 | ||
| Fig. 6 (a) CIE coordinates deviation of EL spectra in response to the current change of PCAN and ADN EL devices (from 10 mA cm−2 to 100 mA cm−2). (b) Operational half lifetime of EL devices (under an initial luminance of 400 cd m−2). | ||
These findings suggest that the greatly improved efficiency and lifetime characteristics arise from a combination of the highly efficient emitting nature of PCAN and its high color purity, the finely tailored electronic energy levels in the EL device, and the fact that the layer interface remains stable during device operation due to PCAN's high thermal and morphological stability.
Although PCAN is comprised of aromatic rings without aliphatic chains, it is readily soluble in common organic solvents. Based on the excellent solubility of PCAN (>100 mg mL−1 in toluene at room temperature), we investigated the possibility of solution-processed deep-blue EL devices with a device structure of ITO (150 nm)/PEDOT:PSS (40 nm)/PCAN (70 nm)/Bphen (30 nm)/LiF (1 nm)/Al (100 nm), where PEDOT:PSS = poly(3,4-ethylenedioxythiophene):poly(styrenesulfonate), Bphen = 4,7-diphenyl-1,10-phenanthroline. This preliminary solution-processed device also showed deep-blue EL emission with CIE chromaticity coordinates (x,y) of (0.159, 0.105). The maximum device performance was recorded at 1.15 cd A−1 luminance efficiency, 0.71 lm W−1 power efficiency, and 1.24% external quantum efficiency at 20 mA cm−2 (see Fig. 7). We believe that further improvements in the device performance can be achieved through energy level adjustment between neighboring layers, charge mobility matching, and so on. We also expect PCAN can be a suitable emitter for the fully solution-processed device if an appropriate processing method is developed in near future. Although the results shown here are not from an optimized device, it is notable that we successfully fabricated a solution-processed EL device using the small molecule PCAN, which shows excellent solubility without dendritic structure and/or aliphatic chains. Furthermore, the most important point is that both the VD device and the solution-processed small molecule device showed the best performance for each device type.
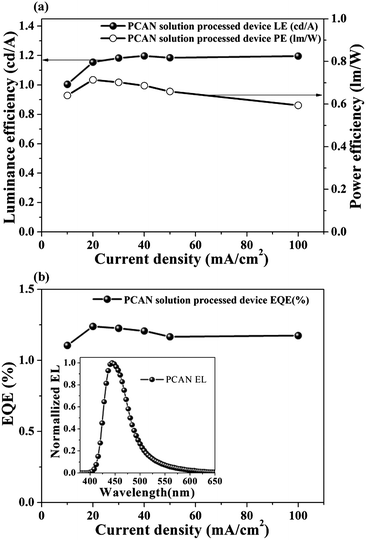 | ||
| Fig. 7 (a) Luminance efficiency and power efficiency of the solution-processed ND blue device with PCAN. (b) EQE of the solution-processed ND blue device (inset—EL spectra of the solution processed device). | ||
Conclusions
In summary, we have developed a multi-processable, deep-blue emitting molecular glass PCAN, which exhibits excellent deep-blue emission, along with high thermal and morphological stability. The highly tilted asymmetric conformation of PCAN efficiently suppresses the close packing of molecules in the solid state, and consequently secures color purity and elevates solubility. The VD ND EL device exhibited not only excellent device performance (4.61% and 3.64 cd A−1) with saturated deep-blue emission (0.151, 0.086), but also a highly improved device lifetime. Furthermore, we successfully demonstrated a solution-processed deep-blue EL device (0.159, 0.105) with high performance (1.24%, 1.15 cd A−1). To the best of our knowledge, this is the first report of a processing-versatile, small molecular deep-blue emitter that shows excellent efficiency in both VD and wet processed devices. We believe that PCAN has strong advantages for the OLED industry owing to its capability to be used in various fabrication processes.Experimental
Synthesis
PCAN was synthesized according to the procedure shown in Scheme 1.:1 v/v). Yield: 71% (4.3 g). 1H NMR (300 MHz, CDCl3, δ): 8.64 (s, 1H), 8.19 (d, J = 7.7, 1H), 7.87 (d, J = 8.3, 1H), 7.63–7.53 (m, 4H), 7.47 (t, J = 7.1, 1H), 7.40–7.35 (m, 3H), 7.29 (t, J = 7.8, 1H), 1.40 (s, 12H).
:7 v/v) and reprecipitation (CH2Cl2/methanol) to give PCAN (1.7 g, 60%). 1H NMR (300 MHz, CDCl3, δ): 8.35 (d, J = 15.5, 1H), 8.13 (t, J = 7.02, 1H), 8.09 (d, J = 8.2, 1H), 8.04 (d, J = 8.2, 1H), 7.87 (d, J = 8.6, 2H) 7.74–7.45 (m, 14H), 7.33–7.20 (m, 7H). 13C NMR (500 MHz, CDCl3, δ): 141.6, 140.6, 138.5, 138.0, 137.1, 135.0, 134.0, 133.9, 131.0, 130.8, 130.7, 130.2, 129.6, 129.5, 129.4, 128.4, 128.3, 127.8, 127.7, 127.4, 127.2, 127.0, 126.5, 126.2, 125.8, 125.4, 125.2, 123.7, 123.5, 123.4, 120.7, 120.4, 110.2, 109.9. MS(FAB, m/z): calcd for C42H27N, 545.21; found, 545.21. Anal calcd for C42H27N: C: 92.44, H: 4.99, N: 2.57; found: C: 92.44, H: 4.98, N: 2.58.
Characterization
Chemical structures were fully identified by 1H NMR (Bruker, Avance-300), 13C NMR (Bruker, Avance 500), GC-Mass (JEOL, JMS-700), and elemental analysis (EA1110, CE Instrument). The glass transition temperatures and melting temperatures of the compounds were determined using DSC under an N2 atmosphere, using a PERKIN ELMER DSC7, TAC7/DX. The decomposition temperatures of the compounds were obtained using TGA under an N2 atmosphere, using a TA instruments Q50 model. UV-Vis spectra were recorded on a SHIMADZU UV-1650PC, while PL spectra were taken using a Varian Cary Eclipse fluorescence spectrophotometer. HOMO levels of organic materials were obtained from the cyclic voltammetry measurements. Cyclic voltammetric measurements were performed using a 273A (Princeton Applied Research) with a one-compartment electrolysis cell consisting of a platinum working electrode, a platinum wire counter-electrode, and a quasi Ag+/Ag electrode as reference. Measurements were performed in a 0.5 mM CH2Cl2 solution with tetrabutylammonium tetrafluoroborate as the supporting electrolyte, at a scan rate of 100 mV s−1. Each oxidation potential was calibrated using ferrocene as a reference. LUMO levels were calculated from the HOMO level and the energy band-gap, which was obtained from the edge of the absorption spectra. Atomic force microscopy (AFM, PSIA, XE-150) was used to characterize the vacuum-deposited films surface morphology. AFM images were obtained in non-contact mode (scan rate and scan size were 0.5 Hz and 5 × 5 μm2, respectively).Device fabrication and evaluation
Current density–voltage–luminance (J–V–L) characteristics, EL spectra, and CIE chromaticity coordinates of the blue OLEDs were measured simultaneously using a Keithley 237 programmable source meter and a SpectraScan PR650 (Photo Research). Assuming Lambertian emission, the external quantum efficiency (EQE) was calculated from the luminance, current density, and electroluminescence spectrum.
Acknowledgements
This research was supported by Basic Science Research Program (CRI; RIAMIAM0209(0417-20090011)) and WCU (World Class University) project (R31-2008-00010075-0) through National Research Foundation of Korea funded by the Ministry of Education, Science and Technology.Notes and references
- (a) C. W. Tang and S. A. VanSlyke, Appl. Phys. Lett., 1987, 51, 913 CrossRef CAS; (b) M. A. Baldo, M. E. Thompson and S. R. Forrest, Nature, 2000, 403, 750 CrossRef CAS; (c) P. L. Burn, S. C. Lo and D. W. Samuel, Adv. Mater., 2007, 19, 1675 CrossRef CAS; (d) C. D. Műller, A. Falcou, N. Reckefuss, M. Rojahn, V. Wiederhirn, P. Rudati, H. Frohne, O. Nuyken, H. Becker and K. Meerholz, Nature, 2003, 421, 829 CrossRef; (e) Y. Sun, N. C. Giebink, H. Kanno, B. Ma, M. E. Thompson and S. R. Forrest, Nature, 2006, 440, 908 CrossRef CAS.
- (a) L. M. Leung, W. Y. Lo, S. K. So, K. M. Lee and W. K. Choi, J. Am. Chem. Soc., 2000, 122, 5640 CrossRef CAS; (b) Y. Y. Lyu, J. Kwak, O. Kwon, S. H. Lee, D. Kim, C. Lee and K. Char, Adv. Mater., 2008, 20, 2720 CrossRef CAS.
- (a) J. Luo, Y. Zhou, Z. Q. Niu, Y. Ma and J. Pei, J. Am. Chem. Soc., 2007, 129, 11314 CrossRef CAS; (b) C. J. Tonzola, A. P. Kulkarni, A. P. Gifford, W. Kaminsky and S. A. Jenekhe, Adv. Funct. Mater., 2007, 17, 863 CrossRef CAS; (c) S. Tao, Y. Zhou, C. S. Lee, X. Zhang and S. T. Lee, Chem. Mater., 2010, 22, 2138 CrossRef CAS; (d) C. C. Chi, C. L. Chiang, S. W. Liu, H. Yueh, C. T. Chen and C. T. Chen, J. Mater. Chem., 2009, 19, 5561 RSC; (e) S. J. Lee, J. S. Park, K. J. Yoon, Y. I. Kim, S. H. Jin, K. Kang, Y. S. Gal, S. Kang, J. Y. Lee, J. W. Kang, S. H. Lee, H. D. Park and J. J. Kim, Adv. Funct. Mater., 2009, 19, 2661 CrossRef; (f) H. T. Shih, C. H. Lin, H. H. Shih and C. H. Cheng, Adv. Mater., 2002, 14, 1409 CrossRef CAS.
- L. Duan, L. Hou, T. W. Lee, J. Qiao, D. Zhang, G. Dong, L. Wang and Y. J. Qiu, J. Mater. Chem., 2010, 20, 6392 RSC.
- J. Shi and C. W. Tang, Appl. Phys. Lett., 2002, 80, 3201 CrossRef CAS.
- (a) J. N. Moorthy, P. Venkatakrishnan, P. Natarajan, D. F. Huang and T. J. Chow, J. Am. Chem. Soc., 2008, 130, 17320 CrossRef CAS; (b) S. W. Culligan, A. C. A. Chen, J. U. Wallace, K. P. Klubek, C. W. Tang and S. H. Chen, Adv. Funct. Mater., 2006, 16, 1481 CrossRef CAS; (c) Y. Li, M. K. Fung, Z. Xie, S. T. Lee, L. S. Hung and J. Shi, Adv. Mater., 2002, 14, 1317 CrossRef CAS; (d) P. I. Shih, C. Y. Chuang, C. H. Chien, E. W. G. Diau and C. F. Shu, Adv. Funct. Mater., 2007, 17, 3141 CrossRef CAS; (e) Y. H. Kim, H. C. Jeong, K. Yang and S. K. Kwon, Adv. Funct. Mater., 2005, 15, 1799 CrossRef CAS; (f) S. K. Kim, B. Yang, Y. Ma, J. H. Lee and J. W. Park, J. Mater. Chem., 2008, 18, 3376 RSC.
- C. H. Wu, C. H. Chien, F. M. Hsu, P. I. Shih and C. F. Shu, J. Mater. Chem., 2009, 19, 1464 RSC.
- S. H. Kim, I. Cho, M. K. Sim, S. Park and S. Y. Park, J. Mater. Chem., 2011, 21, 9139 RSC.
- C. J. Zheng, W. J. Zhao, Z. Q. Wang, D. Huang, J. Ye, X. M. Ou, X. H. Zhang, C. S. Lee and S. T. Lee, J. Mater. Chem., 2010, 20, 1560 RSC.
- C. H. Chien, C. K. Chen, F. M. Hsu, C. F. Shu, P. T. Chou and C. H. Lai, Adv. Funct. Mater., 2009, 19, 560 CrossRef CAS.
- S. K. Kim, B. Yang, Y. Ma, J. H. Lee and J. W. Park, J. Mater. Chem., 2008, 18, 3376 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, J. A. Montgomery, Jr, T. Vreven, K. N. Kudin, J. C. Burant, J. M. Millam, S. S. Iyengar, J. Tomasi, V. Barone, B. Mennucci, M. Cossi, G. Scalmani, N. Rega, G. A. Petersson, H. Nakatsuji, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, M. Klene, X. Li, J. E. Knox, H. P. Hratchian, J. B. Cross, V. Bakken, C. Adamo, J. Jaramillo, R. Gomperts, R. E. Stratmann, O. Yazyev, A. J. Austin, R. Cammi, C. Pomelli, J. W. Ochterski, P. Y. Ayala, K. Morokuma, G. A. Voth, P. Salvador, J. J. Dannenberg, V. G. Zakrzewski, S. Dapprich, A. D. Daniels, M. C. Strain, O. Farkas, D. K. Malick, A. D. Rabuck, K. Raghavachari, J. B. Foresman, J. V. Ortiz, Q. Cui, A. G. Baboul, S. Clifford, J. Cioslowski, B. B. Stefanov, G. Liu, A. Liashenko, P. Piskorz, I. Komaromi, R. L. Martin, D. J. Fox, T. Keith, M. A. Al-Laham, C. Y. Peng, A. Nanayakkara, M. Challacombe, P. M. W. Gill, B. Johnson, W. Chen, M. W. Wong, C. Gonzalez and J. A. Pople, Gaussian 03, Revision D.02, Gaussian, Inc, Wallingford CT, 2004 Search PubMed.
- (a) B. Wei, J. Z. Liu, Y. Zhang, J. H. Zhang, H. N. Peng, H. L. Fan, Y. B. He and X. C. Gao, Adv. Funct. Mater., 2010, 20, 2448 CrossRef CAS; (b) S. Tang, M. Liu, P. Lu, H. Xia, M. Li, Z. Xie, F. Shen, C. Gu, H. Wang, B. Yang and Y. Ma, Adv. Funct. Mater., 2007, 17, 2869 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: DSC and TGA thermograms of PCAN. See DOI: 10.1039/c1jm14482k |
| ‡ Both of these authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2012 |