Metal-free photocatalytic degradation of 4-chlorophenol in water by mesoporous carbon nitride semiconductors†
Received
19th January 2012
, Accepted 28th March 2012
First published on 29th March 2012
Abstract
Mesoporous graphitic carbon nitrides were synthesized by a thermal-induced polymerization of NH4SCN on the surface of silica nanoparticles as the templates. The catalysts thus obtained were used as visible light photocatalysts for the degradation of organic pollutants in water. The physicochemical properties of catalysts were characterized by several techniques. The photocatalytic activity of samples was evaluated by the decomposition of chlorophenol and phenol in aqueous phase. Results demonstrated that mesoporous graphitic carbon nitrides can photocatalytically oxidize and eventually mineralize organic pollutants in aqueous solution under visible light irradiation. The photocatalysts can induce the formation of reactive oxy-species such as H2O2, ˙OH and ˙O2−/˙OOH under visible light irradiation, while keeping a high photocatalytic reactivity during recycling operations. The higher activity of mesoporous carbon nitrides compared to nonporous carbon nitride can be attributed to their enlarged surface area and enhanced light-harvesting effect, facilitating the photogeneration of active oxy-radicals in water.
Introduction
Hydroxylated aromatic compounds (HACs) like phenol and chlorophenol are widely used as pesticides, herbicides and wood preservatives, and thus they have become ubiquitous xenobiotic micropollutants in aquatic environments.1–3 Since HACs are considered to be recalcitrant organic pollutants, considerable research effort has been devoted to the destruction of HACs in aqueous streams. Although many wastewater treatment methods based on physicochemical and biological technologies, such as adsorption by activated carbon4,5 and microbial degradation,6 have been applied for the purification of wastewater containing HACs, these systems suffer from incomplete removal of the organics and the formation of byproducts that are even more toxic than the starting materials.
Compared with the conventional wastewater treatment methods, advanced oxidation processes (AOPs)7 hold immense promise because of their low cost, high efficiency, and deep oxidation power that allows for the mineralization of organic molecules under mild conditions. Among various AOPs, photochemical generation of reactive oxy-species for environmental remediation is of particular interest because the abundant solar radiation can in principle be used to run the reaction in a sustainable manner. Ultraviolet (UV) light has been reported to activate inorganic8 or organic oxidants9 to generate highly-reactive hydroxyl radicals, giving rise to efficient degradation and mineralization of 4-chlorophenol (4-CP) in water. Titanium dioxide (TiO2) has also been broadly applied to couple with UV light to catalyze the formation of hydroxyl radicals from water and oxygen as clean oxidants, being considered a green technique for environmental purification.10 It is well-known that TiO2 photocatalysts have some significant advantages over other semiconductors like ZnO and CdS, since they are nontoxic, inexpensive, stable against photocorrosion, and able to mineralize most organic pollutants under ambient conditions. However, the UV light required for the excitation of TiO2 occupies only ∼4% of the solar spectrum. Therefore, the exploration of an efficient system on the basis of modified TiO2 working with visible light (occupies ∼43% of sunlight) is actively pursued nowadays,11 but it remains a challenge because there are concerns regarding the fast charge recombination as a result of metal/nonmetal doping and also regarding the stability of the modified systems during prolonged operation.12,13 On the other hand, non-TiO2 based materials, such as metal (oxy)nitrides and (oxy)sulfides,14,15 have also been investigated as visible-light responsive photocatalysts, but relying greatly on metal components that are too rare and/or expensive to be broadly used.
In recent years, a metal-free graphitic carbon nitride (g-C3N4) has become a new generation of polymeric semiconductor that is relevant in energy, (photo)catalysis and environmental fields.16–19 g-C3N4 is constructed from N-bridged poly(tri-s-triazine) planes that stack together in a similar way to graphene sheets in graphite. The highly condensed tri-s-triazine ring structure makes the polymer possess high stability with respect to thermal (up to 600 °C in air) and chemical attacks (e.g. acid, base, and organic solvents) and an appealing electronic structure, being a medium-gap semiconductor excitable with visible light (λ < 470 nm). With the proper HOMO and LUMO levels, this metal-free catalyst has been shown previously to be able to photocatalyze water splitting reaction17,20 and selective oxidation of benzene to phenol in a phase-transfer system to avoid overoxidation of phenol.21 The application of polymeric carbon nitride based photocatalysts in environmental cleanup has also been demonstrated for the degradation of dyes22 and colorless light-weight molecules as well.23 However, the given examples using bulk carbon nitrides showed a moderate photocatalytic activity only. Complete mineralization of organics by carbon nitride polymers has not been achieved and the process of degradation was not investigated in detail.
Much effort has consequently been devoted to improving the photocatalytic activity of carbon nitride by modification of its surface and textural structures, for example, by creation of nanosized pores in the carbon nitride matrix to increase surface active sites and to enhance light-harvesting.24 Especially, porous carbon nitride still features aromatic π-electron conjugated structure with surface basic motifs related to uncondensed amino groups, which is believed to promote the adsorption of HACs via Lewis acid–base interaction and π–π interaction, making it a recommended choice for the photocatalytic degradation of HACs.
In this study, mesoporous g-C3N4 (mpg-CN) was synthesized by a hard-templating method using 12 nm silica particles as the templates. The polymeric catalysts were applied as metal-free photocatalysts for the treatment of HACs under visible light illumination. 4-CP and phenol were chosen as model pollutants in aqueous phase. The reactive oxy-species generated during the photocatalytic reactions, together with the potential degradation mechanism, were described in detail.
Experimental section
Preparation
Synthesis of a carbon nitride photocatalyst.
mpg-CN was prepared according to our previous report.25 Briefly, ammonium thiocyanate (NH4SCN) (Sinopharm Chemical Reagent Co. Ltd) and silica nanoparticles (12 nm, Ludox HS40, Aldrich) were mixed and heated to 600 °C under a N2 atmosphere for 2 h, followed by the removal of the template with NH4HF2. The obtained sample was named as mpg-CNr, where r refers to the initial mass ratio of silica to NH4SCN. Bulk g-C3N4 was prepared by heating NH4SCN under the same conditions.
Synthesis of N-doped TiO2 (N/TiO2).
An amorphous TiO2 xeogel was prepared by a sol–gel method as described previously.11c The as-prepared TiO2 xeogel was then heated under flowing NH3 gas (flow rate: 400 mL min−1) at 400 °C for 3 h, followed by naturally cooling to room temperature under NH3 gas.
Fabrication of the working electrode.
The working electrode was coated on fluorine doped tin oxide (FTO) glass,17c which was cleaned by sonication in chloroform, acetone and ethanol for 30 min, respectively. The FTO glass was then rinsed with Millipore water and kept in isopropanol for 24 h. 50 mg of catalyst was mixed with 2 mL of dimethylformamide under sonication for 30 min to obtain a slurry. The slurry was spread onto FTO glass whose side part was previously protected using Scotch tape. After air drying, the electrode was fired at 350 °C for 30 min in air to improve the adhesion. A copper wire was connected to the side part of the FTO glass using conductive tape. Uncoated parts of the electrode were isolated with epoxy resin, and the exposed area of the electrode was 0.25 cm2.
Photocatalytic tests
Photocatalytic activities of samples were evaluated by degradation of 4-CP and phenol (1.2 × 10−4 M) in aqueous solution with irradiation from a 300 W Xe lamp. The wavelength of the incident light was controlled by applying an appropriate cut-off filter (λ > 420 nm). In a Pyrex glass reactor, the pollutant solution (80 mL) and 40 mg carbon nitride catalyst were mixed. After the adsorption–desorption equilibrium was established within 0.5 h, the lamp was turned on to initiate photocatalytic reaction. A 3 mL suspension was sampled at fixed time intervals during the reaction. The suspension was centrifuged to remove the catalyst and then the concentration variation of 4-CP/phenol was examined by HPLC.
Characterization
X-ray diffraction (XRD) patterns were collected using a Bruker D8 Advance X-ray diffractometer (Cu Kα1 irradiation, λ = 1.5406 Å). A Varian Cary 500 Scan UV-Vis spectrophotometer was used to record the UV-Vis diffuse reflectance spectra (DRS) of samples using BaSO4 as the reference. N2-sorption was collected at 77 K using Micromeritics ASAP 2010 equipment. Transmission electron microscopy (TEM) was conducted using a JEOL model JEM 2010 EX instrument. Electron spin resonance (ESR) spectra [signals of radicals spin trapped by 5,5′-dimethylpyrroline-N-oxide (DMPO)] were obtained using a Bruker model ESP 300 E electron paramagnetic resonance spectrometer equipped with a xenon lamp (with a 420 nm filter) as the light source. Electrochemical measurements were conducted with a BAS Epsilon Electrochemical System in a conventional three electrode cell, using a Pt plate as the counter electrode and Ag/AgCl electrode (3 M KCl) as the reference electrode. X-ray photoelectron spectroscopy (XPS) was performed on a Thermo ESCALAB250 instrument with a monochromatized Al Kα line source (200 W). The concentration variations of pollutants (4-CP/phenol) were measured by a HPLC (Waters 2487) system. An Agilent HC-C18(2) reversed phase column was employed. All substances were detected by a UV detector at 280 nm. The mobile phase consisted of a ternary mixture of water, methanol, and acetonitrile (40![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50:10 by volume) and the flow rate was 0.8 mL min−1. A liquid chromatography/mass spectrometry (LCMS) system was equipped with a Zorbax C18 column (150 mm × 4.6 mm, id = 5 μm) and coupled on-line to an LC/MSD Trap XCT ion-trap mass spectrometer (Agilent Technologies, CA, USA). The examination of chloride ions was performed using an ion chromatograph (881 Compact IC, pro). The buffer solution was 3.2 mM Na2CO3/1.0 mM NaHCO3. The total organic carbon (TOC) values were determined using a Shimadzu TOC-Vcph total organic carbon analyzer.
:50:10 by volume) and the flow rate was 0.8 mL min−1. A liquid chromatography/mass spectrometry (LCMS) system was equipped with a Zorbax C18 column (150 mm × 4.6 mm, id = 5 μm) and coupled on-line to an LC/MSD Trap XCT ion-trap mass spectrometer (Agilent Technologies, CA, USA). The examination of chloride ions was performed using an ion chromatograph (881 Compact IC, pro). The buffer solution was 3.2 mM Na2CO3/1.0 mM NaHCO3. The total organic carbon (TOC) values were determined using a Shimadzu TOC-Vcph total organic carbon analyzer.
Results and discussion
Physicochemical properties of mpg-CN
Fig. 1A shows XRD patterns of the mpg-CN samples, showing a strong peak at 27.4° related to a tight inter-layer stacking distance (0.325 nm) of the aromatic planes in the graphite-like carbon nitride structure. The other pronounced peak at 13.1° corresponds to an in-plane structural repeating motif with a repeated distance of 0.675 nm. With an increasing amount of the SiO2 template, the intensity of the peak at 27.4° keeps consistent, while the peak at 13.1° decreases gradually. This is an indication of the weakening of the conjugated interaction of a continuous heptazine network in graphitic carbon nitride. These defects may promote electron localization and then prevent charge recombination.
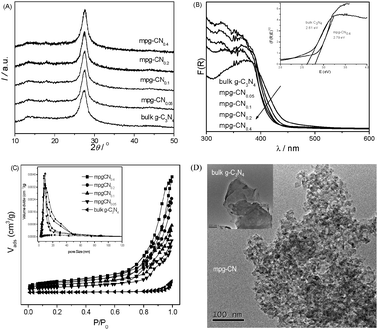 |
| | Fig. 1 Characterization of bulk g-C3N4 and mpg-CN samples. (A) XRD patterns, (B) optical absorption, (C) N2-sorption and pore size distribution, (D) TEM images. | |
Fig. 1B presents the optical absorption spectra of mpg-CN, showing intense absorption bands with steep edges at ca. 470 nm, due to the visible-light induced transition of electrons from the valence band to the conduction band of the carbon nitride semiconductor. The band edges of the materials appear slightly blue shifted compared with that of the bulk sample, due to the quantum confinement effect.24 Thus, the redox ability of the catalyst improves.
The surface area of mesoporous samples was ca. 180 m2 g−1, which is much higher than that of bulk material. The isotherms and Barrett–Joyner–Halenda pore size distributions are shown in Fig. 1C. The isotherms of mpg-CN show typical hysteresis, revealing the existence of mesopores. The surface area and pore volume of mpg-CN samples increase with the increasing amount of initial silica added, as summarized in Table 1. However, when the amount of silica further increases, we cannot obtain polymeric carbon nitride with an intact mesoporous structure. This is due to the deformation of thin pore walls of soft materials during the removal of excess silica template. This enlarged surface area of mpg-CN not only facilitates the mass transfer of reaction species and the light harvesting by the multiple scattering effect of nanopores, but also provides more catalytic active sites for photoredox reaction. Thus, a better photocatalytic activity can be envisaged for mpg-CN over bulk g-C3N4.
Table 1 Physicochemical properties and photocatalytic activity of bulk g-C3N4 and mpg-CN samples for 4-CP and phenol degradation with visible light
| Sample |
SAa/m2 g−1 |
PVb/cm3 g−1 |
K (10−2 min−1) |
[TOC]/[TOC0] (%) |
[Cl−]/mg L−1 |
| 4-CP |
Phenol |
4-CP |
Phenol |
4-CP |
|
Specific surface area.
Pore volume.
|
| bulk g-C3N4 |
11 |
/ |
0.15 |
0.23 |
/ |
/ |
/ |
| mpg-CN0.05 |
88 |
0.35 |
1.59 |
1.23 |
35 |
7 |
4.09 |
| mpg-CN0.1 |
125 |
0.45 |
2.42 |
1.65 |
38 |
18 |
4.15 |
| mpg-CN0.2 |
154 |
0.63 |
3.1 |
2.40 |
43 |
31 |
4.14 |
| mpg-CN0.4 |
176 |
0.77 |
5.26 |
3.42 |
56 |
40 |
4.16 |
Fig. 1D shows typical TEM images of mpg-CN0.4. The inner structure of the existing disordered but well-developed spherical pores with pore diameter close to 12 nm (consistent with the size of the silica template) can be clearly observed. By contrast, the picture of bulk g-C3N4 (inset in Fig. 1D) shows the typical flat layer structure of carbon nitride, without forming any textural structures.
Mesostructured design of a semiconductor is known to endow the materials with small particle size and large specific surface area, which was already demonstrated to facilitate charge collection and separation at the heterogeneous interface. This is indeed confirmed by the enhanced photocurrent intensity and EPR signals of mpg-CN samples over bulk g-C3N4 under visible light illumination (Fig. 2).
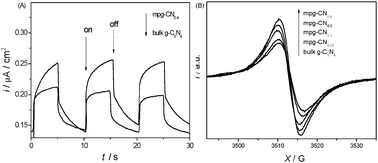 |
| | Fig. 2 Transient photocurrent taken at 0.4 V vs. Ag/AgCl (A) and EPR spectra (B) of mpg-CN and bulk g-C3N4. | |
Photocatalytic degradation of 4-CP and phenol by mpg-CN
The photocatalytic degradation curves of 4-CP and phenol as a function of reaction time are shown in Fig. 3. The results illustrate that mesoporous structure greatly influences the photocatalytic performance of g-C3N4 materials. Bulk g-C3N4 shows a moderate photoreactivity toward the degradation of the pollutants. Only ∼20% of 4-CP and phenol was degraded in 120 min over bulk g-C3N4 under visible light irradiation, which is however much smaller than those of mesoporous samples. mpg-CN0.4 exhibits the highest photocatalytic activity, degrading nearly 100% and 96% of 4-CP and phenol in 60 and 90 min, respectively. The first order reaction kinetic model is adopted for fitting the reaction process and the results demonstrate the linear relationship well. The curves of ln(C0/C) as a function of irradiation time are shown in Fig. 4 and the calculated degradation rate constants are provided in Table 1. As a comparison, the photocatalytic degradation of 4-CP by mesoporous N/TiO2 was also tested, and results showed that only 12% of 4-CP was degraded within 60 min which is much lower than that of mpg-CN.
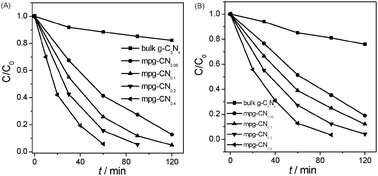 |
| | Fig. 3 Visible-light photocatalytic degradation of 4-CP (A) and phenol (B) over bulk g-C3N4 and mpg-CN samples. | |
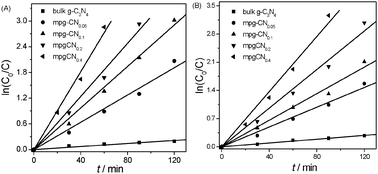 |
| | Fig. 4 Kinetics of 4-CP (A) and phenol (B) decomposition in water over bulk g-C3N4 and mpg-CN samples under visible light irradiation. | |
The formation of Cl− ions in solution as a result of dechlorination of 4-CP by photoexcited mpg-CN is also illustrated in Table 1. After 3 h irradiation, the concentration of Cl− in the solution for all mesoporous samples rises to ∼4.1 mg L−1, close to the theoretical quantity (4.2 mg L−1). This indicates that there is virtually no residual organic chlorine existing in aqueous solution after 3 h reaction. To further testify the mineralizing capability of mesoporous photocatalysts, the removal of total organic carbon (TOC) in the solution was measured. Table 1 also lists the photocatalytic removal of TOC of 4-CP and phenol by mpg-CN after visible light irradiation for 3 h. The mineralization ratio of organic pollutants (defined as [TOC]/[TOC0]) by bulk g-C3N4 is negligible in 3 h reaction, due to the low activity of the bulk sample for the degradation of 4-CP and phenol. Nevertheless, in the case of the mpg-CN0.4 sample, the mineralization ratio of 4-CP and phenol is 56% and 40%, respectively, and it increases with the increase in the specific surface area of mpg-CN.
The main intermediates produced prior to the ring-opening stage in the photodegradation of 4-CP by mpg-CN0.4 were identified by LC-MS techniques. Fig. 5 reports the chromatograms of 4-CP solutions subjected to photocatalytic treatment at different periods of irradiation time, for example, 0, 10 and 40 min. In the dark, there is only one strong peak in the chromatogram corresponding to 4-CP with the mass peak at m/z = 127. When the light was turned on, the intensity of the peak slowly reduced, while other main byproducts in the degradation process appeared. After 10 min of irradiation, one byproduct with the mass peak at m/z = 143 appeared, corresponding to species 1 (4-chloricatechol) or 2 (peroxychlorophenol). The other product with the mass peak at m/z = 157 (species 3 and 4) formed after 40 min of irradiation. With the irradiation time prolonging, the intensity of the two peaks first increased and then slowly reduced. Clearly, we can deduce that the intermediates generated from 4-CP on mpg-CN0.4 are hydroxylate and hyperoxide species. The dechlorination process of 4-CP following the irradiation has also been monitored (see Fig. 6). At the very onset of light irradiation, a large amount of Cl− ions were released, and its concentration in the solution increased as irradiation time prolonged. That is to say, dechlorination and radical reaction occurred simultaneously. This indicates that reactive oxy-species like ˙OH and O2˙− radicals are probably generated in the suspension of mpg-CN0.4, attacking the aromatic ring of 4-CP competitively. But the exact molecular structure of those intermediate species couldn't be confirmed ambiguously, as the attack positions are unclear. Further investigation is needed.
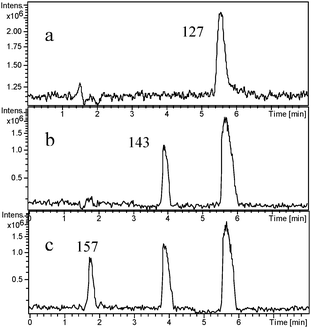 |
| | Fig. 5 LC-MS chromatograms of 4-CP degraded by mpg-CN0.4 under visible light irradiation at different irradiation intervals: (a) original 4-CP solution after adsorption–desorption equilibrium in the dark; (b) after 10 min of irradiation; (c) after 40 min of irradiation. | |
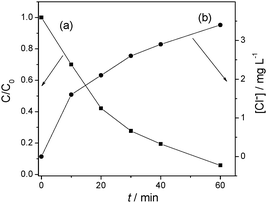 |
| | Fig. 6 Degradation (a) and dechlorination (b) of 4-CP on mpg-CN0.4 as a function of irradiation time. | |
The current photocatalytic system was extended to treat other related aromatic pollutants, such as dichlorophenol 2,6-DCP and 2,4-DCP, that are also widely applied in the chemical industry. As can be seen from Fig. S1 (ESI†), after 60 min of visible light irradiation, only one main byproduct with m/z = 191 was detected in the 2,6-DCP solution, attributable to ˙O2−/˙OOH-substituted chlorophenol. Different from this phenomenon, besides the above mentioned intermediate species, two new peaks at m/z = 177 and 157 were observed from the chromatograms of the 2,4-DCP solution. These additional peaks can be attributed to hydroxylate of the original substrate and completely-dechlorinated hyperoxide.
The summary of photocatalytic performance in terms of the degradation rate, mineralization ratio and generated Cl− is shown in Table S1 (ESI†). After irradiation with visible light for 60 min, 61% of 2,4 DCP and 72% of 2,6-DCP were decomposed, but the decomposed pollutants were not completely oxidized to CO2. With a prolonged illumination time of 180 min, the mineralization ratio reached 40% and more than 90% of Cl− was released into the aqueous solutions. The slightly reduced degradation rate of 2,4-DCP compared to that of 2,6-DCP may be attributed to its relatively-complicated transformation process of intermediates. In brief, using mpg-CN as a photocatalyst, chlorophenol compounds could be effectively degraded and eventually mineralized under ambient conditions.
According to the above results and knowledge that the OH group in the phenolic ring is o, p directing with activation for electrophilic reagents, the early reaction steps in the transformation of 4-CP and 2,4-DCP by mpg-CN0.4 under visible light irradiation are proposed in Scheme 1 and Scheme S1 (ESI†), respectively. From these results, we can draw the following conclusions, namely (a) ˙O2−/˙OOH and ˙OH were generated by photoillumination of mpg-CN and can react with chlorophenol and (b) ˙OH more frequently attacks the ortho position of the phenolic ring.
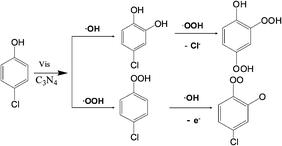 |
| | Scheme 1 Proposed early steps in the transformation pathways of 4-CP under visible light irradiation. | |
Active radicals detection during photodegradation of 4-CP
To confirm the existence of O2˙− and ˙OH radicals, an ESR spin-trap technique (with DMPO) was employed to probe the nature of the reactive oxygen species generated during the visible light irradiation in the reaction system. The ESR signals are shown in Fig. 7. Under our experimental conditions, no ESR signals of O2˙− and ˙OH radicals were observed on bulk g-C3N4 in the dark or even under visible light irradiation. On the other hand, the characteristic sextet peaks of the DMPO–O2˙− adduct and the characteristic quartet peaks of the DMPO–˙OH adduct were clearly observed in the presence of mpg-CN0.4 after being illuminated for 5 min with visible light. Although weak O2˙− signals in the mpg-CN0.4 system can be observed in the dark (note that the operation condition was not in the absolute dark, but under a neutral environment), their intensity enhanced dramatically after light illumination from the Xe lamp. Both O2˙− and ˙OH radicals have been well documented to be responsive for the oxygenation and hydroxylation of aromatics in water, and ultimately leading to the mineralization of organic pollutants.
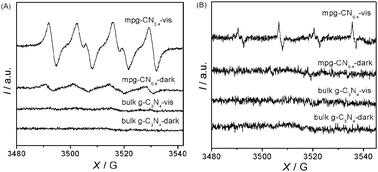 |
| | Fig. 7 ESR spectra of DMPO–O2˙−/˙OOH (A) and DMPO–˙OH (B) adducts in the systems of bulk g-C3N4/DMPO and mpg-CN0.4/DMPO before and after visible light irradiation. | |
To further confirm the formation of ˙OH radicals, a terephthalic acid photoluminescence probing technique (PL-TA) was used for the mpg-CN0.4 and bulk g-C3N4 systems.21,27 As shown in Fig. S2 (ESI†), the fluorescence spectra related to ˙OH radicals were clearly observed for the light-illuminated mpg-CN0.4 system, with their intensity steadily increasing with the irradiation time. In the bulk g-C3N4 system, ˙OH radicals were also detected, but with a very low intensity, which explains its low but not negligible activity for chlorophenol degradation. It can be concluded that polymeric carbon nitride can indeed induce the formation of hydroxyl radicals in aqueous solution under visible light irradiation, consistent with other reports.28
In the next set of experiments, we carried out electrochemical analysis (Mott–Schottky plots) of bulk g-C3N4 and mpg-CN0.4 to determine their electronic band structure.17c,20c,29 Fig. S3 (ESI†) shows typical Mott–Schottky plots in the dark for the samples, suggesting an n-type feature of the organic semiconductors due to the positive slope of the linear plot.29 The flat-band potential derived from these measurements is roughly −1.29 and −1.13 V vs. NHE at pH 7 for bulk g-C3N4 and mpg-CN0.4, respectively. By combining with the band gap estimated from optical absorption, the valence band positions of bulk g-C3N4 and mpg-CN0.4 can be obtained. In spite of the slightly more positive conduction band position, O2˙− can be easily and considerably generated from the interaction of dissolved oxygen and conduction electron in mpg-CN0.4 due to its high surface area. However, both bulk g-C3N4 and mpg-CN0.4 with low valence band position at VVB = 1.32 and 1.66 V vs. NHE at pH 7, respectively, are not appropriate for the direct generation of ˙OH radicals [E0(˙OH/H2O) = 2.27 eV; E0(˙OH/OH−) = 1.99 eV vs. NHE]. Thus, the potential pathway for ˙OH generation in the carbon nitride photocatalytic system is the interaction of light-induced electron, O2˙− and H2O2.
Indeed, the spectrophotometric analysis with N,N-diethyl-p-phenylenediamine (DPD) in the presence of peroxidase (POD)30 confirmed the formation of H2O2 during the degradation of 4-CP by mpg-CN0.4 with visible light. Fig. 8 shows the detection results of H2O2 in water dispersion of mpg-CN0.4 and bulk g-C3N4. Under dark conditions in the presence of the catalyst and light conditions without the catalyst, there were no obvious peaks observed. However, in the light-excited mpg-CN0.4 system, clear peaks with two absorption maxima assigned to the oxidized DPD can be observed (one at 510 nm and the other one at 551 nm). The peak intensity reflecting the quantities of generated H2O2 after irradiation for 30 min is much higher than only exposed to light for 10 min. When using bulk g-C3N4 as a photocatalyst, the same peaks can be seen, but the quantities of H2O2 presented are much lower in this system, obviously. That is to say, in the mpg-CN0.4 and bulk g-C3N4-visible light systems, H2O2 as an intermediate can be generated. Therefore, it not only verified the existence of O2˙− species again but also was an inevitable source of ˙OH species, following the photochemical processes as listed below.
| | | g-C3N4 + hν → (hVB+ + eCB−) | (1) |
| | | eCB− + HO2˙ + H+ → H2O2 | (4) |
| | | H2O2 + eCB− → ˙OH + OH− | (5) |
Photocatalytic stability evaluation
To check the photostability of mpg-CN0.4, the photocatalytic degradation of 4-CP was repeated up to five cycles under the same conditions. The mpg-CN0.4 catalyst after five successive operations still maintained high photocatalytic activity to degrade 4-CP (see Fig. 9). The slightly decreased degradation rate was due to the inevitable loss of some catalyst during the operation for recovery.
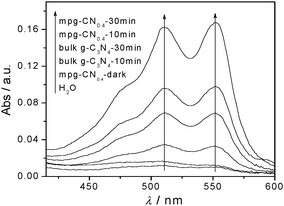 |
| | Fig. 8 Detection of H2O2 in water dispersions of bulk g-C3N4 and mpg-CN0.4 (0.04 g per 80 mL) under visible light irradiation. Curves were obtained by addition of DPD and POD (50 μL) to the dispersions (20 mL) after irradiation with visible light. | |
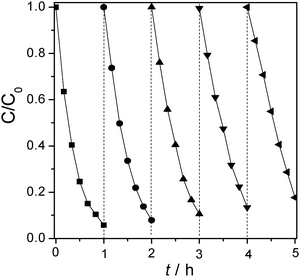 |
| | Fig. 9 Stability test of mpg-CN0.4 for 4-CP degradation under visible light irradiation. | |
The samples before and after degradation of 4-CP were also investigated by XRD and XPS. The XRD patterns (Fig. 10) indicate that there is no remarkable alteration in the crystal structure of the catalyst before and after photoreaction, which was also proved by the XPS spectra (Fig. 11). The BE of C1s and N1s before and after degradation were nearly the same. There is no photocorrosion or photodissolution of catalysts in the photocatalytic reaction, showing great promise of conjugated carbon nitride photocatalysts for metal-free environmental purification of aromatic compounds.
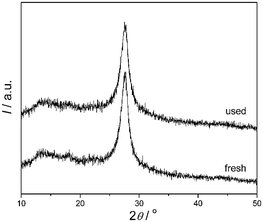 |
| | Fig. 10 XRD patterns of mpg-CN0.4 before and after used for the photocatalytic reaction in 4-CP degradation under visible light. | |
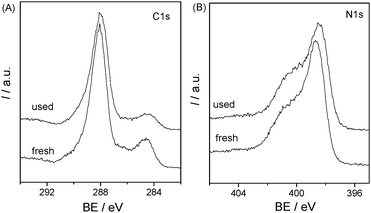 |
| | Fig. 11 XPS spectra of C1s and N1s for mpg-CN0.4 before and after used for the photocatalytic reaction in 4-CP degradation under visible light. | |
Conclusions
In this paper, we have shown that mesoporous graphitic carbon nitride synthesized by a simple thermal-induced polymerization of NH4SCN could effectively degrade 4-chlorophenol and phenol with visible light irradiation under ambient conditions. The physicochemical structure and optical properties of the mesoporous samples were analyzed by a variety of techniques and turned out to be defectuous g-C3N4 with well-templated mesoporous structure and high specific surface area. During the reaction process, the oxy-species of H2O2 and ˙OH play a dominant role in the degradation of pollutants. We expect that the carbon nitride semiconductor and its modifications will be a new kind of organocatalyst for degradation of recalcitrant organic pollutants.
Acknowledgements
This work is financially supported by the National Natural Science Foundation of China (21033003, 21173043, 21173046, 21103095 and U1033603).
Notes and references
- S. K. Khetan and T. J. Collins, Chem. Rev., 2007, 107, 2319–2364 CrossRef CAS.
- Q. Huang and W. J. Weber, Environ. Sci. Technol., 2005, 39, 6029–6036 CrossRef CAS.
- J. W. Park, J. Dec, J. E. Kim and J. M. Bollag, Environ. Sci. Technol., 1999, 33, 2028–2034 CrossRef CAS.
- V. C. Srivastava, M. M. Swamy, I. D. Mall, B. Prasad and I. M. Mishra, Colloids Surf., A, 2006, 272, 89–104 CrossRef CAS.
- L. Khenniche and F. Benissad-Aissani, J. Chem. Eng. Data, 2010, 55, 4677–4686 CrossRef CAS.
- H. L. Jiang, J. H. Tay, A. M. Maszenan and S. T. Tay, Environ. Sci. Technol., 2006, 40, 6127–6142 Search PubMed.
- O. Legrini, E. Oliveiros and A. M. Braun, Chem. Rev., 1993, 93, 671–698 CrossRef CAS.
- C. E. Catalkaya, U. Bali and F. Sengul, Environ. Sci. Pollut. Res., 2003, 10, 113–120 CrossRef.
- S. Sharma, M. Mukhopadhyay and Z. V. P. Murthy, Ind. Eng. Chem. Res., 2010, 49, 3094–3098 CrossRef CAS.
-
(a) C. Li, F. Wang and J. C. Yu, Energy Environ. Sci., 2010, 4, 100–113 RSC;
(b) X. Chen, X. Wang and X. Fu, Energy Environ. Sci., 2009, 2, 872–877 RSC;
(c) T. Ochiai, H. Nanba, T. Nakagawa, K. Masuko, K. Nakata, T. Murakami, R. Nakano, M. Hara, Y. Koide, T. Suzuki, M. Ikekita, Y. Morito and A. Fujishima, Catal. Sci. Technol., 2011, 2, 76–78 RSC;
(d) T. Kinugawa, S. Enami, A. Yabushita, M. Kawasaki, M. R. Hoffmann and A. J. Colussi, Phys. Chem. Chem. Phys., 2011, 13, 5144–5149 RSC;
(e) H. Einaga, S. Futamura and T. Ibusuki, Phys. Chem. Chem. Phys., 1999, 1, 4903–4908 RSC;
(f) Z. Ding, J. Huang and X. Wang, Phys. Chem. Chem. Phys., 2010, 12, 5983–5985 RSC.
-
(a) T. Lv, L. Pan, X. Liu, T. Lu, G. Zhu, Z. Sun and C. Q. Sun, Catal. Sci. Technol., 2012, 2, 754–758 RSC;
(b) Y. Irokawa, T. Morikawa, K. Aoki, S. Kosaka, T. Ohwaki and Y. Taga, Phys. Chem. Chem. Phys., 2006, 8, 1116–1121 RSC;
(c) X. Chen, X. Wang, Y. Hou, J. Huang, L. Wu and X. Fu, J. Catal., 2008, 255, 59–67 CrossRef CAS;
(d) X. Chen and S. Mao, Chem. Rev., 2007, 107, 2891–2959 CrossRef CAS;
(e) D. Chatterjee and S. Dasgupta, J. Photochem. Photobiol., C, 2005, 6, 186–205 CrossRef CAS.
-
(a) R. Asahi, T. Morikawa, T. Okwaki, K. Aoki and Y. Taga, Science, 2001, 293, 269–271 CrossRef CAS;
(b) W. H. Ma, J. Li, X. Tao, J. He, Y. M. Xu, J. C. Yu and J. C. Zhao, Angew. Chem., Int. Ed., 2003, 42, 1029–1032 CrossRef CAS.
-
(a) H. T. Yu, X. Y. Li, X. Quan, S. Chen and Y. B. Zhang, Environ. Sci. Technol., 2009, 43, 7849–7855 CrossRef CAS;
(b) Z. Q. He, L. Xie, J. J. Tu, S. Song and W. P. Liu, J. Phys. Chem. C, 2010, 114, 526–532 CrossRef CAS.
- J. H. Huang, Y. J. Cui and X. C. Wang, Environ. Sci. Technol., 2010, 44, 3500–3504 CrossRef CAS.
- G. Q. Li, N. Yang, X. L. Yang, W. L. Wang and W. F. Zhang, J. Phys. Chem. C, 2011, 115, 13734–13738 CAS.
- A. Thomas, A. Fischer, F. Goettmann, M. Antonietti, J.-O. Müller, R. Schlögl and J. M. Carlsson, J. Mater. Chem., 2008, 18, 4893–4908 RSC.
-
(a) X. Wang, K. Maeda, A. Thomas, K. Takanabe, G. Xin, J. M. Carlsson, K. Domen and M. Antonietti, Nat. Mater., 2009, 8, 76–80 CrossRef CAS;
(b) K. Maeda, X. Wang, Y. Nishihara, D. Lu, M. Antonietti and K. Domen, J. Phys. Chem. C, 2009, 113, 4940–4947 CrossRef CAS;
(c) J. Zhang, X. Chen, K. Takanabe, K. Maeda, K. Domen, J. D. Epping, X. Fu, M. Antonietti and X. Wang, Angew. Chem., Int. Ed., 2010, 49, 441–444 CrossRef CAS;
(d) K. Takanabe, K. Kamata, X. Wang, M. Antonietti, J. Kubota and K. Domen, Phys. Chem. Chem. Phys., 2010, 12, 13020–13025 RSC.
-
(a) F. Su, S. C. Mathew, L. Möhlmann, M. Antonietti, X. Wang and S. Blechert, Angew. Chem., Int. Ed., 2011, 50, 657–660 CrossRef CAS;
(b) F. Su, S. C. Mathew, G. Lipner, X. Fu, M. Antonietti, S. Blechert and X. Wang, J. Am. Chem. Soc., 2010, 132, 16299–16301 CrossRef CAS.
-
(a) Y. Cui, Z. Ding, P. Liu, M. Antonietti, X. Fu and X. Wang, Phys. Chem. Chem. Phys., 2012, 14, 1455–1462 RSC;
(b) S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2009, 25, 10397–10401 CrossRef CAS.
-
(a) J. Zhang, M. Grzelczak, Y. Hou, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Sci., 2012, 3, 443–446 RSC;
(b) J. Zhang, G. Zhang, X. Chen, S. Lin, L. Möhlmann, G. Dolega, G. Lipner, M. Antonietti, S. Blechert and X. Wang, Angew. Chem., Int. Ed., 2012, 51, 3183–3187 CrossRef CAS;
(c) J. Zhang, J. Sun, K. Maeda, K. Domen, P. Liu, M. Antonietti, X. Fu and X. Wang, Energy Environ. Sci., 2011, 4, 675–678 RSC;
(d) K. Kailasam, J. D. Epping, A. Thomas, S. Losse and H. Junge, Energy Environ. Sci., 2011, 4, 4668–4674 RSC;
(e) G. Zhang, J. Zhang, M. Zhang and X. Wang, J. Mater. Chem., 2012, 22, 8083–8091 RSC.
- X. F. Chen, J. S. Zhang, X. Z. Fu, M. Antonietti and X. C. Wang, J. Am. Chem. Soc., 2009, 131, 11658–11659 CrossRef CAS.
- X. C. Wang, X. F. Chen, A. Thomas, X. Z. Fu and M. Antonietti, Adv. Mater., 2009, 21, 1609–1612 CrossRef CAS.
- L. Ge, Mater. Lett., 2011, 65, 2652–2654 CrossRef CAS.
-
(a) X. C. Wang, K. Maeda, X. F. Chen, K. Takanabe, K. Domen, Y. D. Hou, X. Z. Fu and M. Antonietti, J. Am. Chem. Soc., 2009, 131, 1680–1681 CrossRef CAS;
(b) X. Chen, Y.-S. Jun, K. Takanabe, K. Maeda, K. Domen, X. Fu, M. Antonietti and X. Wang, Chem. Mater., 2009, 21, 4093–4095 CrossRef CAS.
- Y. Cui, J. Zhang, G. Zhang, J. Huang, P. Liu, M. Antonietti and X. Wang, J. Mater. Chem., 2011, 21, 13032–13039 RSC.
- E. Androulaki, A. Hiskia, D. Dimotikali, C. Minero, P. Calza, E. Pelizzetti and E. Papacontinou, Environ. Sci. Technol., 2000, 34, 2024–2028 CrossRef CAS.
- J. C. Barreto, G. S. Smith and N. H. P. Strobel, Life Sci., 1995, 56, 89–96 Search PubMed.
- S. C. Yan, Z. S. Li and Z. G. Zou, Langmuir, 2010, 26, 3894–3901 CrossRef CAS.
- D. S. Kong, Langmuir, 2008, 24, 5324–5331 CrossRef CAS.
- H. Bader, V. Sturzenegger and J. Hoigne, Water Res., 1988, 22, 1109–1115 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: 10.1039/c2cy20036h |
|
| This journal is © The Royal Society of Chemistry 2012 |
Click here to see how this site uses Cookies. View our privacy policy here.