Selective hydrogenation of cyclohexenone on iron–ruthenium nano-particles suspended in ionic liquids and CO2-expanded ionic liquids
Jean-Michel
Andanson
a,
Stefan
Marx
a and
Alfons
Baiker
*ab
aDepartment of Chemistry and Applied Biosciences, ETH Zurich, Hönggerberg, HCI CH-8093 Zürich, Switzerland. E-mail: baiker@chem.ethz.ch; Fax: +41 44 632 1163; Tel: +41 44 632 3153
bChemistry Department, Faculty of Science, King Abdulaziz University, Jeddah 21589, Saudi Arabia
First published on 16th April 2012
Abstract
Iron and ruthenium mono- and bimetallic nanoparticles (NPs) with different metal ratios (Fe–Ru 9![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, Fe–Ru 3:1, and Fe–Ru 1:1) have been prepared in imidazolium ionic liquids (ILs) via decarbonylation of the corresponding metal carbonyl precursors. The as-prepared ionic liquid suspended nanoparticles were tested as catalysts for the selective hydrogenation of cyclohexenone to cyclohexanone at 50 °C in a high pressure batch reactor equipped for in situ Attenuated Total Reflection (ATR) infrared spectroscopy. After reaction, supercritical carbon dioxide was applied to extract the products from the reaction mixture, thus facilitating a green approach to solventless transition metal catalysis. The course of the reaction and extraction was followed by ATR-IR spectroscopy. From the IL suspended monometallic NPs, Ru showed much higher activity than Fe. Addition of 50 at% iron to Ru (Fe–Ru 1:1) resulted in a remarkable decrease of the average particle size of the NPs from 2.9 nm to 1.6 nm. Among the different NPs prepared, the bimetallic Fe–Ru 1:1 was most active followed by the pure Ru. Addition of CO2 to the reaction mixture greatly increased the reaction rate (4 times faster with 60 bar of CO2 than without), as demonstrated for the highly active Fe–Ru 1:1 NPs suspended in 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIm][BF4]). At 60 bar of CO2 high selectivity >95% to cyclohexanone, at a TOF of ca. 300 h−1, was achieved and the catalyst could be reused 5 times without noticeable deactivation.
:1, Fe–Ru 3:1, and Fe–Ru 1:1) have been prepared in imidazolium ionic liquids (ILs) via decarbonylation of the corresponding metal carbonyl precursors. The as-prepared ionic liquid suspended nanoparticles were tested as catalysts for the selective hydrogenation of cyclohexenone to cyclohexanone at 50 °C in a high pressure batch reactor equipped for in situ Attenuated Total Reflection (ATR) infrared spectroscopy. After reaction, supercritical carbon dioxide was applied to extract the products from the reaction mixture, thus facilitating a green approach to solventless transition metal catalysis. The course of the reaction and extraction was followed by ATR-IR spectroscopy. From the IL suspended monometallic NPs, Ru showed much higher activity than Fe. Addition of 50 at% iron to Ru (Fe–Ru 1:1) resulted in a remarkable decrease of the average particle size of the NPs from 2.9 nm to 1.6 nm. Among the different NPs prepared, the bimetallic Fe–Ru 1:1 was most active followed by the pure Ru. Addition of CO2 to the reaction mixture greatly increased the reaction rate (4 times faster with 60 bar of CO2 than without), as demonstrated for the highly active Fe–Ru 1:1 NPs suspended in 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIm][BF4]). At 60 bar of CO2 high selectivity >95% to cyclohexanone, at a TOF of ca. 300 h−1, was achieved and the catalyst could be reused 5 times without noticeable deactivation.
Introduction
Ionic liquids (ILs) have attracted considerable interest mainly due to their diversity and unique chemical properties compared to conventional organic solvents. ILs possess very low vapor pressure, exhibit high thermal stability and chemical resistance, offer high polarity,1 and cover a broad viscosity range.2 Thus, ILs have the potential for being adapted to a specific application by choosing the appropriate combination of anions and cations. ILs have been heavily used in catalysis, as covered in various reviews.3 In many cases the IL is employed as a stabilizing and protective agent for metal nanoparticles (NPs).4–8 IL supported metal NPs can be prepared in a simple way by dissolving a metal precursor in the IL followed by reduction of the metal salt, e.g. with hydrogen,4 or thermal decomposition of the corresponding metal carbonyl.5 In the latter case, only CO is produced during the thermal decomposition and no other impurities like halides or organic ligands, stemming from the metal precursor, are present after particle formation. NPs can be stabilized by the IL due to an electrostatic protection in the form of a “protective shell”.9 However, frequently a co-stabilizer is added to the IL because NPs have the tendency to agglomerate when stabilized only by an IL.6 Janiak and co-workers developed synthesis methods for many monometallic NPs in ILs from corresponding carbonyl precursors, in which no co-stabilizer was added. They synthesized exclusively monometallic NPs for a broad range of metals, including Co, Cr, Fe, Ir, Mn, Mo, Os, Re, Ru, Rh, and W,10 and recently reviewed this topic.11Ruthenium NPs stabilized in IL are very active in the hydrogenation of ketones,7 alkenes,12,13 arenes,14 and citral.15 However, ruthenium is an expensive platinum group metal and substitution by a cheap and environmentally benign metal would be an improvement. Recently, de Vries and colleagues reported that iron NPs suspended in THF are active in the hydrogenation of alkenes and alkynes.16 Thus, the substitution of ruthenium by its lighter homologue may be an interesting alternative to the use of monometallic ruthenium catalysts. Iron–ruthenium catalysts have proven to be interesting catalysts for various reactions, such as Fischer–Tropsch synthesis17,18 and the selective hydrogenation of unsaturated aldehydes.19 This prompted us to synthesize a series of mono- and bimetallic particles stabilized in 1-butyl-3-methylimidazolium hexafluorophosphate ([BMIm][PF6]) and 1-butyl-3-methylimidazolium tetrafluoroborate ([BMIm][BF4]). The solventless hydrogenation of cyclohexenone was chosen as a test reaction. The possible products of the hydrogenation of cyclohexenone are shown in Scheme 1. CO2-expanded liquids have gained considerable attention in chemical synthesis.20 Addition of CO2 to ILs can be essential for efficient reactions in ILs due to enhanced mass transfer caused by lowering the viscosity of the IL and increased solubility of gases such as hydrogen.21,22 Furthermore, the reaction mixture can be extracted efficiently by supercritical carbon dioxide (scCO2) from the IL and NPs. In a similar manner as in our previous studies,23,24 we followed the reaction in the liquid phase as well as the scCO2 extraction by in situ Attenuated Total Reflection Infrared (ATR-IR) spectroscopy.
 | ||
| Scheme 1 Hydrogenation of 2-cyclohexenone to various products. 1: 2-cyclohexenone; 2: cyclohexanone; 3: 2-cyclohexenol; 4: cyclohexanol. | ||
Experimental
Synthesis of mono- and bimetallic nanoparticles
Diironnonacarbonyl (Fe2(CO)9) (98%, Acros) and triruthenium-dodecacarbonyl (Ru3(CO)12) (99%, Acros) were stored under an inert atmosphere in a glove box and used as received. After a fast initial pre-drying at room temperature under vacuum, [BMIm][PF6] (Alfa Aesar, 99%) and [BMIm][BF4] (Acros, 98+%) were dried at 150 °C under vacuum (5 × 10−2 mbar) for at least three days to remove residual water. 2-Cyclohexenone (Fluka, 99.5%) was used without further purification. CO2 (Pangas, 3.0) was used as solvent and for supercritical extraction.Janiak et al. synthesized monometallic Fe and Ru NPs from metal-carbonyl precursors in [BMIm][BF4].9 We adopted a similar protocol to the synthesis of bimetallic iron–ruthenium NPs. The dried ILs were introduced into a glove box and the metal carbonyls, Fe2(CO)9 and Ru3(CO)12, were added in corresponding amounts to obtain mono- or bimetallic NPs. The total molar amount of metal was kept approximately constant (ca. 0.4 mmol, Table 1) while the ratio of the two metals was varied. Thus, pure Fe and Ru particles were prepared in addition to mixed Fe–Ru 9:1, Fe–Ru 3:1, and Fe–Ru 1:1 particles.
| Composition | n metal/mmol | m precursor/mg | wt% (metal) | c metal a/mmol ml−1 |
|---|---|---|---|---|
| a mmol metal per ml IL. | ||||
| Fe | 0.367 | 66.8 | 1.01 | 0.25 |
| Fe–Ru 9:1 |
0.352 (Fe) | 64.1 | 0.96 | 0.24 |
| +0.044 (Ru) | 9.3 | +0.22 | +0.03 | |
| =0.396 | =1.18 | =0.27 | ||
| Fe–Ru 3:1 |
0.308 (Fe) | 56.1 | 0.85 | 0.21 |
| +0.109 (Ru) | 23.2 | +0.54 | +0.08 | |
| =0.417 | =1.39 | =0.29 | ||
| Fe–Ru 1:1 |
0.188 (Fe) | 34.2 | 0.51 | 0.13 |
| +0.203 (Ru) | 43.3 | +1.01 | +0.14 | |
| =0.391 | =1.52 | =0.27 | ||
| Ru | 0.397 | 84.6 | 1.91 | 0.27 |
In a typical experiment, 2 g of the IL were mixed with a total amount of ca. 0.4 mmol of metal. Exact values are given in Table 1. The suspension of the precursors in the IL was heated for 18 hours under an inert atmosphere to 180 °C for pure iron particles, 190 °C for Fe–Ru 9:1, and 250 °C for the other compositions. The thermal decomposition of the iron carbonyl precursor started at about 120 °C, while the decomposition of ruthenium carbonyl occurred at slightly higher temperature. For all prepared NPs the resulting mixture was black in colour and highly viscous at room temperature, and only the pure iron NPs showed ferromagnetic behavior. Finally, the reaction mixture was evacuated to remove remaining CO that dissolved in the IL. The samples were stored under an inert atmosphere until their use in the catalytic tests.
Characterization of the NPs
Transmission electron microscopy (TEM) investigations were performed on a Tecnai F30 microscope (FEI, field emission gun operated at 300 kV). The particles were dispersed in acetone and deposited onto a perforated carbon foil supported on a copper grid. The particle size distribution was automatically calculated using ImageJ 1.44 software. The mean particle size was obtained from Gaussian fitting where the full width at half maximum (FWHM) is provided as a measure of the broadness of the particle size distribution. The atomic ratio of the metals in the bimetallic NPs was determined by ICP-MS analysis and corroborated the nominal ratios. Compositions of the NPs are listed in Table 1. Temperature programmed reduction (TPR) measurements were performed with ca. 50 mg of the as-prepared ionic liquid suspended NPs in a quartz reactor using 5 vol% H2 in He at a flow rate of 50 ml min−1 and a heating rate of 1 °C min−1 up to 650 °C.Hydrogenation experiments and in situ ATR-IR
Hydrogenation experiments were performed in a magnetically stirred high pressure stainless steel reactor cell equipped for in situ Attenuated Total Reflection (ATR) infrared spectroscopy. The reactor cell was connected to a high pressure hydrogen supply, a CO2 compressing unit (NWA PM-101), and an expansion unit (NWA PE-103). It was equipped with either a Ge or ZnSe ATR-crystal (with 6 active reflections, Crystran Ltd, U.K.), serving as an internal reflection element, and placed in an Equinox 55 IR spectrometer (Bruker Optics). A detailed description of this set up can be found elsewhere.25 Spectra were recorded in the range between 8000–400 cm−1 with a resolution of 4 cm−1.In a typical experiment, 300 μl of the as-prepared ionic liquid suspended NPs were placed on top of the ATR-crystal and 1 ml of cyclohexenone was added at 50 °C. The reaction cell was then sealed, and flushed with CO2. Mixing of substrate and IL was finished when no change in two subsequent IR spectra was observed. To start the reaction, the cell was pressurized with the desired amount of H2 and CO2, while simultaneously recording the first IR spectrum. IR spectra were then taken repeatedly every 5 minutes. If only hydrogen was used for the reaction, the cell was pressurized with 50 bar of hydrogen. To study the effect of dense CO2 on the reaction kinetics another series of experiments was performed, in which only 30 bar of hydrogen and different pressures of CO2 were applied (30, 60, and 100 bar).
Supercritical CO2 extraction
After the reaction, the extraction was slowly started by flowing compressed CO2. This resulted in a continuous supercritical extraction procedure, during which the pressure was kept between 120 and 130 bar to ensure a high solubility of all organics in the scCO2 phase. The extraction was also followed by ATR-IR spectroscopy, indicating the complete removal of reactants and products in less than 20 minutes. The components of the reaction mixture were collected in a cold trap (acetone, liquid nitrogen) that was connected to the expansion unit. The samples were analyzed with standard GC methods on a HP 6890 equipped with a FFAP column and a FI detector. GC-MS data have been collected on a HP 6890 with an Agilent 5973 mass spectrometer.Results and discussion
Structural properties of Fe–Ru nanoparticles
TEM pictures and histograms showing the particle size distribution of Ru and Fe–Ru 1:1 NPs are presented in Fig. 1. NPs of the latter were considerably smaller (1.65 ± 0.30 nm) than those of the pure Ru (2.91 ± 0.75 nm), in both cases particles showed a narrow size distribution. The limited resolution of the micrographs of the other NPs did not allow accurate determination of their mean particle size. Janiak et al.9 characterised Fe and Ru NPs derived from metal carbonyl precursors in [BMIm][BF4] by means of dynamic light scattering and estimated the mean diameter to be 10.7 ± 2.4 and 2.8 ± 0.6 nm for Fe and Ru, respectively.
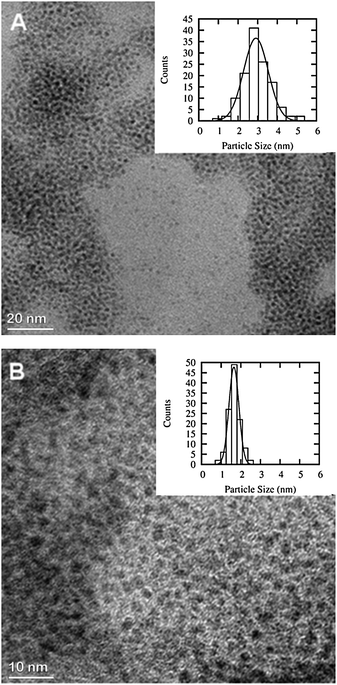 | ||
| Fig. 1 TEM images of Ru (A, particles count 128) and Fe–Ru 1:1 (B, particles count 114). Note the different magnification, the scale bar of the Ru image is 20 nm while it is 10 nm for the Fe–Ru 1:1. | ||
TPR measurements of the NPs suspended in the IL confirmed that the NPs were present as metals, that is in a fully reduced state (zero oxidation state) when applied in the catalytic tests. However, partial oxidation of the NPs cannot be ruled out if the materials are stored over longer periods.
Consideration of the phase diagram of the binary Fe–Ru system indicates complete miscibility (alloy formation) only for Fe–Ru 1:1, while mixtures with 5 to 24 at% of Ru are expected to be biphasic.26 Consequently, the Fe–Ru 9:1 and 3:1 samples are likely not to form alloys. However, it is not clear whether the information extracted from the phase diagram can safely be applied to predict the behavior of bimetallic NPs suspended in IL. Furthermore, surface segregation phenomena in the synthesized Fe–Ru NPs cannot be ruled out due to the different surface free energy of the constituents.18,27 Evidence for iron enrichment on the surface of alloyed Fe–Ru NPs supported on amorphous carbon black was reported by Vannice and colleagues based on CO and H2 chemisorption measurements.27 Although surface segregation of iron cannot be ruled out in our IL stabilized Fe–Ru NPs, this effect seems not to be significant and particularly the formation of a core–shell structure can be excluded because the catalytic behavior of the Fe–Ru 1:1 samples resembled that of the pure Ru NPs (Table 2) which is not expected in the case of extensive iron enrichment on the surface.
| Entry | Catalyst | Time/h | Cycle | Conv. [%] | Sel. [%] | TOF/h−1 | |
|---|---|---|---|---|---|---|---|
| Ketone | Alcohol | ||||||
| 1 | Fe | 1 | 1 | 33 | 82 | 18 | 53 |
| 2 | 1 | 2 | 17 | 83 | 17 | 27 | |
| 3 | 1 | 3 | 12 | 79 | 21 | 21 | |
| 4 | 1 | 4 | 13 | 86 | 14 | 20 | |
| 5 | 20 | 1 | 51 | 64 | 36 | 3 | |
| Fe–Ru | |||||||
| 6 | 9:1 |
1 | 1 | 22 | 93 | 7 | 25 |
| 7 | 1 | 2 | 5 | 83 | 17 | 7 | |
| 8 | 1 | 3 | 7 | 87 | 13 | 9 | |
| Fe–Ru | |||||||
| 9 | 3:1 |
1 | 1 | 72 | 95 | 5 | 74 |
| 10 | 1 | 2 | 64 | 96 | 4 | 65 | |
| 11 | 1 | 3 | 54 | 96 | 4 | 56 | |
| Fe–Ru | |||||||
| 12 | 1:1 |
1 | 1 | 91 | 87 | 13 | 111 |
| 13 | 1 | 2 | 83 | 96 | 4 | 106 | |
| 14 | 1 | 3 | 77 | 96 | 4 | 93 | |
| 15 | Ru | 1 | 1 | 94 | 75 | 25 | 104 |
| 16 | 1 | 2 | 97 | 88 | 12 | 107 | |
| 17 | 1 | 3 | 99 | 88 | 12 | 109 | |
Catalytic properties
The catalytic performance of the mono- and bimetallic NPs stabilized in [BMIm][PF6] has been investigated in the hydrogenation of cyclohexenone (Scheme 1). Hydrogenation with pure Fe NPs at 50 °C gave a moderate conversion of 33% after 1 hour for the first run (Table 2, entry 1). Typically the C–C double bond was hydrogenated first yielding cyclohexanone while the consecutive hydrogenation resulted in the formation of cyclohexanol, indicated by the increase of selectivity to cyclohexanol from 18% (after 1 h) to 36% after 20 hours (Table 2, entries 1 and 5). Kinetic aspects of the reaction are discussed later on using the in situ ATR-IR spectroscopy data. Formation of cyclohexenol has not been observed in any experiments by in situ ATR-IR spectroscopy, GC, or GC-MS analysis. Several recycle experiments have been performed to investigate the stability and reusability of the NPs. Between each cycle, organics were removed by scCO2 followed by addition of new cyclohexenone.The situation changed dramatically when the amount of Ru was increased. Fe–Ru 3:1 particles exhibited a conversion up to 72% for the first experiment and the formation of cyclohexanone was favored as indicated by a selectivity of 95% (Table 2, entry 9). The ratio of ketone to alcohol was almost 5 times higher compared to pure iron particles (Fig. 2). Nevertheless the activity of the catalyst decreased in the following recycle experiments. The conversion dropped by about 10% between the cycles while the selectivity to cyclohexanone remained constant at high level 96%.
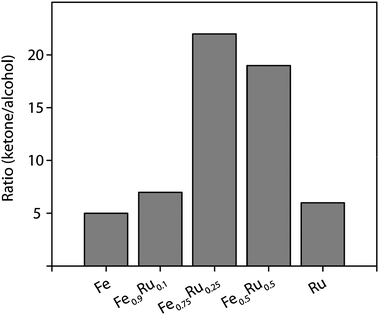 | ||
| Fig. 2 Cyclohexanone:cyclohexanol molar ratio obtained with various NPs. | ||
The activity of Fe–Ru 1:1 was the highest among the Fe–Ru NPs and remained high during the following recycle experiments. The pure Ru NPs showed even slightly higher conversion (94%), but at lower selectivity to cyclohexanone (75%) in the first experiment (Table 2, entry 15). Interestingly upon reuse, both conversion and selectivity increased (Table 2, entries 15–17), which may be attributed to the decontaminating effect of the dense carbon dioxide. This behavior contrasts that of all iron containing catalysts, where significant deactivation was observed upon reuse of the catalysts (Table 2, entries 1–14).
Both the Fe–Ru 3:1 and the Fe–Ru 1:1 NPs exhibited excellent selectivity to cyclohexanone, that is C–C double bond hydrogenation, as indicated in Fig. 2 which shows the molar ratio of cyclohexanone/cyclohexanol achieved with the different catalysts. The amount of cyclohexanone formed was on average around 20 times higher than that of cyclohexanol for any experiment using Fe–Ru 3:1 NPs and for recycle experiments with Fe–Ru 1:1 particles, while for pure Ru it was only around 7 times higher, indicating the significant role of the iron constituent in achieving high selectivity to cyclohexanone. However, the comparison of the selectivities achieved after 1 h reaction time is not conclusive due to the different conversions. A more rigorous comparison of the selectivities of the NPs emerges from the in situ ATR-IR experiments described in the next section, which allowed a comparison of the selectivity at similar conversion levels.
An important factor concerning the applicability of a catalytic system is its recyclability. We used CO2 as solvent to extract organic molecules. The ILs and the NPs are insoluble in the CO2 phase and could be reused after extraction.28 Deactivation of the particles was most pronounced for iron-rich particles. For pure ruthenium particles a slight increase of the activity was observed similar to the observation of Janiak et al. for the hydrogenation of cyclohexene at 90 °C and 10 bar of hydrogen.12 The deactivation of the bimetallic Fe–Ru catalysts strongly depended on the iron content of the NPs, as emerges from Fig. 3 where the relative deactivation from the first experiment to the second is plotted. The reason for this deactivation is not clear yet, it is possibly due to strong agglomeration of Fe NPs.
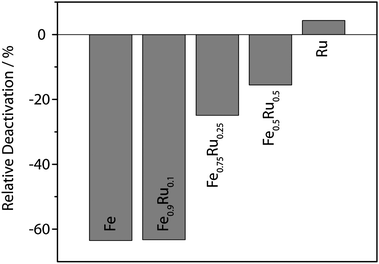 | ||
| Fig. 3 Dependence of relative deactivation of the catalysts from the first to the second run on the iron content. | ||
In situ monitoring of reaction and product extraction
The course of reaction and extraction was followed by in situ ATR-IR spectroscopy. A multivariate analysis of the spectra allowed the extraction of the time-dependent concentration profiles of the individual products. The procedural details of the multivariate analysis were the same as reported elsewhere.24 Typical examples of the IR spectra measured during the reaction, a spectrum deconvolution and concentration profiles of all species are shown in Fig. 4 for the hydrogenation with Ru NPs. In addition to the species indicated in Fig. 4, bands due to CO formed via reverse water gas shift reaction were observed during hydrogenations over Ru and Fe–Ru NPs in dense CO2. The conversion of cyclohexenone to cyclohexanone was complete after 45 min (TOF circa 150 h−1) and was followed by a slower conversion of cyclohexanone to cyclohexanol.![(A) Evolution of the infrared spectra during the reaction of cyclohexenone (520 mg Ru/[BMIm][PF6] with 975 mg of cyclohexenone at 50 °C and 50 bar H2). Arrows show the evolution of the main infrared bands with time in the range of [1800–900 cm−1]. (B) Example of deconvolution of an infrared spectrum by summation of reactant, products, and solvent spectra. Spectra are stacked for clarity reasons. (C) Evolution of the concentrations of reactant, products and solvent with time obtained in situ by the deconvolution of infrared spectra.](/image/article/2012/CY/c2cy00533f/c2cy00533f-f4.gif) | ||
| Fig. 4 (A) Evolution of the infrared spectra during the reaction of cyclohexenone (520 mg Ru/[BMIm][PF6] with 975 mg of cyclohexenone at 50 °C and 50 bar H2). Arrows show the evolution of the main infrared bands with time in the range of [1800–900 cm−1]. (B) Example of deconvolution of an infrared spectrum by summation of reactant, products, and solvent spectra. Spectra are stacked for clarity reasons. (C) Evolution of the concentrations of reactant, products and solvent with time obtained in situ by the deconvolution of infrared spectra. | ||
This observation is essential for a straightforward comparison of selectivity between the different types of NPs. Note that in Table 2 selectivities to cyclohexanone were compared after 1 hour of reaction time, that is at different conversion levels due to the different activity of the NPs. The in situ monitoring allowed us to give a more precise assignment of the selectivity to cyclohexanone, it showed that the selectivity is strongly correlated to the conversion and at a similar conversion the selectivity to cyclohexanone was high for all active catalysts (Fe–Ru 3:1, Fe–Ru 1:1 and Ru). The consecutive hydrogenation to cyclohexanol only started to play a role after most of the cyclohexenone had been hydrogenated. Reaction kinetics observed with mono- and bimetallic NPs in the absence of CO2 are compared in Fig. 5. For the sake of clarity only the concentration of cyclohexanone (main product) is shown. The Fe–Ru 1:1 catalyst was the most active catalyst, slightly more active than the pure Ru NPs. The decrease of the cyclohexanone concentration with the most active catalysts, Fe–Ru 1:1 and pure Ru, after about 40–45 minutes is due to the consecutive hydrogenation of cyclohexanone to cyclohexanol. Interestingly, Fe–Ru 1:1 NPs were even more active than the pure Ru NPs, which would not be expected from the intrinsic activities of these constituents (compare TOFs in Table 2). However, it should be stressed that the Fe–Ru 1:1 NPs were considerably smaller (1.6 ± 0.3 nm) than the pure Ru NPs (2.9 ± 0.7 nm) and thus exposed a higher number of surface atoms per catalyst mass.
![Formation of cyclohexanone followed with time by ATR-IR spectroscopy for Fe (triangle), Fe–Ru 3 : 1 (squares), Fe–Ru 1 : 1 (diamonds) and Ru (circles) NPs. Conditions: 520 mg Ru/[BMIm][PF6] or 484 mg Fe/[BMIm][PF6], 428 mg Fe–Ru 1 : 1/[BMIm][PF6], 473 mg Fe–Ru 3 : 1/[BMIm][PF6], with about 1000 mg of cyclohexenone at 50 °C and 50 bar H2.](/image/article/2012/CY/c2cy00533f/c2cy00533f-f5.gif) | ||
| Fig. 5 Formation of cyclohexanone followed with time by ATR-IR spectroscopy for Fe (triangle), Fe–Ru 3:1 (squares), Fe–Ru 1:1 (diamonds) and Ru (circles) NPs. Conditions: 520 mg Ru/[BMIm][PF6] or 484 mg Fe/[BMIm][PF6], 428 mg Fe–Ru 1:1/[BMIm][PF6], 473 mg Fe–Ru 3:1/[BMIm][PF6], with about 1000 mg of cyclohexenone at 50 °C and 50 bar H2. | ||
Effect of dense carbon dioxide
Dissolving CO2 in ILs can strongly decrease their viscosity,21 and enhance mass transfer and the solubility of hydrogen.22 Thus CO2 increases the availability of hydrogen in the IL and limitations due to transport phenomena are alleviated. To observe the effect of dissolving CO2 in the liquid phase, the hydrogenation of cyclohexenone with the most active catalyst, Fe–Ru 1:1, was tested using different pressure of CO2 at constant H2 pressure (30 bar). While CO2 had a moderate influence on the reactions performed in Fe–Ru 1:1 NPs stabilized by [BMIm][PF6], it strongly affected the activity of Fe–Ru 1:1 NPs suspended in [BMIm][BF4]. The reason for this behaviour is not clear yet. The production of cyclohexanone during the hydrogenation of cyclohexenone at different CO2 pressure over Fe–Ru 1:1 NPs suspended in [BMIm][BF4] is shown in Fig. 6.
![Cyclohexanone formation at 50 °C under 30 bar H2 and different pressure of CO2 followed by in situ ATR-IR spectroscopy. Five consecutive experiments were performed using [BMIm][BF4] stabilized Fe–Ru 1 : 1 NPs; 0 bar (filled circle), 30 bar (triangle), 60 bar (square), 100 bar (diamond), and finally 0 bar (empty circle) a second time. Between each reaction, products were extracted by scCO2 for at least 1 hour.](/image/article/2012/CY/c2cy00533f/c2cy00533f-f6.gif) | ||
| Fig. 6 Cyclohexanone formation at 50 °C under 30 bar H2 and different pressure of CO2 followed by in situ ATR-IR spectroscopy. Five consecutive experiments were performed using [BMIm][BF4] stabilized Fe–Ru 1:1 NPs; 0 bar (filled circle), 30 bar (triangle), 60 bar (square), 100 bar (diamond), and finally 0 bar (empty circle) a second time. Between each reaction, products were extracted by scCO2 for at least 1 hour. | ||
The same catalyst was reused in five consecutive experiments. The first and last experiments were performed under the same conditions (0 bar CO2) and the activity of the catalyst was identical. The TOF for Fe–Ru 1:1/[BMIm][BF4] in experiments without CO2 was lower (ca. 75 h−1) compared to that of Fe–Ru 1:1 NPs suspended in [BMIm][PF6] (ca. 185 h−1). When CO2 was added (Fig. 6), the reaction rate almost doubled at 30 bar and increased by a factor of about 4 at 60 bar of CO2 compared to the same reaction performed without CO2. Interestingly in the presence of 60 bar of CO2, the first hydrogenation step to cyclohexanone was completed after less than 30 min (corresponding to a TOF of ca. 300 h−1), and the consecutive hydrogenation to cyclohexanol was not observed unlike in the previous tests without CO2 over Fe–Ru 1:1/[BMIm][PF6] (Fig. 5). At 100 bar CO2, the cyclohexanone formation rate decreased strongly compared to that without CO2. This is attributed to the fact that above the critical pressure the solubility of cyclohexenone in the CO2 phase increases sharply, resulting in a substantial decrease of the reactant in the IL phase. Thus, the beneficial effects of expanding the IL and facilitating the solubility of hydrogen with increasing CO2 pressure (observed for 30 and 60 bar) are overcompensated by the decrease of the concentration of the substrate in the liquid phase at 100 bar.
Conclusions
Mono- and bimetallic Fe–Ru particles stabilized in [BMIm][PF6] and [BMIm][BF4] have been synthesized from corresponding carbonyl precursors and applied in the hydrogenation of cyclohexenone. In [BMIm][PF6], Ru NPs were much more active than Fe. The addition of Fe to Ru in a ratio of 1:1 resulted in NPs with considerably smaller mean size (1.6 nm) compared to the pure Ru NPs (2.9 nm). The Fe–Ru 1:1 NPs showed an activity higher than that of the pure Ru NPs probably due to their smaller size. These catalysts showed excellent selectivity for the hydrogenation of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, that is to cyclohexanone, in several repeated runs. The beneficial influence of CO2 in the reaction mixture was shown for NPs stabilized in [BMIm][BF4]. With increasing pressure (up to 60 bar) the reaction rate was greatly enhanced due to improved mass transfer and/or hydrogen solubility. The slowdown of the reaction rate at 100 bar is attributed to lowering of the reactant concentration in the IL containing the active NPs due to strong increase of the solubility of the reactant in the scCO2 phase. When adding 60 bar of CO2, the TOF of the selective hydrogenation of cyclohexenone to cyclohexanone over Fe–Ru 1:1 suspended in [BMIm][BF4] was about four times higher than in the absence of CO2 and no consecutive hydrogenation of cyclohexanone to cyclohexanol was observed. The product mixture could be easily separated by extraction with supercritical CO2 from the IL suspended NPs under mild conditions without using toxic, organic solvents or time consuming separation techniques. Thus the combination of ILs and CO2 may offer unique opportunities for green transition metal catalysis, by acting as reaction medium, extraction solvent, and moderator for tuning the properties of the IL.
C double bond, that is to cyclohexanone, in several repeated runs. The beneficial influence of CO2 in the reaction mixture was shown for NPs stabilized in [BMIm][BF4]. With increasing pressure (up to 60 bar) the reaction rate was greatly enhanced due to improved mass transfer and/or hydrogen solubility. The slowdown of the reaction rate at 100 bar is attributed to lowering of the reactant concentration in the IL containing the active NPs due to strong increase of the solubility of the reactant in the scCO2 phase. When adding 60 bar of CO2, the TOF of the selective hydrogenation of cyclohexenone to cyclohexanone over Fe–Ru 1:1 suspended in [BMIm][BF4] was about four times higher than in the absence of CO2 and no consecutive hydrogenation of cyclohexanone to cyclohexanol was observed. The product mixture could be easily separated by extraction with supercritical CO2 from the IL suspended NPs under mild conditions without using toxic, organic solvents or time consuming separation techniques. Thus the combination of ILs and CO2 may offer unique opportunities for green transition metal catalysis, by acting as reaction medium, extraction solvent, and moderator for tuning the properties of the IL.
Acknowledgements
Financial support of this work by the Swiss National Science Foundation and the Foundation Claude & Guliana is kindly acknowledged. Thanks are due to Dr Frank Krumeich and the Electron Microscopy Center of ETH Zurich (EMEZ) for the TEM investigations, Dr Matthias Beier for valuable discussions and Dr Robert Büchel for performing the TPR measurements.Notes and references
- H. Olivier-Bourbigou, L. Magna and D. Morvan, Appl. Catal., A, 2010, 373, 1–56 CrossRef CAS; C. Chiappe, M. Malvaldi and C. S. Pomelli, Pure Appl. Chem., 2009, 81, 767–776 CrossRef.
- A. P. Froba, H. Kremer and A. Leipertz, J. Phys. Chem. B, 2008, 112, 12420–12430 CrossRef.
- T. Welton, Coord. Chem. Rev., 2004, 248, 2459–2477 CrossRef CAS; V. I. Parvulescu and C. Hardacre, Chem. Rev. (Washington, DC, U. S.), 2007, 107, 2615–2665 Search PubMed; J. Muzart, Adv. Synth. Catal., 2006, 348, 275–295 CrossRef; M. Haumann and A. Riisager, Chem. Rev. (Washington, DC, U. S.), 2008, 108, 1474–1497 Search PubMed; J. Durand, E. Teuma and M. Gomez, C. R. Chim., 2007, 10, 152–177 CrossRef; P. Sledz, M. Mauduit and K. Grela, Chem. Soc. Rev., 2008, 37, 2433–2442 RSC; K. Bica and P. Gaertner, Eur. J. Org. Chem., 2008, 3235–3250 CrossRef.
- G. S. Fonseca, G. Machado, S. R. Teixeira, G. H. Fecher, J. Morais, M. C. M. Alves and J. Dupont, J. Colloid Interface Sci., 2006, 301, 193–204 CrossRef CAS.
- E. Redel, J. Kramer, R. Thomann and C. Janiak, J. Organomet. Chem., 2009, 694, 1069–1075 CrossRef CAS.
- J. Dupont and J. D. Scholten, Chem. Soc. Rev., 2010, 39, 1780–1804 RSC.
- G. S. Fonseca, J. D. Scholten and J. Dupont, Synlett, 2004, 1525–1528 CAS.
- P. Migowski and J. Dupont, Chem.–Eur. J., 2007, 13, 32–39 CrossRef CAS; G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner, S. R. Teixeira and J. Dupont, Chem.–Eur. J., 2003, 9, 3263–3269 CrossRef; J. Dupont, G. S. Fonseca, A. P. Umpierre, P. F. P. Fichtner and S. R. Teixeira, J. Am. Chem. Soc., 2002, 124, 4228–4229 CrossRef; V. Cimpeanu, M. Kocevar, V. I. Parvulescu and W. Leitner, Angew. Chem., Int. Ed., 2009, 48, 1085–1088 CrossRef.
- J. Kramer, E. Redel, R. Thomann and C. Janiak, Organometallics, 2008, 27, 1976–1978 CrossRef.
- C. Vollmer, E. Redel, K. Abu-Shandi, R. Thomann, H. Manyar, C. Hardacre and C. Janiak, Chem.–Eur. J., 2010, 16, 3849–3858 CrossRef CAS; E. Redel, R. Thomann and C. Janiak, Chem. Commun., 2008, 1789–1791 RSC; E. Redel, J. Krämer, R. Thomann and C. Janiak, J. Organomet. Chem., 2009, 694, 1069–1075 CrossRef; D. Marquardt, Z. Xie, A. Taubert, R. Thomann and C. Janiak, Dalton Trans., 2011, 40, 8290–8293 RSC; D. Marquardt, C. Vollmer, R. Thomann, P. Steurer, R. Mülhaupt, E. Redel and C. Janiak, Carbon, 2011, 49, 1326–1332 CrossRef.
- C. Vollmer and C. Janiak, Coord. Chem. Rev., 2011, 255, 2039–2057 CrossRef CAS.
- C. Vollmer, E. Redel, K. Abu-Shandi, R. Thomann, H. Manyar, C. Hardacre and C. Janiak, Chem.–Eur. J., 2010, 16, 3849–3858 CrossRef CAS.
- L. M. Rossi, G. Machado, P. F. P. Fichtner, S. R. Teixeira and J. Dupont, Catal. Lett., 2004, 92, 149–155 CrossRef CAS.
- M. H. G. Prechtl, M. Scariot, J. D. Scholten, G. Machado, S. R. Teixeira and J. Dupont, Inorg. Chem., 2008, 47, 8995–9001 CrossRef CAS; E. T. Silveira, A. P. Umpierre, L. M. Rossi, G. Machado, J. Morais, G. V. Soares, I. L. R. Baumvol, S. R. Teixeira, P. F. P. Fichtner and J. Dupont, Chem.–Eur. J., 2004, 10, 3734–3740 CrossRef.
- P. Meric, K. M. K. Yu, A. T. S. Kong and S. C. Tsang, J. Catal., 2006, 237, 330–336 CrossRef CAS.
- P. H. Phua, L. Lefort, J. A. F. Boogers, M. Tristany and J. G. de Vries, Chem. Commun., 2009, 3747–3749 RSC.
- G. L. Ott, T. Fleisch and W. N. Delgass, J. Catal., 1979, 60, 394–403 CrossRef CAS; M. C. Bahome, L. L. Jewell, K. Padayachy, D. Hildebrandt, D. Glasser, A. K. Datye and N. J. Coville, Appl. Catal., A, 2007, 328, 243–251 CrossRef.
- G. L. Ott, T. Fleisch and W. N. Delgass, J. Catal., 1980, 65, 253–262 CrossRef CAS.
- B. Bachiller-Baeza, A. Guerrero-Ruiz, P. Wang and I. Rodríguez-Ramos, J. Catal., 2001, 204, 450–459 CrossRef CAS.
- P. G. Jessop and B. Subramaniam, Chem. Rev. (Washington, DC, U. S.), 2007, 107, 2666–2694 CAS.
- G. Laurenczy and P. J. Dyson, Z. Naturforsch., B: Chem. Sci., 2008, 63, 681–684 CAS.
- M. Solinas, A. Pfaltz, P. G. Cozzi and W. Leitner, J. Am. Chem. Soc., 2004, 126, 16142–16147 CrossRef CAS.
- F. Jutz, J. M. Andanson and A. Baiker, J. Catal., 2009, 268, 356–366 CrossRef CAS.
- J. M. Andanson, F. Jutz and A. Baiker, Appl. Spectrosc., 2010, 64, 286–292 CrossRef CAS.
- J. M. Andanson and A. Baiker, Chem. Soc. Rev., 2010, 39, 4571–4584 RSC.
- E. Raub and W. Plate, Z. Metallkd., 1960, 51, 477–481 CAS; M. Fallot, C. R. Hebd. Seances Acad. Sci., 1937, 205, 227 Search PubMed.
- M. Kaminsky, K. J. Yoon, G. L. Geoffroy and M. A. Vannice, J. Catal., 1985, 91, 338–351 CrossRef CAS.
- M. C. Kroon, J. van Spronsen, C. J. Peters, R. A. Sheldon and G. J. Witkamp, Green Chem., 2006, 8, 246–249 RSC; M. T. Reetz, W. Wiesenhofer, G. Francio and W. Leitner, Adv. Synth. Catal., 2003, 345, 1221–1228 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |