Salt-induced size-selective separation, concentration, and preservation of zwitterion-modified gold nanoparticles†
Han
Yang
a,
Xiaoping
Heng
a,
Weiyu
Wang
a,
Jiawen
Hu
*a and
Weiqing
Xu
b
aState Key Laboratory of Chemo/Biosensing and Chemometrics, College of Chemistry and Chemical Engineering, Hunan University, Changsha, 410082, China. E-mail: jwhu@hnu.edu.cn
bState Key Laboratory of Supramolecular Structure and Materials, Jilin University, Changchun, 130012, China
First published on 10th February 2012
Abstract
We report salt-induced size-selective separation, concentration, and preservation of Au nanoparticles, which are based on their size-dependent critical coagulation concentration and reversible aggregation properties imparted by zwitterion ligand.
The physical and chemical properties of nanoparticles (NPs) are highly dependent on particle size, shape, and crystallinity.1 To achieve size- and/or morphology-selected samples of NPs, delicate size- and/or shape-controlled synthesis strategies have been developed.2,3 On the other hand, the challenges faced in controlling particle size and shape prompt increasing efforts for size- and shape-selective separation from the usually polydispersed NPs.4–13 The principle of particle separation relies on the size-dependent differences. These differences can be size,4 magnetism,5 migration velocity in electric6 and centrifuging fields7 and so on, namely, the differences which originate from the NPs themselves. They, as well, can be generated or amplified by the ligand, such as the melting temperature of DNA,8 dissolution of the ligand-capped NPs,9,10 or interparticle interactions.11–13 The flexibility of chemical composition and functions in ligands provide plenty of choices for the separation of polydispersed NPs.
Among them, salt-induced precipitation, which relies on finely tuning interparticle interactions,11–13 is relatively straightforward for high-throughout separation. The colloidal stability of charge-stabilized NPs is explained in terms of interparticle van der Waals attraction (VV) and electrostatic repulsion (VE) according to the Derjaguin–Landau–Verwey–Overbeek (DLVO) theory.14 Usually, the colloidal particles are stable. As VE is larger than VV; the sum of VE and VV thus forms a potential barrier (higher than the Brown motion), which restrains particle aggregation. When salt is introduced into the media, VE can easily be shielded by counterions. When the salt concentration is above the critical coagulation concentration (CCC), VV starts to dominate so that the NPs aggregate. Even the charged molecule such as 3-mercaptopropionic acid11 was introduced so as to improve the charges on the NPs, i.e., increasing the VE, the CCC of solely charge-stabilized NPs is still low and their size-dependent CCC difference is faint. Thus, salt is seldom exploited for the size-selective separation of charge-stabilized NPs.11 Recently, nucleotides12 and thiol-PEG-carboxy13 have been used to confer both negative charges and steric hindrance to the NPs. Steric hindrance is a non-charge repulsion, inert to salt shielding, as it originates from the interactions of the polymer chains anchored on the particles. Due to its conferring, the NPs of different sizes aggregate and redisperse reversibly at distinctly different salt concentrations.12,13 These results hint that the introduction of a non-charge repulsion into the whole interparticle interactions can improve the size-dependent CCCs of the NPs, and thus can facilitate size-selective separation.
It has been reported that zwitterion-modified 4 nm Au NPs show great stability in 3 mol dm−3 NaCl solution.15 We, hereby, use the same zwitterion disulfide ligand to confer another non-charge repulsion, hydration repulsion,16 between the NPs. The zwitterion-modified NPs show distinct size-dependent CCC and reversible aggregation properties, which allow for size-selective separation, concentration, and long-term preservation of the NPs. There are several advantages for zwitterion ligands in particle separation. They are small, easily synthesized molecules so that the cost for separation would be low. Different zwitterion ligands have been used to stabilize NPs of different sizes.15,17 It is therefore possible to separate NPs in a wide range of sizes, meanwhile, with improved separation resolution, by systematically tuning the ligand structure. Finally, the zwitterion ligand has emerged as an excellent alternative to PEGylation because of its biocompatibility, resistance to non-specific adsorption of protein, and monolayer-type coverage with minimum thickness.18 The zwitterion ligand-modified NPs thus posses not only high stability, but also minimized hydrodynamic diameters, biocompatibility, and improved immunity to protein adsorption, which favour greatly their downstream applications such as in biology, i.e. cells.19
Unprotected, citrate-stabilized 4, 16, and 70 nm Au NPs were used to demonstrate the method developed in the present study. The synthesis of the 4, 16 and 70 nm Au NPs20,21 and zwitterion disulfide (Scheme S1)15 are detailed in the electronic supplementary information (ESI†). Surface modification of the Au NPs by the zwitterion is based on the large affinity of thiol and its derivatives for the Au surface via the formation of a strong Au–S bond. This surface bonding configuration forms a strongly polarized dipole layer around the Au NPs, schematically shown in Scheme S2 (ESI†), and makes the particle surface extremely hydrophilic. The polarized dipole layer induces the organization and orientation of the solvent (water) molecules around the Au NPs, and provides a new repulsive hydration force (VH) between the particles.16 For NPs especially with a diameter less than 10 nm, this non-DLVO force is more apparently dependent on particle size, because the particle size is comparable in dimension to that of the adsorbed organic layer.22 Furthermore, zwitterion-modified 4 nm Au NPs are remarkably stable in 3 mol dm−3 NaCl solution.15 This fact indicates that as far as NaCl is concerned, the VH is insensitive to its concentration. As a result, the zwitterion-modified Au NPs behave in a distinctly different way from the citrate-stabilized NPs in NaCl solutions, as demonstrated below.
Table 1 summarizes the CCCs for unprotected, citrate-stabilized and zwitterion-modified Au NPs, which were determined from their corresponding UV-vis spectral sequences upon the addition of varying amount of NaCl (shown in Fig. S1, ESI†). For the citrate-stabilized Au NPs, the 70 nm Au NPs have the smallest CCC, and are most unstable; whereas the 4 nm Au NPs have the largest CCC. However, the largest CCC is just of 50 mmol dm−3 and only 2.7 times that of the smallest CCC for particles ranging from 4 to 70 nm in radius. Once particle aggregation was induced by salt, the particle aggregates were unable to be redispersed in water, as revealed by Fig. S2 (ESI†) and in ref. 23. Therefore, the aggregation is irreversible. As introduced previously, the stability of the charge-stabilized NPs is determined by the sum of interactions of the VE and the VV . For spheric particles, VV = Aa/12H0, where A, a, and H0 represent the Hamaker constant, particle radius, and distance between the surfaces of two particles, respectively.14 This formula stipulates that larger NPs have a higher VV between them, and thus are less stable than the smaller NPs. After shielding the VE, the particles can sufficiently approach each other due to the lack of steric hindrance, and this largely increases the VV. In this case, even the VE between particles was recovered by desalting, and it is unable to overcome the largely increased VV to redisperse the particle aggregates.
| Size/nm | CCC/mol dm−3 | |
|---|---|---|
| unmodified | modified | |
| 4 | 0.050 | > 3.000 |
| 16 | 0.033 | 0.120 |
| 70 | 0.018 | 0.045 |
For the zwitterion-modified Au NPs, however, their CCC is distinctly size-dependent, especially for particles in the range of 4–16 nm. The smallest zwitterion-modified, 4 nm Au NPs, are still stable in 3 mol dm−3 NaCl solution, as observed by us and by Rouhana et al.15 Additionally, as can be seen from Fig. S3 (ESI†), the UV-vis spectrum of the redispersions of their particle aggregates recovered to that of the original dispersion, indicating the reversibility of the process. To further demonstrate this reversibility, Fig. 1 shows the integrated areas of the surface plasmon peaks of the supernatants and redispersions of particle aggregates as a function of aggregation and dispersion cycle. Except for the 4 nm Au NPs, the cycles for the 16 and 70 nm Au NPs can be repeated at least 5 times. These size-dependent CCC and reversible aggregation properties are inherent to the nature of the zwitterion. Upon adsorption, the zwitterion ligand confers a new VH (hydration force) into the whole interparticle interactions. Our experiments confirmed that whilst the zeta potentials of zwitterion-modified Au NPs did decrease to some extent upon adsorption, they did not approach zero (neutral). This means electrostatic repulsion VE still exists in between the particles. Therefore, compared with solely citrate-stabilized Au NPs whose total interparticle interaction is V = VE + VV, the zwitterion-modified Au NPs have a new term and their total interparticle interaction becomes V = VE + VH + VV. For larger particles, whose attractive VV is larger and repulsive VH is weaker, particle aggregation is initiated just by partially screening VE. For middle sized particles, its VV becoming smaller and VH becoming stronger, the initiation of particle aggregation requires VE to be further screened. For the smallest 4 nm Au NPs, their VV is smallest and VH is strongest. Even when the VE is completely screened by salt, the VH alone is able to counteract the VV and provides high stability to the 4 nm Au NPs against the 3 mol dm−3 NaCl solution. Thus, the introduction of the VH distinctly improves the dependence of CCC on particle size. As there is a hydrated layer around the zwitterion-modified Au NPs, the nearest distance at which particles can approach each other is at the second minimum potential well rather than at the primary minimum potential well. That is, the salt-induced aggregates are the product of flocculation rather than coagulation, and thus they can be redispersed in water, more correctly flocculated, again by the addition of salt.
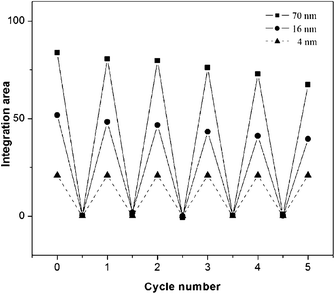 | ||
| Fig. 1 Salt-induced aggregation of different sized, zwitterion-modified Au NPs and redispersion of the aggregates upon dilution. For the 4 nm Au NPs, they cannot be induced to aggregate by salt so that dashed curve is used for clear differentiation. | ||
Particle separation involves modification of the particle mixtures with the zwitterion disulfide and tuning salt concentration in between the CCCs of different sized Au NPs. Due to their lower stability, the larger particles selectively aggregate, and can be easily separated from the smaller ones. Fig. 2a and 2b show the time-dependent UV-vis spectra of 4/16 nm and 16/70 nm binary particle mixtures upon the addition of 0.5 and 0.09 mol dm−3 of NaCl, respectively. For both binary mixtures, a new broad peak appears in the long wavelength range. This peak gradually red-shifts, decreases in absorption intensity, and disappears as the standing time of the mixtures is prolonged. These time-dependent UV-vis spectral sequences vividly show the nucleation, growth, and sedimentation of particle aggregates. In the end, particle separation was accomplished either by standing the mixtures for a long time (i.e. 12 h), or, more quickly, by low-speed centrifuging after standing the mixtures for a short time (i.e. 3 h). The supernatants containing the smaller Au NPs were collected through a syringe. With the concentrated aggregates at the bottom, when needed, they were further centrifuged twice in the NaCl solution of the same concentration that initiated the aggregation. This step removed residual supernatants. The final separated Au NPs dispersions show distinct differences in surface plasmon position and colour (Fig. S4a and S4b, ESI†). Fig. 3a–c and 3d–f show the TEM images of the two binary particle mixtures before and after separation, respectively. As is seen, the separated larger Au NPs contain only < 10% of smaller particles, illustrating good efficiency of the separation.
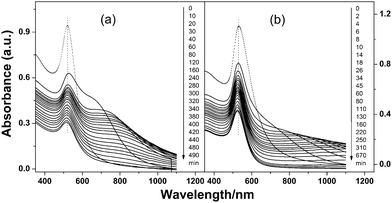 | ||
Fig. 2 Time-dependent UV-vis spectra of zwitterion-modified binary Au NP mixtures (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, in volume): (a) 4/16 and (b) 16/70 nm binary mixtures upon the addition of 0.5 and 0.09 mol dm−3 NaCl, respectively. :1, in volume): (a) 4/16 and (b) 16/70 nm binary mixtures upon the addition of 0.5 and 0.09 mol dm−3 NaCl, respectively. | ||
 | ||
| Fig. 3 Representative TEM images of zwitterion-modified binary Au NP mixtures before and after size-selective separation: (a) 4/16 nm binary Au NP mixtures and separated (b) 4 and (c) 16 nm Au NPs; (d) 16/70 nm binary Au NP mixtures and separated (e) 16 and (f) 70 nm Au NPs. | ||
To further demonstrate the separation, we also tried separating ternary mixtures. Fig. 4a and 4b show the time-dependent UV-vis spectra of 4/16/70 nm ternary particle mixtures upon the addition of 0.09 and then 0.5 mol dm−3 NaCl. Apart from a slight decrease in absorption intensity due to dilution, the spectral sequence is very similar to that shown in Fig. 2. Similarly, the final separated Au NPs dispersions show distinct differences in surface plasmon position and colour (Fig. S4c, ESI†). Therefore, separation of 4, 16 and 70 nm Au NPs was possible, as is further confirmed by the TEM images shown in Fig. 5.
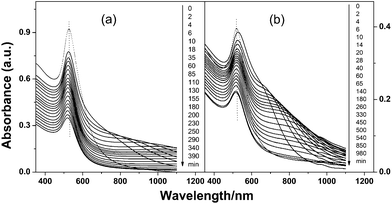 | ||
| Fig. 4 Time-dependent UV-vis spectra of zwitterion-modified 4/16/70 nm Au NP ternary mixtures upon the addition of (a) 0.09 and then (b) 0.5 mol dm−3 NaCl. | ||
 | ||
| Fig. 5 Representative TEM images of a zwitterion-modified ternary Au NP mixture before and after size-selective separation: (a) 4/16/70 nm ternary Au NP mixtures and separated (b) 4, (c) 16, and (d) 70 nm Au NPs. | ||
We noted that the original and the separated zwitterion-modified Au NPs both show highly reversible aggregation properties. The salt-induced aggregates can easily be redispersed in water just by shaking by hand. Usually, concentration involves energy-consuming evaporation. The high stability and reversible aggregation of the zwitterion-modified Au NPs, in contrast, allow for an energy-saving, salt-induced concentration strategy. The dilute dispersions can be concentrated to a wanted concentration just by adding salt into the dilute dispersions and then separating some water. When needed, the concentrated dispersions can be recovered into dilute dispersions by diluting and shaking. This would largely facilitate the storage, transportation, and endpoint applications of the NPs. In one experiment, we stored the 16 and 70 nm Au NPs as aggregates in salt solutions for more than 2 months without losing this reversible aggregation property.
In conclusion, zwitterion-modified Au NPs show interesting, size-dependent CCC and reversible aggregation properties, which allow for the salt-induced size-selective separation of Au NPs and concentration of dilute Au NPs dispersions. Au NPs in concentrated dispersions can be preserved long-term, and once needed, can subsequently be recovered into well-dispersed dilute dispersions without losing the properties of single, isolated particles. The results of this study, therefore, will have significance in the fractionation, storage, and transport of NPs for commercial applications.
Acknowledgements
This study was financially supported by NSFC (20603008, 20873037, and J0830415) and Open Projects of State Key Laboratory of Supramolecular Structure and Materials (Jilin University) (SKLSSM200902).References
- M. A. El-Sayed, Acc. Chem. Res., 2001, 34, 257–264 CrossRef CAS.
- J. C. Liu, F. He, T. M. Gumn, D. Y. Zhao and C. B. Roberts, Langmuir, 2009, 25, 7116–7128 CrossRef CAS.
- T. S. Ahmadi, Z. L. Wang, T. C. Green, A. Henglein and M. A. El-Sayed, Science, 1996, 272, 1924–1926 CAS.
- (a) A. Akthakul, A. I. Hochbaum, F. Stellacci and A. M. Mayes, Adv. Mater., 2005, 17, 532–535 CrossRef CAS; (b) S. F. Sweeney, G. H. Woehrle and J. E. Hutchison, J. Am. Chem. Soc., 2006, 128, 3190–3197 CrossRef CAS.
- C. T. Yavuz, J. T. Mayo, W. W. Yu, A. Prakash, J. C. Falkner, S. Yean, L. L. Cong, H. J. Shipley, A. Kan, M. Tomson, D. Natelson and V. L. Colvin, Science, 2006, 314, 964–966 CrossRef.
- (a) M. Hanauer, S. Pierrat, I. Zins, A. Lotz and C. Sönnichsen, Nano Lett., 2007, 7, 2881–2885 CrossRef CAS; (b) F. K. Liu, F. H. Ko, P. W. Huang, C. H. Wu and T. C. Chu, J. Chromatogr., A, 2005, 1062, 139–145 CrossRef CAS; (c) I. Arnaud, J. P. Abid, C. Roussel and H. H. Girault, Chem. Commun., 2005, 787–788 RSC.
- (a) L. Bai, X. J. Ma, J. F. Liu, X. M. Sun, D. Y. Zhao and D. G. Evans, J. Am. Chem. Soc., 2010, 132, 2333–2337 CrossRef CAS; (b) X. M. Sun, S. M. Tabakman, W. S. Seo, L. Zhang, G. Y. Zhang, S. Sherlock, L. Bai and H. J. Dai, Angew. Chem., Int. Ed., 2009, 48, 939–942 CrossRef CAS.
- (a) J. S. Lee, S. I. Stoeva and C. A. Mirkin, J. Am. Chem. Soc., 2006, 128, 8899–8903 CrossRef CAS; (b) J. S. Lee, D. S. Seferos, D. A. Giljohann and C. A. Mirkin, J. Am. Chem. Soc., 2008, 130, 5430–5431 CrossRef CAS.
- C. B. Murray, D. J. Norris and M. G. Bawendi, J. Am. Chem. Soc., 1993, 115, 8706–8715 CrossRef CAS.
- M. C. McLeod, M. Anand, C. L. Kitchens and C. B. Roberts, Nano Lett., 2005, 5, 461–465 CrossRef CAS.
- C. L. Wang, M. Fang, S. H. Xu and Y. P. Cui, Langmuir, 2010, 26, 633–638 CrossRef CAS.
- W. T. Zhao, L. Lin and I.-M. Hsing, Langmuir, 2010, 26, 7405–7409 CrossRef CAS.
- Y. H. Zheng, C. H. Lalander and U. Bach, Chem. Commun., 2010, 46, 7963–7965 RSC.
- P. C. Hiemenz and R. Rajagopalan, Principles of Colloid and Surface Chemistry, Marcel Dekker, New York, 3rd edn, 1997 Search PubMed.
- L. L. Rouhana, J. A. Jaber and J. B. Schlenoff, Langmuir, 2007, 23, 12799–12801 CrossRef CAS.
- M. Manciu and E. Ruckenstein, Langmuir, 2001, 17, 7061–7070 CrossRef CAS.
- (a) Q. Jin, J. P. Xu, J. Ji and J. C. Shen, Chem. Commun., 2008, 3058–3060 RSC; (b) X. S. Liu, H. Y. Huang, Q. Jin and J. Ji, Langmuir, 2011, 27, 5242–5251 CrossRef CAS; (c) K. Matsuura, K. Ohno, S. Kagaya and H. Kitano, Macromol. Chem. Phys., 2007, 208, 862–873 CrossRef CAS; (d) R. Tatumi and H. Fujihara, Chem. Commun., 2005, 83–85 RSC.
- (a) S. F. Chen, J. Zheng, L. Y. Li and S. Y. Jiang, J. Am. Chem. Soc., 2005, 127, 14473–14478 CrossRef CAS; (b) Z. G. Estephan, P. S. Schlenoff and J. B. Schlenoff, Langmuir, 2011, 27, 6794–6800 CAS.
- A. M. Smith and S. M. Nie, J. Am. Chem. Soc., 2008, 130, 11278–11279 CrossRef CAS.
- R. T. Tom, V. Suryanarayanan, P. G. Reddy, S. Baskaran and T. Pradeep, Langmuir, 2004, 20, 1909–1914 CrossRef CAS.
- G. Frens, Nat. Phys. Sci., 1973, 241, 20–22 CAS.
- J. F. Zhou, D. A. Beattie, J. Ralston and R. Sedev, Langmuir, 2007, 23, 12096–12103 CrossRef CAS.
- W. T. Zhao, T. M. H. Lee, S. S. Y. Leung and I.-M. Hsing, Langmuir, 2007, 23, 7143–7147 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental details, and supporting figures. See DOI: 10.1039/c2ra00828a |
| This journal is © The Royal Society of Chemistry 2012 |