Methods and options for the heterologous production of complex natural products
Haoran
Zhang
,
Brett A.
Boghigian
,
John
Armando
and
Blaine A.
Pfeifer
*
Department of Chemical & Biological Engineering, Science & Technology Center, Tufts University, 4 Colby Street, Medford, MA 02155, USA. E-mail: blaine.pfeifer@tufts.edu
First published on 9th November 2010
Abstract
Covering: 1990 to 2010
Heterologous biosynthesis has emerged as a viable route to access the beneficial properties of natural products. This development primarily owes to the difficulties encountered when using traditional methods of natural product discovery and production. However, the process of heterologous biosynthesis also presents a number of challenges that have produced an array of creative solutions and noteworthy success stories. In this review, a historical perspective will be presented, together with an analysis of the experimental approaches thus far used to address the unique issues associated with heterologous natural product biosynthesis.
 Haoran Zhang | Haoran Zhang is completing his doctorate at Tufts University after having received his B.S. and M.S. degrees from Xiamen University. His research at Tufts has focused on establishing and optimizing the biosynthesis of erythromycin A from E. coli. He has also made efforts to heterologously produce marine natural products using E. coli as a surrogate host. |
 Brett Boghigian | Brett Boghigian is completing his doctoral research at Tufts University after having completed his B.S. and M.S., also at Tufts. His research has focused on applying metabolic and process engineering to the heterologous production of complex natural products. Specifically, he has combined computational and experimental approaches to rationally improving the heterologous production titers of both polyketide and isoprenoid natural products using E. coli. |
 John Armando | John Armando is completing a dual B.S/M.S. degree at Tufts in chemical and biological engineering. His research is focused on further expanding the metabolic capabilities of heterologous host systems to better accommodate complex natural product pathways. |
 Blaine Pfeifer | Blaine Pfeifer received his bachelor's degree in 1997 from Colorado State University in chemical engineering. He pursued graduate work the following year under the direction of Chaitan Khosla at Stanford University. His doctoral thesis focused upon the production of complex polyketide and nonribosomal peptide compounds using E. coli as a heterologous host. After receiving his PhD in 2002, and following a postdoctoral position at MIT, he began as an assistant professor at Tufts University in the chemical and biological engineering department. Since that time, his laboratory has continued to work on problems related to heterologous natural product biosynthesis, with particular focus on successful gene transfer to and metabolic engineering of the recipient hosts. |
1 Introduction
Natural products continue to fascinate both basic and applied scientists due to the complex biochemical pathways that give rise to them and their incredible therapeutic range. Natural products may be grouped into several classes; this review will focus on the polyketide, nonribosomal peptide, and isoprenoid groups. Fig. 1 highlights examples from these featured groups. The enzymology, biochemistry, and protein structural detail behind natural product biosynthesis have been the focus of numerous research programs. This interest stems from the array of unique biosynthetic steps used to produce equally diverse chemical architectures. Briefly, though there are different subdivisions of biosynthetic mechanisms, polyketide and nonribosomal peptide biosynthesis often features large enzymes (referred to as megasynthases or megasynthetases, depending on the biosynthetic context) characterized by a modular or multi-domain nature. Polyketide biosynthesis can be further subdivided into type I, type II, and type III systems, with the type I and II systems sharing similarities and nomenclature with fatty acid synthases. Isoprenoid biosynthesis features terpene synthases capable of cyclizing poly-isoprene units. For each natural product class to be featured in this review, biosynthesis may also include several tailoring transformations such as glycosylation, hydroxylation, methylation, halogenation, or reduction before the final natural compound is produced. Readers are directed to several recent reviews that further detail the biosynthetic processes underlying these natural product classes.1–9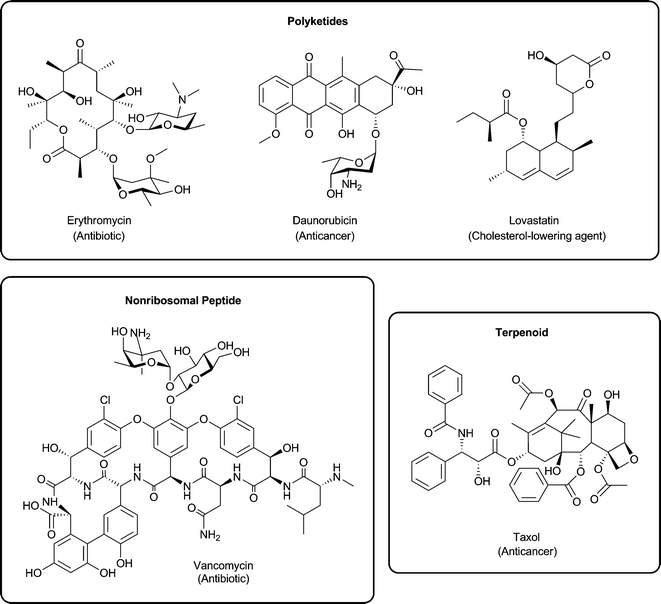 | ||
| Fig. 1 Established therapeutic natural products in three major classes: (a) polyketides are represented by erythromycin, daunorubicin, and lovastatin; (b) non-ribosomal peptides are represented by vancomycin; and (c) isoprenoids are represented by Taxol. | ||
Besides providing a rich environment for genetic- and biochemical-based research, the key attraction of the study of natural product biosynthesis is the impressive range of therapeutic value associated with the final compounds produced. This is partially captured in Fig. 1, but the compounds featured are only a fraction of those that have been instrumental in transforming modern medicine. For example, analysis of small molecule compounds introduced into the clinic from 1981–2006 indicates that 76% of antibacterials and 78% of anticancer agents were natural products or derived from natural products.10 Beyond the impact natural products have had in antibacterial and anticancer treatment, they have also found applications as anti-cholesterol, antimalarial, antiparasitic, and antifungal agents.
The therapeutic potential of natural products has also attracted the interest of applied scientists and engineers eager to access the associated medicinal value. This same interest was the basis for significant industrial investment in natural product programs. As a result, since the 1940s, a community of scientists and engineers has propelled efforts to understand and overproduce clinically-relevant natural products. This review will look at the emerging frontiers for natural product biosynthesis with a particular focus on heterologous biosynthesis. In particular, the review will detail the steps required to transfer the responsible genetic material to a given host and establish natural product formation (Fig. 2). Special emphasis will be placed on guiding methodology and current experimental tools available to complete this task. Several excellent reviews provide similar summaries of heterologous biosynthesis.11–14 Here, we intend to update and expand the analysis across additional natural product classes and host systems. However, to be somewhat succinct, the review has been constrained to a select number of hosts and natural product systems and excludes certain natural product types (such as flavonoids, stillbenes, alkaloids, carotenoids, and others) and hosts (such as plant systems, myxobacteria, and others). We direct the reader to other reviews featuring aspects of heterologous biosynthesis in relation to these compounds and hosts.15–23 In addition, although mentioned peripherally, the review will not comprehensively address the steps involved in natural product screening; compound chemical characterization; host isolation; gene cluster identification, sequencing, analysis, or synthesis; and the metabolic and process engineering needed to optimize heterologous production once achieved.
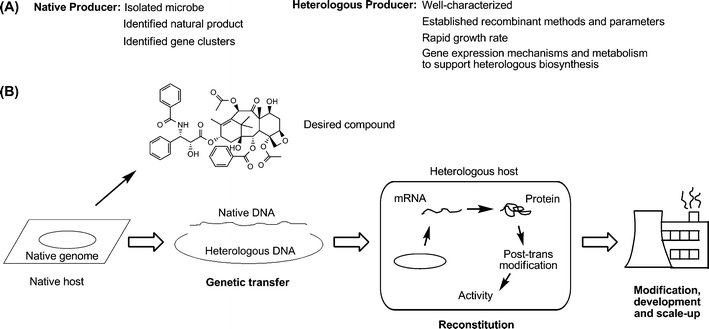 | ||
| Fig. 2 (a) A comparison of the typical characteristics between native and heterologous hosts. (b) A generalized workflow for establishing natural product biosynthesis in a heterologous host. First, a native host is identified as a producer of a natural product of interest. Second, the genes responsible for producing the product are identified, isolated, and integrated within a heterologous host whether it be through an artificial replicon or within the host's own chromosome(s). Third, expression of the heterologous genes is initiated, producing soluble protein and the product of interest. Last, this host and process are scaled up and/or further modified for large-scale production. | ||
2 Heterologous biosynthesis
The following sub-sections will present the components, motivation, and general experimental considerations for heterologous natural product biosynthesis. Specific examples and case studies will follow. To further focus the review, these cases will feature those efforts carried out from 1990 onward.2.1 Native hosts
Natural products are produced by environmental organisms that may be prokaryotic or eukaryotic in origin. Referencing Fig. 1, Taxol is produced by the Pacific Yew tree, Taxus brevifolia.24 Erythromycin is produced by the soil bacterium Saccharopolyspora erythraea.25 Similarly, daunorubicin and vancomycin derive from the actinomycetes Streptomyces peucetius and Amycolatopsis orientalis (formerly Streptomyces orientalis), respectively.26,27 This sampling highlights traditional environmental sources for medicinal natural products. In particular, the soil-dwelling actinomycetes are renowned for their output of therapeutically useful natural products. It has been estimated that ∼3,000 antibiotics have been identified from the actinomycetes and 90% of those from the Streptomyces genus.28Starting from the earliest isolated natural products, there was practical interest in extending their medicinal properties for widespread use. The most famous example here is penicillin.29 Spurred in large part by the need to treat the injured of World War II, the successful mass production of penicillin was used as a blueprint for similar efforts to capitalize upon the therapeutic properties of other natural products as they emerged. This process began with natural product discovery which usually involved an extensive effort to screen environmental samples for a desired bioactivity. Because antibacterial assays were (and still are) simple to implement, this class of natural products emerged first. As other screens or assays developed, additional bioactivities were also identified. Many times, these activities were found in addition to the antibiotic properties of a given natural product. Once a promising natural product and its accompanying host were identified, there was a need to scale production for eventual application. It could be claimed that this need was an early harbinger of the heterologous biosynthesis concept, because the native producers of many therapeutically-relevant natural products were not process-friendly. For example, native hosts typically exhibited slow growth rates, fastidious nutrient requirements, and low production titers that made mass production challenging from a time and cost perspective. Nonetheless, and perhaps due to the limited molecular biology protocols at this point in time, process development efforts were built around the native host. In parallel, strain improvement programs were dedicated to increasing titers from the native producer, primarily relying on mutagenesis and screening.30 The result was a gradual improvement in natural product production, and the success associated with penicillin and other early compounds ushered in a “golden age” (ranging from approximately 1940–1960) for natural products that featured both industrial and academic commitment to the field.
However, over time the relative ease of natural product discover began to diminish.31,32 In particular, it became more difficult to isolate new natural products possessing new forms of bioactivity.33 The situation spurred a number of changes in academic and industrial perspectives. From an industrial viewpoint, the lack of readily available new natural products, coupled with the substantial time, effort, and cost associated with environmental screens, provided a significant obstacle in terms of justifying continual research in this direction.34–37 At the same time, the continual use of already existing antibacterial natural products was inevitably perpetuating resistance mechanisms in the target bacterial populations.38–40 Therefore, from a global perspective, there is a continual need for new natural products, yet there is waning interest to initiate this search from industry. However, there are still both basic and applied reasons for continuing the search for new natural products, and though several strategies have now been pursued towards this goal, one of the simplest approaches is to expand the search for new compounds.
A recent frontier in the search for new natural products has been the marine environment.38,41–46 For example, the marine actinomycete Salinispora tropica has received particular interest, given that approximately 10% of its >5 Mb genome is dedicated to secondary metabolism and because a particularly notable metabolite generated by this species is the potent anticancer hybrid nonribosomal peptide-polyketide salinosporamide A.47–49 The cited reviews summarize and testify to the potential of this environment for the isolation of structurally and medicinally diverse new natural products. With this search comes a range of new native host systems that offer even more challenges regarding process scale and development. In many cases, compounds have been isolated from higher-order organisms (ascidians, sponges, mollusks, etc.) that may or may not involve the symbiotic involvement of microbial producers.50 Further, the references above have pointed to the need to include growth medium components which help to replicate the marine environment, highlighting the familiar challenge of satisfying the fastidious nature of many natural product producers.
Although there are several reasons for the emergence of heterologous natural product biosynthesis, the challenges detailed above provided much of the initial impetus. The prospect of producing a medicinally-relevant natural product through a host that offers technical convenience with regard to process development was considered an ideal situation for accessing the potential of natural products. Of course, as will be detailed below, this goal is not without its own challenges, but the promise and potential of natural products provide the motivation towards overcoming these concerns.
2.2 Choosing a heterologous host
Before different options are presented, this sub-section will provide heuristics in choosing a heterologous host. Natural product discovery efforts may identify a native host system that is incapable of providing large-scale access to the medicinal value of the associated natural product. This or a similar motivation would spur an interest in heterologous biosynthesis. Regardless of the reason, once a situation has been established that merits heterologous biosynthesis, the first choice is which heterologous system to use. Often, the decision comes down to technical convenience. Here, organisms like E. coli are at a great advantage simply because they are well-characterized and simple to culture.To begin, one must also recognize the origin, type, and complexity associated with the natural product of interest. First, it is important to consider the native host system and the associated biology. Host-specific biology then serves as the first heuristic in choosing a heterologous host. For example, a natural product derived from a eukaryotic system will most likely benefit from reconstituted production within a heterologous eukaryotic host with the logic that the similarities in gene expression and cellular environment would better suit the natural product pathway to be reconstituted. The same logic could be applied to prokaryotic systems where the differences between Gram-positive and Gram-negative native and heterologous hosts may affect eventual heterologous production. Second, one needs to assess the availability of genetic transfer techniques associated with the chosen heterologous host. Although numerous methods and protocols have been developed to facilitate the introduction of foreign DNA into heterologous systems, host-specific technical barriers still exist especially given the unique features common to natural product gene clusters.
Finally, besides biological similarities between the native and heterologous hosts and readily available genetic transfer protocols as factors for heterologous host selection, it is important to understand what is known about the candidate heterologous host's native metabolism. If the candidate heterologous host already possesses the ability to produce a natural product, there then exists a level of confidence that the chosen heterologous host will be able to support the metabolic requirements of a foreign natural product biosynthetic scheme. In addition, many common heterologous hosts have now been sequenced, and this information also provides clues about the native capabilities of heterologous biosynthetic support. At the same time, an overabundance of natural product capabilities within a heterologous host may provide unwanted “crosstalk” between the pathway of interest and those native to the heterologous host, such that biosynthetic precursors are sub-optimally supplied to the desired heterologous pathway or, alternatively, natively produced compounds complicate heterologous product analysis. These criteria will be further emphasized in the specific examples below used to illustrate the attempts and successes with heterologous natural product biosynthesis.
2.3 Early heterologous host systems
We will now provide additional motivations for heterologous biosynthesis and the initial candidates for heterologous hosts that were identified. Traditional discovery programs, if successful, would identify a host system producing a therapeutic natural product. As mentioned earlier, while successful in narrowing the search to the exact producer of a valuable compound, the situation was often difficult to then economically improve or scale commercial production of the natural compound. One solution to this dilemma is to establish a universal host as the heterologous system for multiple newly discovered natural products. In other words, a universal, well-characterized heterologous host would support the process development opportunities that were difficult to achieve with native production hosts. To accomplish this, there would need to be two components: a well-characterized host system and knowledge of the genetic material required for heterologous biosynthesis.Because the actinomycetes (and particularly, the Streptomyces genus) were recognized as a prolific source of therapeutic compounds, early heterologous host choices included Streptomyces coelicolor and Streptomyces lividans. Largely through the efforts of Sir David Hopwood and colleagues at the John Innes Centre, these hosts had been extensively characterized, and this effort included the development of a set of recombinant DNA techniques to facilitate the steps needed for eventual heterologous gene transfer. It is of note that many of the technical and scientific advances associated with heterologous natural product biosynthesis have coincided with general advances in molecular biology protocols and recombinant DNA technology. In the case of heterologous natural product biosynthesis, the emerging recombinant DNA technology no doubt helped establish S. coelicolor as a potential heterologous host. These same technical developments also contributed to the second requirement for heterologous biosynthesis. Namely, as DNA identification and sequencing technology progressed, it became increasingly technically feasible to identify the gene sequences responsible for natural product biosynthesis. Fortuitously, many natural product pathways were found clustered genetically within the chromosomes of native producers. This is particularly true for polyketide and nonribosomal peptide products, and the clusters typically feature genes responsible for polyketide or nonribosomal peptide biosynthesis, product tailoring (glycosylation biosynthesis and transfer, etc.), regulation, and self-resistance (i.e., antibiotic protection). The availability of recombinant protocols and gene sequence information for early natural product clusters allowed Streptomyces-based hosts to emerge as early candidates for heterologous biosynthesis.
There is an additional subtlety associated with the choice of Streptomyces host systems and their ability to support heterologous natural product biosynthesis. This distinction particularly applied to polyketide natural products traditionally associated with actinomycetes. However, as introduced above, the analogy also extends to other types of natural product classes. Specifically, the new host must also supply the newly-introduced biosynthetic pathway with the needed intracellular substrates required for final product formation. As such, the requirements for a heterologous host must now be amended. For successful heterologous biosynthesis, one must have available the sequence information and/or genetic material for a particular natural product and a well-characterized host with (1) associated recombinant DNA tools and (2) the metabolic capability to support the biosynthetic process.
In principle, successful transfer and reconstituted biosynthesis would then eliminate the need to develop a new production process for every new natural product. Instead, the knowledge associated with the new host would allow more consistency in process development. Through the course of gene cluster identification and more basic biochemical analysis, models for natural product biosynthesis began to emerge. As detailed briefly above, these models vary depending upon the natural product class in question, but in regard to the technical steps of natural product heterologous biosynthesis, details of the biosynthetic process have repercussions regarding the relative ease in reconstituting biosynthesis. As an example, polyketide and nonribosomal peptide biosynthesis is often associated with megasynthase enzymes that can easily surpass 300 kDa in size and require post-translational modification. Moreover, these enzymes can associate into higher-order super structures which often exceed 1 MDa in size. In addition, biosynthesis of the pharmacologically-active polyketide, nonribosomal peptide, or isoprenoid product may require the coordinated activity of several to >20 enzymes. As such, a heterologous host must be capable of accommodating both the individual gene size (especially for those encoding multi-domain megasynthases) and total gene number associated with eventual biosynthesis. Furthermore, as before, the host must support the metabolic- and enzymatic-specific requirements of the newly introduced pathway. Parenthetically, as details of complex natural product biosynthesis were determined, it became clear that final compound structure could be altered by modifying the genes/proteins responsible for biosynthesis. Hence, another rationale for heterologous biosynthesis was to exert a level of control over the biosynthetic process for the purpose of diversifying natural product structure through use of the molecular biology tools available to the new host.51,52
2.4 Emerging hosts and the accompanying molecular biology tools
Though Streptomyces strains were good first options for heterologous polyketide biosynthesis, other hosts offered even more potential. This was primarily because of the relatively slow growth kinetics associated with Streptomyces strains when compared to workhorse bacterial hosts like E. coli. The same rapid growth rate that made E. coli an attractive heterologous host option also led to an incredible number of experimental techniques that ranged from molecular biology protocols to scaled bioreactor processes for biological product manufacture. As a result, organisms such as E. coli, B. subtilis, and S. cerevisiae provided key elements of an ideal heterologous host: they were all well-characterized and detailed protocols were available to aid genetic transfer, product formation, and process development. However, a key drawback to these new host systems was the question of viable support for complex natural product formation. Could these convenient hosts allow the production of active megasynthase protein complexes? Could these hosts provide the needed metabolic precursors to support biosynthesis? Could they accommodate the often sizable number of foreign genes needed for biosynthesis? These and other questions needed to be addressed before successful heterologous biosynthesis could be achieved.However, one cannot overlook the advances in molecular biology and gene expression that have emerged with the use of these new hosts. The number of total and successful attempts at heterologous biosynthesis through these new hosts is growing, thanks in part to the increasing number of gene expression parameters (promoters, operators, plasmids, and strains) available to answer some of the questions raised in the last paragraph. In addition, the speed at which DNA can be sequenced and synthesized is further expediting heterologous biosynthetic attempts, first, by rapidly identifying promising gene clusters of interest and, second, by allowing custom genetic content design for subsequent transfer to the host of choice. Such tools are intrinsically tied to well-characterized strains, and this, in part, can be traced back to the simple advantage these new hosts possess in the form of rapid growth kinetics. As a result, the previously daunting challenges raised in the last paragraph are becoming more manageable as the genetic and metabolic engineering tools continue to improve with these new host options.
Before specific examples are presented, an overview of common gene transfer approaches will be provided. General comments will be presented regarding the options available for facilitating genetic cluster transfer between native and heterologous host systems. At the outset, every heterologous biosynthesis attempt is presented with the challenge of genetic transfer. The complexity behind natural product pathways can result in two primary obstacles to this step: 1) the number of genes needed for complete biosynthesis and 2) the size of the individual genes within this total number. These constraints complicate common molecular biology strategies used for heterologous gene transfer and have emphasized the need for alternative approaches and strategies.
As mentioned, genetic information (through genetic pathway identification and DNA sequencing and synthesis improvements) has provided a number of relevant natural product pathways with many more anticipated in the future. For example, sequencing of Saccharopolyspora erythraea and Streptomyces avermitilis revealed the expected gene clusters for erythromycin and avermectin in addition to >20 other clusters (per organism) putatively capable of producing secondary metabolite products.53,54 Once identified, a plan must be formulated for genetic transfer to a heterologous host. Traditional molecular biology protocols would suggest cloning or expression plasmids be used for this purpose. However, when faced with the prospect of transferring individual genes >10 kb or total genetic material >50 kb in size, common steps (ligations, transformations, etc.) or plasmid stability may become problematic. As such, one must consider non-traditional uses of familiar plasmids or altogether different options.
The dilemma of stably cloning, transferring, and maintaining large genes or gene clusters has previously been addressed through the use of specialized plasmids capable of harboring large DNA fragments. Simpler options include cosmid or fosmid vectors.55,56 In some cases, conventional cloning vectors have been redesigned to stably maintain up to 300 kb of foreign DNA.57 Alternatively, P1-derived artificial chromosomes (PACs), bacterial artificial chromosomes (BACs), or yeast artificial chromosomes (YACs) have demonstrated the capability of stably maintaining 100–300 kb genetic inserts, sizes similar to many natural product gene cluster lengths.58–61 Many of these vectors have been developed for E. coli; however, they have also seen utility within other heterologous biosynthetic efforts and will be highlighted, where appropriate, in the specific examples below.
Another option for harboring natural product gene clusters within a heterologous host is within the host's own chromosome. Both well-characterized and sequenced prokaryotic systems and more complicated eukaryotic hosts have been used in this context. This alternative would appear to present a stable final location for heterologous natural product gene clusters; however, regardless of the final option, challenges in gene transfer and localization will accompany the large gene clusters associated with most complex natural products.
The final plasmid or chromosomal locations must be accompanied by tools to enact genetic transfer. Here again, the starting point is the traditional in vitro methodology associated with standard molecular biology. These tools have been continually refined since their introduction in the mid-1970s and advances now allow for rapid manipulation of DNA, including impressive steps in PCR amplification that allow the direct isolation of genes >10 kb and a range of mutational techniques. There have also been a number of systems developed for plasmid-based insertion into the chromosomes of target heterologous hosts.62–65 These are just a few examples of the methods available, and others will be featured in specific cases below. In addition, two powerful advances that are expected to play a significant role in the design and transfer of natural product gene clusters to heterologous hosts include the capability to synthesize specific genes and the use of PCR-based recombination techniques. Efficient and inexpensive gene synthesis is now allowing the directed production of genes and DNA elements of impressive length, perhaps best highlighted by work at the J. Craig Venter Institute on reconstructing the Mycoplasma mycoides genome.66 Supporting these efforts are specialty academic institutes and private companies capable of supplying rapidly sequenced or precisely-designed synthesized DNA to customers. The situation allows one to quickly move from natural product native host to genetic material for heterologous transfer while simultaneously allowing for a higher degree of engineering in gene cluster design. Likewise, precise recombination techniques have offered new options in site-specific transfer of heterologous gene clusters.67,68 These and other methods will now be presented in the context of the various natural product classes and heterologous host systems and within case-specific examples.
3 Heterologous production of polyketides
3.1 Streptomyces coelicolor
Tetracycline and erythromycin were early examples of polyketides successfully developed as therapeutic products. This precedent helped establish an initiative for continual discovery and development of new polyketides. Heterologous biosynthesis also emerged, with S. coelicolor established as an early surrogate host platform. Hopwood, Cohen, and coworkers published a series of papers in the late 1970s and early 1980s that introduced recombinant tools associated with S. coelicolor (in addition to other Streptomyces spp. such as S. lividans), thus, setting the stage for the bacterium to serve as a viable heterologous host for polyketide biosynthesis.69–71 This development coincided with methods to isolate, decipher, and manipulate emerging polyketide gene clusters for eventual heterologous gene transfer.72,73 Hence, the situation allowed the experimental testing of heterologous biosynthesis: genetic material was available and a well-characterized host had emerged as an early expression/production platform. S. coelicolor naturally produces the polyketide actinorhodin (in addition to several other polyketide products), and, as a result, there was reason to believe that the host would metabolically support the production of heterologous polyketide compounds. Since 1990, there have been numerous examples of S. coelicolor used in heterologous polyketide biosynthesis, both for the purpose of understanding particular biosynthetic pathways and for the purpose of generating established or new therapeutic compounds.By 1990, protocols existed for heterologous transfer of foreign genes to S. coelicolor. Of particular utility was a method reliant upon protoplast transformation, although methods for conjugation also emerged.74–76 In addition, several plasmids were developed for use with the protoplast transformation method.77–79 These plasmids derived from those found naturally within S. coelicolor or S. lividans and have been modified over time to allow a finer level of control when used in heterologous expression attempts. As a result, newly deduced polyketide natural product gene clusters could now be readily packaged and transferred to a well-characterized S. coelicolor heterologous host.
Heterologous transfer would then allow the expanding tools and knowledge available with S. coelicolor to aid in understanding or accessing the desired natural product. However, it should be mentioned that most transfer attempts were from other Streptomyces hosts. As such, S. coelicolor was at an intrinsic advantage for accepting similar GC-rich genetic material (and most likely accounting for any potential codon bias), supporting gene expression and post-translational processes, and metabolically supplying the required substrates (primarily acyl-CoA compounds) for biosynthesis. Considering the enormous number of biologically active polyketides derived from Streptomyces, choosing a member of this family as an early heterologous host was only logical.
The last two decades have seen a steady output of polyketides using S. coelicolor as a heterologous host. These examples have fully utilized and expanded upon the recombinant tools available for heterologous production. Efforts have relied on either cosmids or expression vectors for gene transfer (with the former case taking advantage of the similarities in gene expression between the native and S. coelicolor hosts; this was particularly apparent in an example in which heterologous biosynthesis was observed for S. coelicolor but not S. lividans, presumably because the original host shared a closer gene expression relationship to S. coelicolor80). Both low- and high-copy plasmids have been utilized together with actI, mel (constitutive), aphI (constitutive), ermE (constitutive), tipA (thiostrepton-inducible), PnitA, gylP1/P2 (glycerol-inducible), and tcp830 (tetracycline-inducible) promoter systems.81–98 To reduce potential intra- or inter-plasmid gene recombination events (especially for large PKS or NRPS genes containing homology sequences), codon optimization and alternative gene subunit complementation strategies were successfully used for S. coelicolor.99 Further means to influence gene dosage and transcript levels included the use of a helper plasmid capable of increasing copy levels of the accompanying expression plasmid and specific integrative designs that often utilized the ΦC31 phage recognition capabilities.100–104 Integrative methods have also been championed based upon potential biosynthetic inhibitory properties of self-replicating plasmids; these plasmids may also require antibiotic pressure throughout process development, a prospect that becomes prohibitively costly at large scale for any host utilizing selectable plasmids.105 Also, owing to the often sizable gene clusters associated with natural product biosynthetic pathways, BAC plasmids have been designed for pathway capture and heterologous transfer.106–109 A recent study extended earlier work by using a copy-up BAC plasmid to aid in pathway construction and conjugal integration using the less common (but compatible with ΦC31) ΦBT1 attB site.65,110,111 As a result, an impressive number of heterologous polyketide products have been produced including 6-deoxyerythronolide B, granaticin, epothilones A and B, medermycin, 6-methylsalicylic acid, novobiocin, and several type II polyketide products.112–120 Additional details will be included within the erythromycin case study presented below.
3.2 Streptomyces lividans
S. lividans and S. coelicolor have been the most common choices as hosts for heterologous polyketide biosynthesis (although there are many other Streptomyces hosts that we attempt to summarize in Fig. 6; some of these possess case-specific advantages that would suggest their use in a particular heterologous biosynthetic effort121). Many of the same protocols, plasmids, and recombinant options developed for S. coelicolor are applicable to S. lividans. However, in addition to integrative vectors based upon ΦC31, a separate integrative plasmid, known as pSAM2, is available for use in S. lividans.122–124S. lividans also exhibits certain technical characteristics that are advantageous when compared to S. coelicolor. Namely, unlike S. coelicolor, S. lividans does not require the use of unmethylated DNA during transformation protocols and has been shown to provide higher numbers of transformants when compared to popular S. coelicolor strains.125 Recently, the popular T7 RNA polymerase-promoter system has been implemented in S. lividans (with similar potential to be introduced to S. coelicolor) and used to generate 6-methylsalicylic acid.126–128 As with S. coelicolor, many successful examples which feature S. lividans as a heterologous host arise from the transfer of genetic material from other Streptomyces hosts, often genomic DNA packaged into cosmids. In addition, certain methods, such as tra gene-mediated conjugal transfer, have been established to transfer chromosomal gene clusters from other Streptomyces spp. to S. lividans.129 These examples again indicate the native capabilities of S. lividans to support heterologous biosynthesis and the close relationship between S. lividans and other Streptomyces bacteria.3.3 Escherichia coli
When compared to S. coelicolor or S. lividans, E. coli offers unquestioned advantages in terms of growth kinetics and recombinant tools to aid in heterologous polyketide biosynthetic efforts. In fact, it would be difficult to succinctly profile all of the molecular biology methods available for E. coli (or B. subtilis and S. cerevisiae). Instead, the tools that have been used so far will be highlighted through examples presented here and in this section's case study.Given the critical role E. coli played in establishing modern molecular biology protocols, there is reason to believe that researchers considered E. coli as an early potential candidate host in natural product heterologous biosynthesis. However, there are of course marked differences in cell morphology, physiology, metabolism, and regulatory mechanisms between E. coli and the actinomycetes responsible for the majority of polyketide compounds. As such, any advantages E. coli possessed relative to host options like S. coelicolor or S. lividans were quickly diminished when viewed from this more global perspective.
Nonetheless, the allure of establishing heterologous polyketide production in E. coli was strong enough to attract leaders in the polyketide field. An early success was the production of 6-methylsalicylic acid (6-MSA) from an iterative type I polyketide synthase originally from Penicillium patulum.130 In this case, a T5 promoter was used to drive expression of the 6-methylsalicylic acid synthase (6-MSAS) gene under the control of an inducible lac operator. Highlighting some of the challenges associated with the E. coli system, the native holo-acyl carrier protein synthase was unable to correctly post-translationally modify the 6-MSAS. In addition, 6-MSAS insolubility was observed resulting in associated inactivity. The first problem was solved by the inclusion (on a separately selectable and compatible plasmid) of a recently available promiscuous phosphopantetheinyl transferase termed Sfp from B. subtilis.131,132 The second problem was partially addressed by lowering the post-induction culture temperature to 30 °C. It was suspected that problems of protein folding were due to the E. coli bacterial environment used to express a native eukaryotic gene and because of the large (190 kDa) multidomain nature of 6-MSAS.
Spurred by this early effort, more ambitious attempts at E. coli-derived polyketide production began. The first successful production of a modular type I polyketide product was completed in 2001 (to be profiled below).133 This work resulted in a new E. coli production host (termed BAP1) capable of post-translationally modifying both polyketide synthase (PKS) acyl carrier protein (ACP) and nonribosomal peptide synthetase (NRPS) peptidyl carrier protein (PCP) domains. It was later found that phosphopantetheinylation of polyketide synthases expressed in a BAP1 derivative reached a high level (>99%), further validating this strain for heterologous natural product biosynthesis.134 In addition, the host was engineered to supply propionyl-CoA and (2S)-methylmalonyl-CoA, common polyketide biosynthetic precursors that are uncommon E. coli metabolites. In the 6-MSA case above, the required precursors were acetyl- and malonyl-CoA, compounds known to exist within the framework of E. coli primary metabolism. These examples highlight both advantages and disadvantages of E. coli (or similar heterologous hosts generally devoid of natural product biosynthetic capabilities). Here, the lack of native polyketide products implies that E. coli does not have the metabolism to support more complex heterologous polyketide pathways. Alternatively, E. coli can be viewed as an attractive “clean host” which does not have any molecular bias. In other words, there were no native pathways to either contribute to or detract from targeted heterologous products. In addition, there was little concern of natural polyketide products complicating heterologous production analytical or purification efforts.
The clean host designation associated with E. coli also requires more consideration when designing heterologous production efforts. Unlike Streptomyces hosts, more thought is needed regarding gene design, expression parameters, and post-translational steps (including self-resistance to antibacterial products). As one example, the GC-rich genes associated with Streptomyces sources clearly differ from the codon usage associated with native gene expression. Alternatively, the E. coli expression options may simplify the regulatory aspects of heterologous polyketide production; however, there is also the concern of cellular burden, given the need to engineer so many different aspects of foreign polyketide biosynthesis. These points will be further explored in the context of this section's case study.
3.4 Fungal hosts
Fungal systems will now be introduced and profiled in the context of heterologous polyketide biosynthesis. Eukaryotic hosts such as Sacharomyces cerevisiae, Aspergillus niger, Aspergillus oryzae, and Aspergillus nidulans (with the latter three classified as filamentous fungi) hold an advantage when considering the heterologous production of compounds originating from eukaryotic sources. In particular, Aspergillus spp. have been identified as encoding for an impressive number of natural product clusters.135 As such, there has been renewed interest in developing these hosts as heterologous platforms.136–139 The remainder of this sub-section will be dedicated to examples from filamentous fungi, though S. cerevisiae has been used successfully in a couple of cases. Namely, both 6-MSA and a polyketide intermediate termed triketide lactone (from the 6-deoxyerythronolide B pathway to be profiled below) were produced through S. cerevisiae, with the latter example requiring significant effort to introduce the metabolic pathways needed to provide the propionyl- and (2S)-methylmalonyl-CoA substrates.130,140,141For filamentous fungi, there has been considerable effort placed on developing the recombinant tools associated with these hosts. Once isolated, DNA coding for a particular fungal polyketide product may need to be adjusted for heterologous expression. For example, in the case of 6-MSA production in S. coelicolor, the MSAS gene was altered to remove intron sequences and further tailored to aid S. coelicolor gene expression. Although these concerns may be mitigated when using similar filamentous fungi heterologous hosts, they must still be considered in addition to the common issues of providing the correct intracellular precursors, gene expression elements, and post-translational modification steps. Once prepared for transfer, polyketide-coding genetic material is usually introduced using protoplast fusion or electroporation transformation protocols, and plasmids used for transfer are more commonly integrated into the chromosome (especially when compared to the traditionally autonomous replicating systems associated with E. coli).142,143 Promoters used in heterologous production attempts include amyB (inducible by starch or maltose), trpC (constitutive for A. nidulans), and alcA (cyclopentanone-inducible).144–146 Successful examples include the production of 6-MSA, melanin, squalestatin, 3-methylorcinaldehyde, and monacolin J (a late-stage intermediate of lovastatin biosynthesis).147–150
As with the actinomycetes, many fungal systems, especially the Aspergillus genus, produce a variety of complex secondary metabolites, which may or may not be generated under typical growth conditions. For example, Aspgerillus niger contains a predicted 17 nonribosomal peptide coding regions, 34 polyketide coding regions, and 7 hybrid nonribosomal peptide-polyketide coding regions within its 33.9 Mb genome.151 The crosstalk and regulatory mechanisms within these potential hosts may play a significant role in determining successful heterologous biosynthetic attempts; however, emerging recombinant strategies may be used (similar to previous cases with Streptomyces hosts) to either eliminate or harness these conditions for biosynthetic success.
3.5 Case study: Erythromycin
The first case study will focus on heterologous erythromycin biosynthesis (Fig. 3). This case, in addition to those to be featured later, provides an excellent example of the thought processes that must accompany a heterologous biosynthetic effort. In 1952, erythromycin was isolated from a soil bacterium eventually designated Saccharopolyspora erythraea.25 Production processes were built around this organism and were steadily improved over the years to allow titers of at least 7 g/L.152 Interestingly, these titers were still roughly an order of magnitude lower than those of other classically optimized antibiotic bioprocesses such as penicillin (70 g/L) and salinomycin (60 g/L).153 Efforts to rapidly engineer the biosynthetic process were challenging, spurring development of the heterologous hosts and approaches that were described above. With the identification and sequencing of the gene cluster responsible for erythromycin,154–156 attempts began in earnest to heterologously produce this compound. The first host option was S. coelicolor.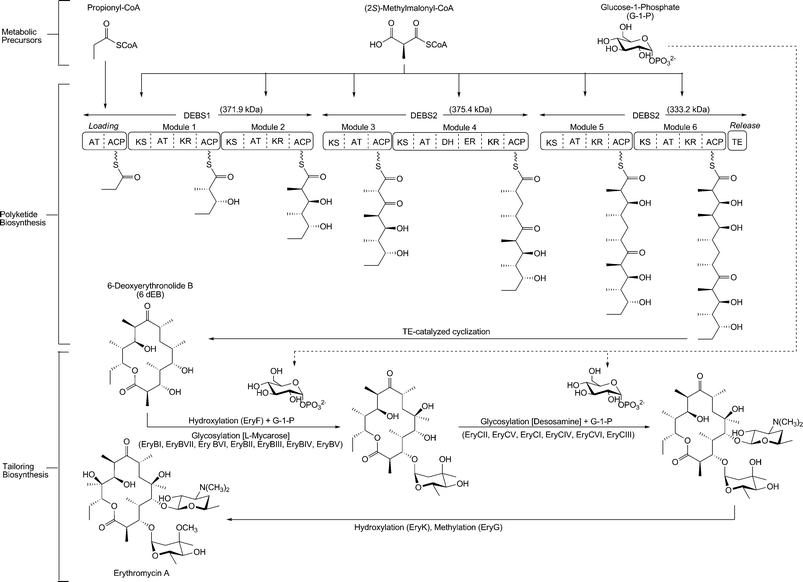 | ||
| Fig. 3 A schematic of erythromycin biosynthesis. A molecule of propionyl-CoA is primed on the loading domain of the DEBS1 enzyme. Polyketide formation requires six (2S)-methylmalonyl-CoA substrates and NADPH as a cofactor in the reductive steps on DEBS1, DEBS2, and DEBS3. The thioesterase domain of DEBS3 is responsible for cyclization and release of the polyketide from the PKS complex. Glucose-1-phosphate is used as a substrate for the EryB and EryC pathways to generate and attach the two sugars (L-mycarose and D-desosamine, respectively) to the aglycone 6dEB core. Abbreviations: AT = acyl transferase; ACP = acyl carrier protein; KS = ketosynthase; KR = ketoreductase; DH = dehydratase; ER = enoyl reductase; TE = thioesterase. | ||
At the time, S. coelicolor had gained momentum as a heterologous host for polyketide compounds. In particular, the strain CH999 developed by Khosla and coworkers had proven very effective in overproducing aromatic polyketide compounds.118 CH999 was designed based upon the actII-ORF4 regulatory mechanism (sourced from the actinorhodin cluster) that controlled gene expression from actI and actIII promoters during the growth transition to secondary metabolism; hence, production of heterologous compounds commenced in a semi-natural manner.157 As mentioned earlier, many polyketide clusters contain regulatory genes that are often implicitly included during heterologous biosynthetic efforts (i.e., through native cluster introduction using cosmid or BAC vectors). Other times, more explicit measures must be taken to express positive regulators that impact reconstituted biosynthesis.158 In the case of CH999, a regulation system had been considered when designing the recombinant strategy. This is also the case for non-Streptomyces heterologous hosts, as gene expression regulation is typically under the control of designed recombinant systems developed for the host of choice.
The CH999 strain had been modified so that an ermE (erythromycin resistance) gene replaced the native actinorhodin gene cluster, reducing unwanted crosstalk between this native pathway and those to be introduced, while simultaneously providing the resistance mechanism that would allow full heterologous erythromycin biosynthesis. CH999 also possessed a mutation that blocked production of another acetate-derived secondary metabolite pigment called undecylprodigiosin. (Additional native polyketide clusters have since been identified in S. coelicolor post-sequencing, and the elimination of these clusters should further alleviate concerns over metabolic crosstalk and product contamination.14,159) To this host, an E. coli–S. coelicolor shuttle vector was introduced carrying the genes coding for three polyketide megasynthases (each >300 kDa) collectively referred to as deoxyerythronolide B synthase (DEBS) and individually referred to as DEBS1, 2, and 3.112
In an excellent early example demonstrating the challenges of working with genes the size of those for DEBS and the tools available for their manipulation, Kao et al. used a recently-developed homologous recombination technique to sidestep traditional molecular biology when dealing with genes of this size (and the lack of unique restriction sites).62 More specifically, regions homologous to the ends of the DEBS operon were used to capture all three genes through a double cross-over event that relied on both temperature- and antibiotic-selection methods to aid the desired recombination events. Once completed, the shuttle vector carrying the DEBS genes was introduced into S. coelicolor and 6-deoxyerythronolide B (6dEB, the polyketide precursor to erythromycin) production was observed. In this case, CH999 provided an accommodating intracellular environment, as would be expected, to aid the production of a polyketide pathway from a close bacterial relative. However, it should be noted that to increase product titers, exogenous propionate was added to the culture medium, presumably to boost the levels of intracellular propionyl- and (2S)-methylmalonyl-CoA, which may have been suboptimal considering that the dominant native polyketide compounds produced by S. coelicolor (such as actinorhodin) were derived from acetate units; thus, it could be argued that S. coelicolor exhibited a compound bias in this example.
S. lividans was also tested as a heterologous host. In particular, a system was implemented where three individual plasmids were used to introduce the three DEBS genes into S. lividans K4-114, a strain designed similar to S. coelicolor CH999 in which the actinorhodin gene cluster was removed.123 Production at levels similar to those reported for the S. coelicolor CH999 system was recorded when two of the plasmids replicated autonomously while the third integrated into the host chromosome. Variations on this theme were explored, including high- and low-copy number plasmids, using compatible and, presumably, incompatible plasmid origins of replication, and localizing multiple DEBS genes to the host chromosome. Of these strategies, production titers peaked when using two low-copy plasmids with the same origin of replication (but separately selectable) carrying the DEBS1 and 2 genes and an integrative vector carrying DEBS3. Similar titers were achieved when using two integrative vectors (including pSAM2) carrying the DEBS2 and 3 genes. This example demonstrates an additional level of flexibility when using S. lividans. As will be outlined below, the use of multiple plasmids was instrumental in allowing for erythromycin biosynthesis in E. coli. It is interesting to note that the addition of exogenous propionate is not mentioned in this work, indicating that S. lividans may provide an innate advantage over CH999 in substrate supply to fuel DEBS.
Having established 6dEB production from either S. coelicolor or S. lividans, there was still the need to convert this polyketide intermediate to full erythromycin. However, as opposed to building upon 6dEB biosynthesis in CH999 (as would be expected given the incorporation of the ermE resistance gene), purified 6dEB produced from CH999 production cultures was exogenously fed to mutants of S. erythraea, allowing subsequent conversion to full erythromycin. Using this strategy, numerous rationally designed 6dEB derivatives were generated using the Streptomyces strains and these analogs were then converted to erythromycin derivatives via converter S. erythraea strains.160,161
Heterologous erythromycin biosynthesis was also attempted using E. coli. The rationale for choosing E. coli as a heterologous host has been previously introduced. In this case, there was also precedent to be set by attempting such a challenging reconstitution problem in this host system. If successful, it was assumed the example would inspire similar efforts at complex natural product biosynthesis. In addition to the drastic differences in cell type when compared to Streptomyces hosts, the regulatory mechanisms intrinsic to and manipulated for polyketide production in Streptomyces would be completely absent within E. coli. As such, an all-inclusive approach was needed to allow successful erythromycin production.
The earliest efforts to generate erythromycin from E. coli began with attempts at functional protein production. A notable early example was the attempt to produce DEBS3 using the T7 expression system.162 Here, it was observed that the resulting DEBS3 protein (though successfully produced and in a partially active, properly folded, and dimerized state) was not correctly post-translationally modified through 4′-phosphotantetheinylation. Nonetheless, this example set the stage for later efforts employing a T7 promoter to drive expression. Later, biochemical analysis of individual modules of the DEBS system also used E. coli as an expression host. In these studies, Sfp was co-produced to allow successful in vivo post-translational modification prior to protein purification and in vitro analysis.131,132,163 By this time, commercial strains and plasmids taking advantage of the T7 system were available and used in these studies to overproduce DEBS components.126 Besides containing the powerful T7 RNA polymerase which would seem to be particularly well-suited to the expression of the large genes often featured in complex natural product biosynthesis,164 the strains selected as expression hosts were also touted as being deficient in certain intracellular proteases which would of course aid in foreign gene expression and would potentially provide an advantage when compared to other host options. Successful production of active DEBS proteins prompted experiments to further probe E. coli's utility in supporting erythromycin biosynthesis.
The first target molecule was the erythromycin intermediate 6dEB. Production of 6dEB required the coordinated expression of DEBS1, 2, and 3 and a cellular environment capable of then supporting biosynthesis. To meet the latter requirement, additional heterologous metabolism was introduced to the E. coli cell. A metabolic pathway was designed such that exogenously added propionate was converted intracellularly to propionyl-CoA and then (2S)-methylmalonyl-CoA, two substrates that, if present in the native BL21(DE3) host, were at concentrations insufficient to support eventual 6dEB biosynthesis. This pathway was partially implemented using the same technology highlighted above in engineering the DEBS genes for introduction into S. coelicolor. Namely, plasmid-based homologous recombination was used to introduce the sfp gene into the prp operon of E. coli. The prp operon contained genes responsible for propionate catabolism.165,166 Clearly, consumption through innate cellular metabolism would be undesirable if propionate were to be channeled to 6dEB formation. Hence, the operon was eliminated except for one gene, prpE, coding for a propionyl-CoA synthetase capable of converting propionate to propionyl-CoA. The integrated sfp, as described above, allowed for in vivo post-translational modification of the DEBS enzymes, with the additional benefit of being chromosomally localized so as to free space for additional expression plasmids. Both the integrated sfp and remnant prpE were placed under inducible (lac operator) T7 promoters.
The CH999 S. coelicolor system featured a shuttle plasmid containing all three DEBS genes. The size of these genes makes normal in vitro molecular biology steps challenging, namely because of the lack of available restriction sites and associated complications with standard ligation and transformation protocols. Hence, recombination techniques were used in constructing the DEBS-CH999 shuttle vector. For E. coli, this problem was addressed by re-designing standard T7 expression plasmids such that more than one gene was expressed per plasmid. Doing so allowed for the expression of DEBS2 and 3 on one plasmid (designed as an operon with one inducible T7 promoter) and DEBS1 and a propionyl-CoA carboxylase167 (PCC, from S. coelicolor) on another separately selectable plasmid (designed such that both the PCC and DEBS1 genes were under separate inducible T7 promoters). The PCC was required for the completion of the metabolic pathway to support intracellular substrate provision by converting propionyl-CoA to (2S)-methylmalonyl-CoA. Prior to consolidating the three DEBS genes onto two expression plasmids, expression of the individual genes was confirmed. Once introduced into the newly designed E. coli strain (BAP1), it was possible to coordinate gene expression and eventual production of 6dEB by the addition of IPTG and propionate to the culture medium.
Using this system, 6dEB production from E. coli was accomplished with a key adjustment in post-induction temperature.133 Production was only observed at 22 °C, and this lower temperature most likely had a positive impact on protein folding or higher-order association of the DEBS enzymatic complex. Later, newly developed recombineering methods were used to integrate the three DEBS genes into the chromosome of BAP1.168 This accomplishment was notable for its application of λ-Red recombination to genes ∼10 kb in size. Although 6dEB production was markedly reduced as a result of reduced gene dosage, activity was observed at a higher post-induction temperature (37 °C), indicating the potential beneficial effects related to protein folding/association as a result of chromosomal gene expression.
Having accomplished 6dEB biosynthesis, efforts then turned towards full erythromycin production. In principle, this goal required an additional 17 heterologous enzymes to generate erythromycin A (the most naturally abundant and clinically-relevant form of erythromycin). Two separate efforts were initiated using either an analogous pathway from Micromonospora megalomicea or a hybrid pathway with tailoring biosynthetic genes from S. fradiae, S. erythraea, and S. venezuelae.169,170 Why these pathways were used instead of the original S. erythraea pathway is not known, although the authors indicate that these particular pathways do result in successful gene expression in E. coli, implying that there were early challenges in establishing active expression of the original S. erythraea tailoring genes. (A similar swapping of gene homologs was used to obtain 3-amino-5-hydroxybenzoic acid biosynthesis in E. coli from similarly designed and regulated T7-driven operons.171) During these attempts, effort again was made to ensure individual gene expression before constructing biosynthetic operons for introduction into E. coli. In the course of these studies, N-terminal leader sequences, specific to the pET expression plasmids, positively influenced gene expression for several tailoring genes. These cassettes were then used in operon construction. Both of these approaches relied upon designed operons and multiple expression plasmids to introduce the required genes into E. coli. This possibility is much more plausible with E. coli as the transformation protocols (in particular, electroporation) and multiple available plasmid options do not generally exist for either S. coelicolor or S. lividans. These efforts led to the production of 6-deoxyerythromycin D and erythromycin C using E. coli. Both efforts also took advantage of accompanying commercially-available chaperone protein folding systems available for E. coli to support eventual biosynthesis. Hence, this approach provides another E. coli tool used in aiding aspects of protein folding.
More recently, an attempt was made to reconstitute the entire S. erythraea pathway within E. coli.172 Here, it should be noted that of the studies dedicated to reconstituting erythromycin biosynthesis in E. coli, nearly all used the native Saccharopolyspora, Streptomyces, or other original gene sequences, indicating the ability of E. coli to accept and coordinately express significantly different (though still bacterial-based) genetic material. (In an interesting related study, the DEBS genes were impressively synthesized and expressed within E. coli with subsequent protein levels showing marked improvements; however, there was a concomitant reduction in 6dEB levels hinting at an imbalance in metabolic substrates for either protein production or polyketide biosynthesis (or both) and pointing to the need for future genetic, protein, and metabolic engineering.173) For the study focused on reconstituting the S. erythraea pathway within E. coli, a combination of plasmid-specific gene leader sequences and chaperone co-expression together with individual and coordinated (operon) gene expression and protein activity assays led to the production of erythromycins B and D. Through careful examination of the designed operons and intermediate products, erythromycin A biosynthesis was accomplished using a separate plasmid carrying eryK (encoding the terminal P450 hydroxylation step). In this way, an additional boost in gene dosage facilitated the ultimate goal of producing erythromycin A. It should be noted that in all of the E. coli efforts, uncommon usage of plasmids, including those with incompatible origins of replication (yet separately selectable) and the inclusion of up to six different plasmids, allowed the eventual single cell conversion of propionate to erythromycin A. Regardless of the methods, the reconstitution of the full erythromycin pathway in E. coli stands as a model for the possibilities in heterologous biosynthesis using this host.
4 Heterologous production of nonribosomal peptide and hybrid nonribosomal peptide-polyketide compounds
4.1 Streptomyces spp.
Although the medicinal contribution of nonribosomal peptides is undeniable, there have been relatively few examples of heterologous production efforts to date. The β-lactams, vancomycin, and, more recently, daptomycin are well-recognized antibiotics, whereas several other compounds exhibit properties ranging from anticancer to immunosuppressive activities.7 These examples each derive from original environmental hosts, and successful production reflects the sufficient titers or combinatorial biosynthetic capabilities from native hosts.174,175 However, heterologous expression is often employed when production from the native host is insufficient. This is likely the reason for the heterologous production of thiocoraline in S. lividans.176 In this work, the gene cluster was obtained from two actinomycete marine microorganisms and designed for chromosomal integration and expression from the ermE promoter. Other examples used strategies related to the methods introduced for Streptomyces coelicolor and lividans to produce compounds that have included epothilone, daptomycin, and capreomycin.108,115,177,1784.2 E. coli
E. coli does naturally produce a nonribosomal peptide (the siderophore enterobactin).179 This capability suggests that E. coli may have more potential as a heterologous host for nonribosomal peptides than for polyketides, and this may reflect the fact that primary amino acids are common building blocks for nonribosomal peptides, although it should be mentioned that over 300 nonproteinogenic amino acids or other precursors have also been identified in nonribosomal peptide products.180 At present, E. coli has been used successfully to produce a number of heterologous nonribosomal peptide and hybrid nonribosomal peptide-polyketide compounds. Examples include yersiniabactin, epothilone (to be profiled below), early intermediates of tyrocidine, and echinomycin.181–184 In three of these examples (yersiniabactin, epothilone, and echinomycin), many of the same strategies and gene expression parameters used were described in the E. coli erythromycin case study. It is worth mentioning that Watanabe and coworkers (for echinomycin biosynthesis) constructed multi-gene plasmids in a non-operon fashion with each gene cassette carrying its own promoter, rbs, and terminator. Presumably, this allowed for better gene expression from smaller transcripts. Gruenwald and coworkers (tyrocidine intermediates) also tested promoters other than T7, which included lac, T5, and trc. There has not been a systematic evaluation of promoters for complex polyketide or nonribosomal peptide biosynthesis (though a more extensive effort has been devoted to isoprenoid compounds; please see below), and the work by Gruenwald et al. is one of the few efforts that has explored other options.In this sub-section, we will highlight another tool for heterologous biosynthesis: the feeding of exogenous substrates to influence precursor supply and biosynthesis. This strategy has been mentioned earlier in the context of propionate addition to growth medium as a way to later increase intracellular propionyl-CoA and, subsequently, (2S)-methylmalonyl-CoA levels. Efforts have also been made to influence malonyl-CoA levels through the addition of glycerol to the growth medium.130 In work related to nonribosomal peptides, the first example was the feeding of salicylate in the production of yersiniabactin.181 A precursor-directed strategy towards epothilone production was conducted by Boddy et al. in which synthetic precursors of epothilone were fed to later-stage biosynthetic enzymes.185 This strategy, in particular, is useful if difficulties are encountered in reconstituting in vivo activity of biosynthetic enzymes or providing suitable precursors. Taking advantage of the modular nature of polyketide or nonribosomal peptide systems allowed the alteration of DEBS such that the loading di-domain was replaced with a nonribosomal peptide synthetase di-domain from the rifamycin synthase. Doing so allowed the production of a hybrid molecule when exogenous benzoate was added to the medium.133 Finally, in a nice example of combinatorial biosynthesis and synthetically-derived precursor feeding, Watanabe et al. generated new derivatives of echinomycin.186
4.3 Bacillus subtilis
Bacillus subtilis stands as an attractive option for the heterologous production of natural products. In particular, the bacterium's natural capacity to produce nonribosomal peptides would indicate that it would be suitable as a heterologous host for this natural product class.7,187,188 In addition, the organism's rapid growth rate, capability to become naturally competent, and thorough characterization offers a range of available recombinant techniques to aid a particular heterologous biosynthetic effort.However, the one apparent shortcoming of B. subtilis as a heterologous host is the lack of autonomous plasmids to facilitate gene transfer and heterologous expression. This problem is made worse by plasmids containing large inserts, as is likely for complex natural product genetic systems.189 The absence of such plasmids may explain the relatively few efforts to date in using the bacterium as a host system. Instead, a diverse range of integrative vectors exist for introducing foreign DNA.190
Integrative vectors featured prominently in a seminal effort by Eppelmann and coworkers to heterologously produce the antibiotic bacitracin using B. subtilis.191 Akin to the strategies for S. coelicolor, the authors were interested in using B. subtilis as a universal host for the numerous peptide products produced nonribosomally through various, and sometimes genetically intractable, Bacillus strains. In the case of bacitracin, the original host was B. licheniformis. The authors began by considering the net loss or gain of chromosomal genetic material. As such, they first removed the 26 kb native surfactin gene cluster (with the exception of the sfp gene). This step was also expected to aid later detection efforts for the desired heterologous product. What was unknown was whether the new B. subtilis host was metabolically optimal to support bacitracin biosynthesis, though this concern was mitigated by the close relationship between native and heterologous hosts. The authors then systematically introduced the 49 kb bacitracin cluster through repetitive double crossover homologous recombination steps that alternated between several antibiotic markers. This was done in a series of events with the 49 kb cluster integrated in two steps, which might suggest that there is a limit to the size of the integrated DNA fragment. However, separate research since the publication of this example suggests that the integrative transfer of foreign DNA ≥100 kb is possible.192,193 Through the course of the work, the authors used both an IPTG-inducible spac promoter and native promoters associated with the bacitracin gene cluster. The native promoters directed expression within the heterologous B. subtilis host, further emphasizing and taking advantage of the similarities between native and heterologous hosts in this case. It was assumed that the use of native promoters would similarly not disrupt any regulatory or typical expression steps in the reconstituted production effort. NRPS gene expression and bacitracin production were ∼50% higher in the heterologous host. This is an impressive result considering that the majority of heterologous production events result in reduced titers (at least initially) of the desired compound.194
Based upon the success of this work, the same authors developed a series of dual expression vectors for the express purpose of aiding nonribosomal peptide heterologous production attempts.195 The vectors were designed to shuttle between E. coli and B. subtilis. In E. coli, they existed extra-chromosomally; whereas, within B. subtilis, they integrated into the same surfactin locus used previously. Expression was driven by a lac-operator controlled T5 promoter functional in both hosts. Thus, these particular vectors not only allowed for convenient cloning steps in E. coli, but they also allowed heterologous production to be tested in both hosts. Heterologous biosynthesis was attempted with a model NRPS from the surfactin system. The subsequent analysis indicated comparable protein levels between the two systems, even though one featured chromosomal gene expression while the other featured expression from a multi-copy expression plasmid. In addition, when analyzed by SDS-PAGE, the protein product from B. subtilis showed improved structural integrity when compared to the product from E. coli which showed a degradation pattern. In this comparison, however, the authors were quick to point out that there may have been a bias in the comparison considering that the genetic material used as the model system derived from a Bacillus source. Notwithstanding this caveat, these vectors would appear to be of great utility when considering heterologous efforts using B. subtilis.
4.4 Pseudomonas putida
Pseudomonas putida has also been tested as a heterologous host in the context of nonribosomal peptide and nonribosomal peptide-polyketide biosynthesis. Led by the efforts of Müller and colleagues, P. putida was considered a promising heterologous option because of several innate capabilities. First, though it is Gram-negative, the host does possess the ability to produce natural products and also possesses a GC content similar to actinomycetes and myxobacteria, both established and emerging natural product producers. In addition, the strain has demonstrated the capability to post-translationally modify ACP and PCP domains through the action of a native 4′-phosphopantetheinyl transferase. The strain is also well-characterized, exhibits a rapid growth rate, and features well-developed accompanying molecular biology tools. As such, the strain was tested for heterologous production of myxochromide S, myxothiazol, and epothilone.196–198In the course of these studies, Müller's group also applied several new techniques to the cloning and transfer of large gene clusters. First, in conjunction with Francis Stewart, Red/ET recombineering was used to rapidly assemble complete gene clusters from genetic material captured by cosmids. (The same general technique has greatly simplified the ability to manipulate the chromosomes of model hosts such as E. coli and has also been adapted to Streptomyces spp.199,200) As the authors pointed out, this step allows for rapid gene assembly when compared to traditional in vitro molecular biology steps which can become cumbersome when dealing with sizable gene clusters typically associated with complex natural product systems. The authors used both conventional Red/ET recombination protocols and triple recombination attempts that further aided the assembly process. Triple recombination is a genetic manipulation protocol in which a short PCR-generated fragment can be used to link two other, substantially larger, DNA elements; the process minimizes the possibilities of PCR-generated errors within the larger DNA fragments. Overall, this approach coupled the recent emergence of E. coli-based Red/ET technology for gene cluster construction with the heterologous production advantages of P. putida. Gene transfer by conjugation, followed by chromosomal integration by homologous recombination, allowed the efficient introduction of the desired gene clusters. The inclusion of only one initiating xylS/Pm repressor-promoter system (toluic-acid-inducible) was used to successfully drive gene expression of the entire clusters. In the case of myxochromide S, heterologous production titers were in excess of those from the native host.
Finally, Müller's group also studied the use of transposition as a method of introducing sizable natural product gene clusters into Myxococcus xanthus and P. putida. (Earlier work by Baltz and coworkers also studied the use of transposition and transduction in streptomycetes.201) Success was achieved for both hosts with an emphasis on improved chromosomal localization using transposition as opposed to homologous recombination. This creative strategy allowed the incorporation of the myxochromide S (∼30 kb) and epothilone (∼60 kb) gene clusters in both organisms. In this study, the Tn5 promoter was used to drive heterologous gene cluster expression.
4.5 Case study: Epothilone
The second case study will feature the nonribosomal peptide-polyketide hybrid product epothilone. Similar to the case study for erythromycin, heterologous production efforts for epothilone will be discussed for S. coelicolor and E. coli. The complexity of the epothilone biosynthetic pathway served to highlight several new approaches for functional reconstitution in heterologous hosts.The epothilones exhibit impressive anticancer properties but are produced at low levels from the slow-growing native producer Sorangium cellulosum. Hence, the situation offered a perfect opportunity for heterologous biosynthesis. The first effort relied on S. coelicolor and was completed by Kosan Biosciences.115 At the time, tremendous success had been achieved with the CH999 S. coelicolor strain and accompanying recombinant plasmids. Therefore, the same system was used in an attempt to produce epothilone-based compounds. The epothilone biosynthetic process is illustrated in Fig. 4. The native building blocks were expected to be available within S. coelicolor. A more significant concern was the need to introduce a 55 kb biosynthetic pathway featuring a megasynthase four modules in length (765 kDa). Introducing the gene cluster was accomplished by transforming two separately selectable plasmids, one a low-copy replicating plasmid (containing epoA, B, C, D) and the other an integrative plasmid (containing epoE, F, K). Both clusters of epo genes utilized the actI promoter in accordance with the expression system designed for CH999. This approach allowed for successful production of epothilones A, B, C and D, and again demonstrated the capabilities of the S. coelicolor heterologous host.
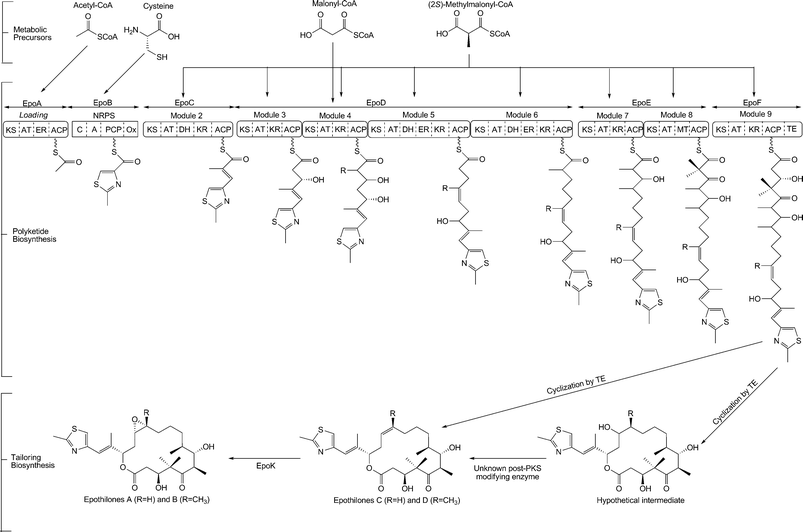 | ||
| Fig. 4 A schematic of epothilone biosynthesis. A molecule of acetyl-CoA is primed on the loading domain of the EpoA enzyme. The hybrid polyketide-nonribosomal peptide chain requires one molecule of cysteine on the NRPS EpoB, and one molecule of malonyl-CoA and eight molecules of (2S)-methylmalonyl-CoA substrates on EpoC, EpoD, EpoE and EpoF. The thioesterase domain on EpoF is responsible for cyclization and release of the hybrid chain from the NRPS-PKS complex. Epothilone A is generated by an EpoK-catalyzed epoxidation. | ||
As expected, the production of epothilone compounds using the significantly different E. coli heterologous host was more complicated. In particular, successfully reconstituting biosynthesis from a ∼55 kb cluster featuring a ∼22 kb gene was a substantial challenge. As a result, the E. coli epothilone biosynthesis effort employed several strategies directly aimed at overcoming the challenges of gene transfer and active expression.
The first attempt used a precursor-directed approach in which the last four modules of the epothilone cluster were expressed from three different T7 expression plasmids.185 Next, Mutka and co-workers introduced the entire epothilone gene cluster taking care to address many of the anticipated problems in subsequent biosynthetic reconstitution. First, the authors synthesized the entire cluster so as to eliminate any expression problems through codon bias within E. coli. This step was a good early example of leveraging emerging technology, in this case, advances in megasynthase gene synthesis.202 Next, a combination of low post-induction temperature (15 °C), the use of the arabinose-inducible PBAD promoter (as an alternative to the commonly used T7 system and in conjunction with a strain modified to eliminate arabinose catabolism and aid arabinose transport), and the co-expression of E. coli-specific chaperonins allowed for the successful expression of the majority of the epo genes. The exception was the 22 kb epoD. Impressively, the authors overcame the lack of expression by dividing the gene in two and using docking domains from the stigmatellin PKS system to ensure protein association of the newly-divided EpoD. Docking domains normally allow inter-enzyme communication within the context of specific natural product biosynthesis schemes.163,203 In this example, the communication capabilities were used in an engineered fashion by designing the docking domains as part of the smaller EpoD subunits, effectively overcoming the need to divide the enzyme. (In an analogous effort, Watanabe et al. divided the 530 kDa RifA protein using docking domains from the DEBS system.163,171 Expression in this example was driven by T7 promoters and allowed for the production of active protein products able to communicate via the introduced docking domains, resulting in an early intermediate of rifamycin.) The authors used a derivative of strain BAP1 to provide the needed (2S)-methylmalonyl-CoA substrate. Combining all of these steps allowed for the production of epothilones C and D.182
5 Heterologous production of isoprenoids
In this section, the focus on isoprenoids is a departure from the polyketide and nonribosomal peptide compounds commonly referred to as complex natural products.204 However, we feel the inclusion of this section is particularly timely as there have been a series of recent reports on the heterologous production of isoprenoid compounds with significant medicinal potential. This work has commonly used either E. coli or S. cerevisiae as heterologous host systems. Because these cellular systems have previously been introduced, this section will feature case studies related to the production of artemisinin (antimalarial) and Taxol (anticancer) through heterologous biosynthesis. Of particular relevance is that both natural products to be profiled originate from plant hosts. Therefore, as opposed to the cases presented above, this section will focus on the challenges in reconstituting biosynthesis of plant-derived natural compounds.5.1 Artemisinin
There are an estimated 350–500 million clinical malaria episodes annually caused by the parasites Plasmodium falciparum and Plasmodium vivax, for which artemisinin is considered an excellent therapy. Due to the fact that this disease afflicts the population of third-world countries, the production cost of artemisinin is of central importance for widespread usage. Total synthesis is possible but the process is costly and results in low yields. As such, production from natural sources has been the method of choice for product supply. Unfortunately, this process suffers from a dependence on the original plant producer, Artemisia annua, and the need for efficient extractions from this source. As such, the situation presented another opportunity for heterologous biosynthesis.205The first report towards heterologous artemisinin was the production of the precursor amorphadiene in E. coli.206 In contrast to modular polyketide or nonribosomal peptide systems, isoprenoid biosynthesis does not usually involve large enzymes. However, there may be the need to introduce multiple genes as part of the overall pathway. E. coli represents a relatively clean host for isoprenoid biosynthesis. Although there are native mechanisms for isoprenoid biosynthesis in E. coli, the innate metabolic pathways needed for precursor supply are considered insufficient to support heterologous biosynthetic efforts and, as a result, there is little concern of an overabundance of native isoprenoid compounds contaminating heterologous production efforts. However, this situation necessitated metabolic engineering to ensure sufficient precursor supply for eventual biosynthesis.
To this end, Martin and coworkers favored heterologous transfer of the yeast mevalonate pathway into E. coli, as opposed to engineering the native DXP pathway (Fig. 5), for which there was a concern about unknown regulatory elements that might hamper over-production.206 Once a pathway was established for precursor supply, the first committed step towards artemisinin was introduced. This step was catalyzed by the amorphadiene synthase from Artemisia annua. Without direct access to the amorphadiene synthase gene, gene synthesis was employed to both optimize codon usage within a heterologous host and to provide de novo genetic material for heterologous transfer. This study featured the use of lac operators and both lac and trc promoters. It is interesting to contrast these gene expression elements with those (primarily T7-based) that have been used for polyketide and nonribosomal peptide heterologous expression attempts in E. coli. Also in this study, conventional K12 E. coli strains were used as opposed to the B strains that commonly accompany the T7 expression system.
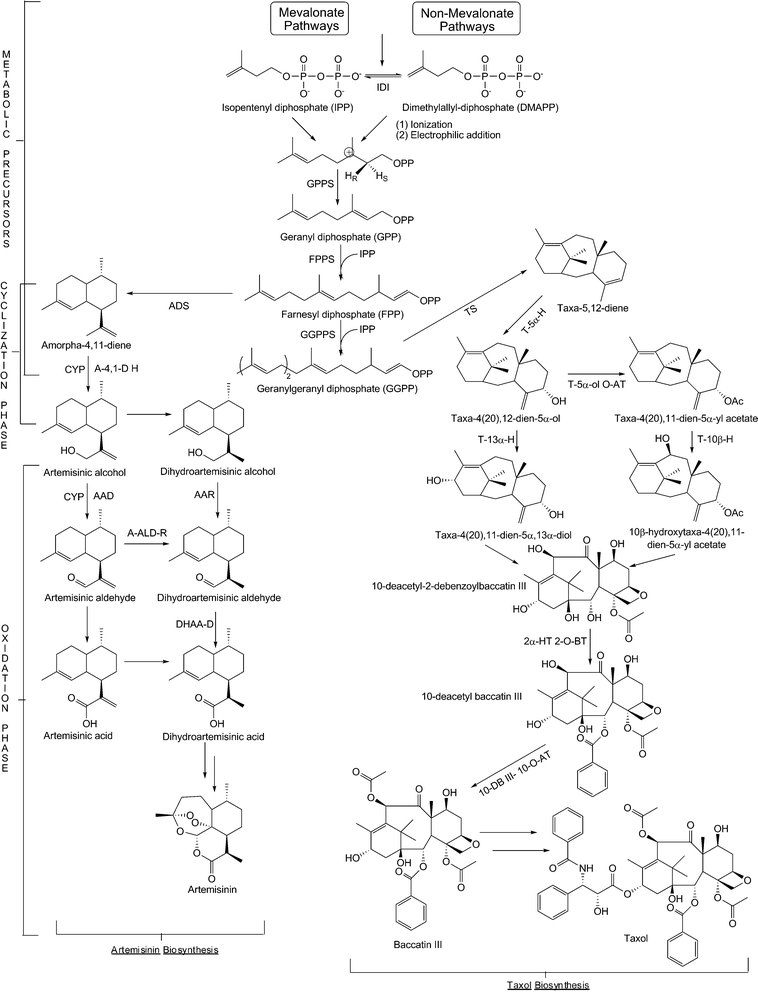 | ||
| Fig. 5 A schematic of artemisinin and Taxol biosynthesis. First, either the mevalonate or the non-mevalonate pathways generate the two universal C5 precursors for isoprenoid natural products, isopentenyl diphosphate and dimethylallyl diphosphate. These two can be interconverted through the action of isopentenyl diphosphate isomerase. One molecule of dimethylallyl diphosphate and one of isopentenyl diphosphate are condensed to give the C10 geranyl diphosphate by a geranyl diphosphate synthase. The C15 farnesyl diphosphate is generated from one molecule of geranyl diphosphate and one of isopentenyl diphosphate by a farnesyl diphosphate synthase. Lastly, the C20 geranylgeranyl diphosphate is generated from one molecule of farnesyl diphosphate and one of isopentenyl diphosphate by a geranylgeranyl diphosphate synthase. The first committed steps towards artemisinin and Taxol biosynthesis produce amorphadiene and taxadiene by cyclization of the C15 and C20 intermediates by an amorphadiene synthase and a taxadiene synthase, respectively. Amorphadiene and taxadiene both undergo significant oxidations on their cyclic cores to generate the final molecules. Abbrevations: IPP = isopentenyl diphosphate; DMAPP = dimethylallyl diphosphate; IDI = isopentenyl diphosphate isomerase; GPP = geranyl diphosphate; FPP = farnesyl diphosphate; GGPP = geranylgeranyl diphosphate; GPPS = geranyl diphosphate synthase; FPPS = farnesyl diphosphate synthase; GGPPS = geranylgeranyl diphosphate synthase; ADS = amorphadiene synthase; CYP = cytochrome P450; A-4,11-D H = amorpha-4,11-diene hydroxylase; AAD = artemisinic alcohol dehydrogenase; A-ALD-R = artemisinic aldehyde reductase; AAR = artemisinic alcohol reductase; DHAA-D = dihydroartemisinic aldehyde dehydrogenase; T-5α-H = taxadiene 5α hydroxylase; T-5α-ol O-AT = taxdien-5α-ol O-acetyltransferase; T-13α-H = taxadiene 13α hydroxylase; T-10β-H = taxane 10β-hydroxylase; 2α-HT 2-O-BT = 2-α-hydroxytaxane 2-O-benzoyltransferase; 10-DB III-10-O-AT = 10-deacetylbaccatin III 10-O-acetyltransferase. | ||
The next step towards artemisinin production was the functional production of plant P450 enzymes within E. coli. Natively, plant-derived P450 enzymes are found membrane-bound, which hints at the challenges likely to be encountered when attempts are made to heterologously express such genes in E. coli. However, numerous strategies have been developed to overcome the lack of heterologous expression/activity and many of these were systematically employed by Chang et al. during the production of artemisinic acid in E. coli.207 Namely, codon optimization, gene truncation, 5′-region modification, reductase partner inclusion and modification, promoter variation (T7, PBAD, lac, tac), and strain variation (BL21(DE3), DH1, DH10B) were tested for improvements in final activity and product formation. Although it may not be possible to simply apply the best case scenario here for future attempts at other plant-derived P450 gene expression/activity studies, the range of options pursued will most likely be applicable and lead to improvements in most cases.
A key alternative option to reconstituting biosynthesis of plant-derived natural products is the use of a eukaryotic host, and the same group that spearheaded artemisinic acid production in E. coli studied production in S. cerevisiae in parallel. Using this route, the first heterologous production of artemisinic acid was actually completed before the success with E. coli. To do so, amorphadiene synthase was placed into an expression plasmid (2μ autonomously replicating) under the control of an inducible GAL1 promoter. The cytochrome P450–reductase pair, isolated through a probing of closely-related plant species, was then introduced on a separately selectable 2μ expression vector also using an inducible GAL1 promoter, with the gene sequences unaltered from their plant-derived origins. Lastly, over-expression of a truncated form of a 3-hydroxy-3-methylglutaryl-CoA reductase (tHMGR), over-expression of upc2-1, and down-regulation of ERG9 (encoding a squalene synthase, the beginning of the essential sterol biosynthetic pathway) using a methionine-repressible promoter (PMET3) improved amorphadiene titers nearly 500-fold; these steps were metabolic engineering attempts to reduce unwanted regulatory or crosstalk mechanisms. Successful production was achieved with little effort invested in optimizing gene expression conditions (at least when compared to E. coli), implying that yeast might be the host of choice moving forward for either total or semisynthetic production of artemisinin.208
5.2 Taxol
Led by the work of Croteau, early efforts to identify the exact biosynthetic steps towards Taxol's production began in the early to mid-1990s.209–211 These early efforts identified the taxadiene synthase and cytochrome P450 hydroxylase needed to make the first two dedicated precursors of the Taxol pathway (Fig. 5). Additional biosynthetic enzymes and intermediates were subsequently identified, followed by the landmark effort to generate a cDNA library from Taxus cuspidata.212–222 The cDNA library either identified or suggested candidates to complete a hypothetical (yet incomplete) pathway of Taxol biosynthesis, as outlined in Fig. 5.Early genes identified were often tested for expression through convenient hosts such as E. coli and S. cerevisiae.216,223 Though such efforts were primarily driven by the interest to better biochemically characterize the enzymes of the Taxol biosynthetic pathway, information also began to accrue regarding the feasibility of reconstituting protein activity through these hosts.
At the same time, a variety of methods were being explored for the production of Taxol. Although the total chemical synthesis of Taxol has been achieved by several routes, the low yields obtained made this approach economically infeasible for large-scale production.224 Semi-synthetic approaches were developed to convert a key intermediate extracted from Yew needles, simultaneously providing a viable production process and minimizing the environmental impact that resulted from initial isolation attempts requiring destructive extraction from Yew bark.225 Alternative efforts were focused on the use of plant cell culture for Taxol production. Several Taxus species have been studied in the context of Taxol cell culture production, with numerous strategies used to boost production.226 Examples include feeding methyl jasmonate, rapid removal of the Taxol product, and immobilizing the plant cells.227–229 However, despite significant advances, plant cell cultures still suffer from low yields, high production costs, and unwanted by-products.226 Heterologous plant hosts were also tested, with taxadiene successfully produced through Arabidopsis thaliana and tomato fruit.230,231 It was discovered that in addition to plant-derived Taxol production, certain Taxus-associated fungi also have the ability to produce Taxol.232–234 One particular strain, Nodulisporium sylviform, was subjected to rounds of mutagenesis to improve Taxol yield,234,235 with production making only modest gains from already low titers (314 to 393 μg/L). Although fungi-derived production does provide an alternative biological route to Taxol, thus far, quantities produced are still well below those from plant-dependent routes with slight, if any, biological advantages provided by the fungal hosts.
The drawbacks associated with each attempted production route and the lure of designed analog production provided arguments for heterologous Taxol biosynthesis through convenient microbial hosts. It was then no surprise to see the first efforts made at Taxol intermediate production in E. coli and yeast, relying on the native cellular pathways to provide the precursors needed for biosynthesis. In the case of E. coli, the taxadiene synthase gene was heterologously expressed, again using the popular T7 system. However, researchers observed exclusive inclusion body formation and no protein activity regardless of whether K12 or B strains were used or the post-induction temperature was modified; soluble and active protein was obtained when a thioredoxin fusion construct was expressed at lowered post-induction temperatures (20 °C).223 Later, efforts were made to improve gene expression by removing a leader N-terminal region (required for plant plastid localization) and including an additional argU gene to aid the codon bias expected for E. coli.236,237 Huang et al. accomplished in vivo production of taxadiene from E. coli by also co-expressing certain DXP pathway genes.237 This theme would continue for E. coli-derived Taxol intermediates with cues taken from an extensive analysis of the DXP pathway to support lycopene production.238
Research also began to test heterologous production in yeast. S. cerevisiae was tested for expression of the first eight genes (after having removed plastidial domains) thought to be responsible for front-end Taxol intermediates. The first five steps were then evaluated for in vivo heterologous reconstitution. To do so, separately selectable autonomous plasmids were used, one of which carried two genes. Initial efforts relied primarily on inducible GAL promoters and one constitutive GPD promoter. Later, this system was exclusively switched to GAL promoters. Using this arrangement, the first two committed Taxol intermediates (taxadiene and taxadien-5α-ol) were quantified from production cultures.239 Building on this work, Engels et al. focused on improved taxadiene production. Specifically, separately selectable plasmids were again used, but this time with a codon-optimized taxadiene synthase under the control of a PGK promoter. In addition, care was taken to avoid biosynthetic overlap with native yeast sterol production (similar to the steps taken for artemisinic acid production described above). Here, it should be noted that although yeast may provide a more accommodating environment for plant-derived genes and intrinsic support for the biosynthesis of isoprenoid compounds, the host displays a disadvantage in native metabolism interacting with and/or contaminating heterologous production efforts. To further address this, the yeast mevalonate pathway, which feeds into both native sterol and heterologous taxadiene biosynthesis, was adjusted to eliminate feedback inhibition resulting from sterol production and by supplying a separate geranylgeranyl diphosphate (GGPP) synthase capable of decoupling the two pathways. In addition, a separate modification allowed for exogenous uptake of sterols; this step was expected to down-regulate native sterol metabolism that would otherwise detract from heterologous taxadiene production. Notably, each metabolic engineering step was designed to eliminate crosstalk between native and heterologous pathways.240
To conclude this sub-section, very recent work will be profiled reporting on the production of the first two Taxol pathway intermediates in E. coli at unprecedented titers.241 In this work, careful attention and an array of modifications were first made to ensure sufficient expression of the first two genes in the overall pathway. This involved codon optimization, 5′-region modifications (both to remove plastidial domains and to improve cytochrome P450 gene expression), gene fusions, and lowered temperature expression. These same techniques have been highlighted throughout previous case studies, but their combination in this case allowed for a comprehensive attempt to facilitate biosynthetic gene expression. However, the drastic improvements in titers resulted from the careful balancing between precursor supply and biosynthetic conversion. (Previous studies for amorphadiene had indicated that improper balancing may negatively impact cell viability and product titers.206) To accomplish this, a combination of promoters (trc, T5, and T7) and gene copy variants (through chromosomal- or plasmid-borne gene expression) allowed an optimization of product biosynthesis. However, as alluded to above, the native DXP pathway was engineered to supply the needed isoprenoid precursors. Success in this example provides another point of debate between using E. coli or S. cerevisiae as a long-term host for attempts at reconstituting the remainder of the Taxol pathway.
6 Summary
Certain situations may not merit heterologous biosynthesis, such as when sufficient production is provided by the native host. When this is not the case, the background and descriptions above serve to highlight successful heterologous production examples and the reasoning and experimental tools behind their implementation. Heterologous natural product biosynthesis has proven capable of producing complex compounds from nearly every natural product group. The examples cited in this review serve to provide guidelines and heuristics, but it is important to emphasize that every future case will require specialized attention. However, growing evidence is presented to indicate that success is achievable with a wide range of heterologous host choices. Moreover, the approaches above summarize certain experimental steps that have been repeatedly successful. As such, a key goal is to give the reader a broad perspective of the methods that have been successful and that might similarly be successful in future heterologous production attempts. Table 1 summarizes both the options available and accomplishments associated with specific hosts. These host choices may each have advantages and disadvantages specific to a targeted natural product. The ultimate choice will revolve around the heuristics outlined above, convenience, and the individual researchers' prior experience.| Microbe | Microbe information | Genome information | Heterologous host | Key example of metabolite produced heterologously | Largest gene cluster expressed | Largest heterologous gene expressed | Largest number of genes expressed and product of the expression | |
|---|---|---|---|---|---|---|---|---|
| Advantages | Disadvantages | |||||||
| Bacillus subtilis | Gram-positive, rod-shaped, doubling time 1 h | 4.2 Mb, 43.5% GC, 4100 ORFs; (strain 168) | Naturally competent, readily available integrative vectors | No native plasmid systems; potentially limited metabolic support | Bacitracin191 | Bacitracin (49 kb)191 | bacC (19 kb)191 | Bacitracin (9 genes)191 |
| Escherichia coli | Gram-negative, rod-shaped, doubling time 20 min | 4.6 Mb, 50.8 GC%, 4288 ORFs, (strain K-12 MG1655) | Readily available genetic engineering tools; rapid growth cycle; established bioreactor protocols | Does not support PKS or NRPS post-translational modification; limited metabolic support | Erythromycin,169,172 epothilone115 | Epothilone (55 kb)182 | epoD (21.7 kb)182 | Erythromycin (23 genes)169,172 |
| Pseudomonas putida | Gram-negative, rod-shaped, doubling time 1 h | 6.18 Mb, 61.6% GC, 5437 ORFs; (strain KT2440) | Supports PKS and NRPS post-translational modification | Potentially limited metabolic support | Myxochromide S,196 epothilone198 | Epothilone(58 kb)198 | epoD (21.7 kb)198 | Epothilone (7 genes)198 |
| Saccharomyces cerevisiae | Eukaryotic, budding yeast, doubling time 1.5 h | 12.5 Mb, 38% GC, 5884 ORFs; (strain S288c) | Plasmid systems available, excellent native recombineering system | Does not support PKS or NRPS post-translational modification; limited metabolic support | 6-methylsalyclic acid,140 dihydromonacolin L245 | — | lovB (10 kb),245pcbAB (12.3 kb)246 | Taxadien-5α-acetoxy-10β-ol (5 genes)239 |
| Aspergillus nidulans | Filamentous fungus, doubling time 1 h | 30 Mb, 50.3 GC%, 9546 ORFs; (strain FGSC A4) | Supports PKS and NRPS post-translational modification; native metabolic support | Slow growth cycle | 6-methylsalyclic acid,144 monacolin J150 | Monacolin J (37 kb)150 | tenS (12.7 kb)243 | Monacolin J (lovastatin intermediate) (10 genes)150 |
| Streptomyces coelicolor | Gram-positive, forms spore and mycelium, produces natural product, doubling time 2 h | 8.7 Mb, 72.1 GC%, 7825 ORFs; (strain A3(2)) | Established recombinant DNA techniques; native metabolic support | Slow growth cycle; need for unmethylated DNA during genetic transfer | Caprazamycin,244 epothilone115 | Epothilone (55 kb)115 | epoD (21.7 kb)115 | Granaticin (37 genes)113 |
| Streptomyces lividans | Gram-positive, forms spore and mycelium, produces natural product, doubling time 4 h | 8.2 Mb, 71 GC%, 7551 ORFs; (strain TK24) | No need for unmethylated DNA for transformation; established recombinant DNA techniques; native metabolic support | Slow growth cycle | Validamycin,242 daptomycin108 | Thiocoraline (53 kb)176 | dptBC (22 kb)108 | Clorobiocin (27 genes)117 |
We also want to emphasize that there are additional host choices available. In an attempt to more broadly capture the alternative options in heterologous natural product biosynthesis, Fig. 6 plots the heterologous natural product efforts across the three classes of natural products discussed above. Here, we have included other hosts in an attempt to present those omitted from the discussions above and to highlight emerging trends in heterologous biosynthetic efforts.
 | ||
| Fig. 6 Cumulative trends in heterologous hosts used for natural product production of polyketides, nonribosomal peptides, isoprenoids, and their intermediates/derivatives. Data were generated from the literature over the time period indicated, and each data point represents a publication presenting results of a novel heterologously produced natural product (including novel methods of producing natural products previously reported; for example 6-MSA through S. cerevisiae and E. coli).243,263–361 Omitted were papers focused upon gene expression (for the purpose of recombinant protein products or enzyme biochemical characterization) or optimization of a previously produced heterologous compound. Hybrid polyketide-nonribosomal peptide compounds were included in the nonribosomal peptide category totals. Extrapolation curves to 2015 were calculated based upon trend slopes over the 5 years from 2005 to 2010. Due to the volume of data produced during the analysis, the results may not be completely comprehensive, but they serve to illustrate current and future trends within the heterologous natural product biosynthetic field. (a) Overall trends of heterologous natural product production of the three classes highlighted in this review. (b) Trends in heterologous hosts for polyketide production. (c) Trends in heterologous hosts for nonribosomal peptide production. (d) Trends in heterologous hosts for terpenoid/isoprenoid production. | ||
7 Emerging themes and future directions
Having addressed the past and current methodologies for heterologous natural product biosynthesis within a range of commonly used hosts, this final section will attempt to predict future directions in the field. Highlighted below will be emerging approaches and their relation to heterologous biosynthesis.The environment holds an unknown level of chemical diversity in the form of natural products. There is every reason to believe this diversity holds tremendous therapeutic potential. Supporting this view is the current track record of successful compounds isolated from the soil and marine environments. However, the challenges in isolating and harnessing the native producers have been a primary driver in the development of heterologous biosynthesis programs. Natural product discovery and screening efforts have suffered over time from rediscovery of the same natural products and natural product producers. Besides leading to a situation of diminished discovery and a lack of new compounds, it is now generally accepted that the majority of environmental microbes will not survive in artificial, man-made culture environments. It is estimated that over 99% of environmental microbes are un-cultivatable outside their native environments.247 Hence, the tantalizing potential of molecular diversity is crippled by lack of access to the native producers.
Several approaches have been developed to address this access concern. First, with the use of consensus sequence information resulting from the genetic and biochemical similarities between certain natural product classes,248 environmental microbial sources can be narrowed through screening made more efficient by probing for those cellular populations putatively possessing natural product pathways. In essence, this reduces the total number of microbes to be screened and narrows the search to those considered most promising.249 However, this approach still does not guarantee successful culturing in the lab or during process scale up. Alternative approaches have used creative culturing techniques to facilitate the successful growth of environmental microbes. In these cases, unique laboratory environments are generated to replicate those found in nature including access to reconstructed growth environments and co-culturing.250,251 In an approach that embraces the idea of heterologous biosynthesis to provide access to natural products, the field of metagenomics has emerged in which environmental DNA (eDNA) is captured from a field sample, packaged into a suitable host plasmid, and either completely sequenced or first transferred to a heterologous host for screening of any encoded natural product activity.252,253 In this example, the challenges in microbial cultivation are by-passed in favor of capturing the genetic material potentially encoding the desired natural product bioactivity. Finally, the number of fully sequenced genomes is growing at a staggering pace.254 As highlighted before for selected hosts, the resulting genomic information has revealed a surprising number of “silent”, “cryptic”, or “orphan” coding regions that serve as potential sources of additional natural products.255–258
In the scheme of natural product heterologous biosynthesis, the above-mentioned research areas help provide access to the DNA encoding for pathways of interest. In the case of metagenomics, this access is aided by the fact that certain natural product pathways are found genetically clustered (though this does not hold for most plant-derived natural product systems). Once a new host organism or an accompanying genetic pathway has been identified, the host-specific gene transfer/reconstitution methods outlined above can be implemented towards heterologous biosynthesis. In addition to those host systems previously profiled, an alternate recent approach has been the re-design (typically through removal of the primary natural product biosynthetic pathway) of established production systems such as S. erythraea and S. fradiae to then serve as heterologous hosts, with the advantage of a highly-manipulated cellular background capable of accommodating biosynthesis.259,260
Once heterologous biosynthesis steps have begun or are completed, additional areas of research can be subdivided into biosynthetic mechanics and metabolic support. Natural product gene clusters may contain very large (>10 kb), numerous (>20), notoriously foreign (CytP450), and non-optimized (containing rare codons) genes that will tax any heterologous host's gene expression machinery. As highlighted throughout case studies in this review, methods to aid gene expression could then focus on a range of promoter, plasmid, or operon designs in addition to control of gene expression and culture conditions. More generally, thought must be given to every stage of the protein's life to ensure that active enzyme is present in sufficient quantities and for an adequate period of time for the purpose of natural product biosynthesis. The process will no doubt be fine-tuned empirically through the currently available tools developed through recombinant protein production and those to be developed for recombinant natural product biosynthesis. Like heterologous protein production, there will most likely be no general approach applicable to all cases. Instead, guidelines will accompany the growing number of options to facilitate the overall process.
Overcoming the challenges of gene expression and biosynthetic mechanics then gives way to intracellular support. Future challenges associated with more complex natural products will include providing the most convenient heterologous hosts with the capabilities to support biosynthesis. As mentioned, the intrinsic intracellular environment was the key early advantage to systems like S. coelicolor. If E. coli, B. subtilis, or others are to truly become universal hosts for complex natural products, they must be metabolically engineered to broadly support biosynthesis. This primarily means providing the substrates of the biosynthetic process, which may require the same gene transfer and expression refinement associated with natural product biosynthetic reconstitution. However, while direct precursors are the most obvious need, they are by no means the only metabolic support required. When one analyzes the biochemical steps from precursor to final product, there exist a number of cofactors and auxiliary components that contribute to overall biosynthesis. These must also be present in non-rate-limiting quantities to optimize eventual biosynthesis. Towards this end, heterologous natural product biosynthesis is now reaching the stage in which traditional modes of metabolic engineering may be applied to optimize cellular production. Metabolic engineering has been defined as “the directed improvement of product formation or cellular properties through the modification of specific biochemical reactions or introduction of new ones with the use of recombinant DNA technology.”261 This definition may be further subdivided into heterologous metabolic engineering (genetic transfer and reconstitution, the focus of this review) and pathway metabolic engineering, where the principles and methods of metabolic engineering may now be applied towards heterologous biosynthesis optimization once product formation has been accomplished. Optimization approaches have included numerous computational and experimental predictive and analytical methods to characterize and improve biosynthesis. Given the relatively recent success of heterologous complex natural product biosynthesis, these tools are just beginning to be applied, but they promise to further add to the success thus far achieved and they are particularly well-suited to the heterologous hosts highlighted above.262 Such approaches, in addition to -omics global characterization techniques and more traditional process engineering steps, will be paramount to maximizing natural product output from heterologous hosts. As emphasized throughout this review, heterologous natural product biosynthesis derives from situations that limit access to or control of natural product medicinal value. Heterologous production at minimal titers does not solve this problem. However, the genetic, metabolic, and process engineering strategies mentioned above serve as powerful routes to realize the full potential of heterologous biosynthesis.
One of the driving goals of the natural products field is to increase access to the medicinal potential of natural products. Heterologous biosynthesis sprung from this objective. Here, we have reviewed the origins of this quest with a special emphasis on the heterologous hosts chosen and their associated experimental tools to aid the gene transfer and reconstitution processes. Future research will continually fuel these efforts with additional promising therapeutic natural product gene clusters that push the limits of the heterologous biosynthetic approach. Looking further, there will be numerous opportunities to continually engineer, fine-tune, and improve heterologous biosynthesis. With dedication to these efforts, there is no reason to believe the impact of natural products is in decline, but simply in a state of re-invention, with the promise of even more therapeutic impact in the future.
8 Acknowledgements
The authors gratefully acknowledge support from the Milheim Foundation (2006-17), the NSF (IIP-0924699), and the NIH (GM085323) for support towards ongoing projects related to heterologous biosynthesis.9 References
- C. Khosla, J. Org. Chem., 2009, 74, 6416–6420 CrossRef CAS.
- A. Koglin and C. T. Walsh, Nat. Prod. Rep., 2009, 26, 987–1000 RSC.
- C. Hertweck, Angew. Chem., Int. Ed., 2009, 48, 4688–4716 CrossRef CAS.
- K. J. Weissman and R. Muller, ChemBioChem, 2008, 9, 826–848 CrossRef CAS.
- E. S. Sattely, M. A. Fischbach and C. T. Walsh, Nat. Prod. Rep., 2008, 25, 757–793 RSC.
- M. A. Marahiel, J. Pept. Sci., 2009, 15, 799–807 CrossRef CAS.
- E. A. Felnagle, E. E. Jackson, Y. A. Chan, A. M. Podevels, A. D. Berti, M. D. McMahon and M. G. Thomas, Mol. Pharmaceutics, 2008, 5, 191–211 CrossRef CAS.
- J. Kirby and J. D. Keasling, Annu. Rev. Plant Biol., 2009, 60, 335–355 CrossRef CAS.
- K. Watanabe, Biosci., Biotechnol., Biochem., 2008, 72, 2491–2506 CrossRef CAS.
- D. J. Newman and G. M. Cragg, J. Nat. Prod., 2007, 70, 461–477 CrossRef CAS.
- S. C. Wenzel and R. Muller, Curr. Opin. Biotechnol., 2005, 16, 594–606 CrossRef CAS.
- I. Fujii, Nat. Prod. Rep., 2009, 26, 155–169 RSC.
- E. Rodriguez, H. G. Menzella and H. Gramajo, Methods Enzymol., 2009, 459, 339–365 CAS.
- R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2010 Search PubMed.
- Z. L. Fowler and M. A. Koffas, Appl. Microbiol. Biotechnol., 2009, 83, 799–808 CrossRef CAS.
- J. A. Chemler and M. A. Koffas, Curr. Opin. Biotechnol., 2008, 19, 597–605 CrossRef CAS.
- M. K. Julsing, A. Koulman, H. J. Woerdenbag, W. J. Quax and O. Kayser, Biomol. Eng., 2006, 23, 265–279 CrossRef CAS.
- P. C. Lee and C. Schmidt-Dannert, Appl. Microbiol. Biotechnol., 2002, 60, 1–11 CrossRef CAS.
- G. Sandmann, Arch. Biochem. Biophys., 2001, 385, 4–12 CrossRef CAS.
- M. E. Kolewe, V. Gaurav and S. C. Roberts, Mol. Pharmaceutics, 2008, 5, 243–256 CrossRef CAS.
- S. C. Wenzel and R. Muller, Mol. BioSyst., 2009, 5, 567–574 RSC.
- S. C. Roberts, Nat. Chem. Biol., 2007, 3, 387–395 CrossRef CAS.
- C. Schmidt-Dannert, Curr. Opin. Biotechnol., 2000, 11, 255–261 CrossRef CAS.
- M. C. Wani, H. L. Taylor, M. E. Wall, P. Coggon and A. T. McPhail, J. Am. Chem. Soc., 1971, 93, 2325–2327 CrossRef.
- J. M. McGuire, R. L. Bunch, R. C. Anderson, H. E. Boaz, E. H. Flynn, H. M. Powell and J. W. Smith, Antibiot. Chemother., 1952, 2, 281–284 Search PubMed.
- K. J. Stutzman-Engwall and C. R. Hutchinson, Proc. Natl. Acad. Sci. U. S. A., 1989, 86, 3135–3139 CrossRef CAS.
- H. Nishimura, S. Okamoto, K. Nakajima, M. Shimohira and N. Shimaoka, Ann. Rep. Shionogi Res. Lab, 1957, 7, 165–171 Search PubMed.
- M. G. Watve, R. Tickoo, M. M. Jog and B. D. Bhole, Arch. Microbiol., 2001, 176, 386–390 CrossRef CAS.
- A. L. Demain and R. P. Elander, Antonie van Leeuwenhoek, 1999, 75, 5–19 CrossRef CAS.
- R. H. Baltz, Antonie van Leeuwenhoek, 2001, 79, 251–259 CrossRef CAS.
- R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2006, 33, 507–513 CrossRef CAS.
- J. Berdy, J. Antibiot., 2005, 58, 1–26 CrossRef CAS.
- J. Clardy, M. A. Fischbach and C. T. Walsh, Nat. Biotechnol., 2006, 24, 1541–1550 CrossRef CAS.
- S. J. Projan, Curr. Opin. Microbiol., 2003, 6, 427–430 CrossRef.
- F. E. Koehn and G. T. Carter, Nat. Rev. Drug Discovery, 2005, 4, 206–220 CrossRef CAS.
- K. M. Overbye and J. F. Barrett, Drug Discovery Today, 2005, 10, 45–52 CrossRef.
- F. von Nussbaum, M. Brands, B. Hinzen, S. Weigand and D. Habich, Angew. Chem., Int. Ed., 2006, 45, 5072–5129 CrossRef CAS.
- C. Walsh, Nat. Rev. Microbiol., 2003, 1, 65–70 CrossRef CAS.
- M. A. Fischbach and C. T. Walsh, Science, 2009, 325, 1089–1093 CrossRef CAS.
- D. I. Andersson and D. Hughes, Nat. Rev. Microbiol., 2010, 8, 260–271 Search PubMed.
- J. W. Blunt, B. R. Copp, M. H. Munro, P. T. Northcote and M. R. Prinsep, Nat. Prod. Rep., 2010, 27, 165–237 RSC.
- H. Rahman, B. Austin, W. J. Mitchell, P. C. Morris, D. J. Jamieson, D. R. Adams, A. M. Spragg and M. Schweizer, Mar. Drugs, 2010, 8, 498–518 CrossRef CAS.
- C. Olano, C. Mendez and J. A. Salas, Mar. Drugs, 2009, 7, 210–248 CrossRef CAS.
- T. A. Gulder and B. S. Moore, Curr. Opin. Microbiol., 2009, 12, 252–260 CrossRef CAS.
- J. L. Fortman and D. H. Sherman, ChemBioChem, 2005, 6, 960–978 CrossRef CAS.
- D. J. Newman and G. M. Cragg, Curr. Med. Chem., 2004, 11, 1693–1713 CAS.
- D. W. Udwary, L. Zeigler, R. N. Asolkar, V. Singan, A. Lapidus, W. Fenical, P. R. Jensen and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2007, 104, 10376–10381 CrossRef CAS.
- Y. Liu, C. Hazzard, A. S. Eustaquio, K. A. Reynolds and B. S. Moore, J. Am. Chem. Soc., 2009, 131, 10376–10377 CrossRef CAS.
- A. S. Eustaquio, R. P. McGlinchey, Y. Liu, C. Hazzard, L. L. Beer, G. Florova, M. M. Alhamadsheh, A. Lechner, A. J. Kale, Y. Kobayashi, K. A. Reynolds and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2009, 106, 12295–12300 CrossRef.
- L. Fieseler, U. Hentschel, L. Grozdanov, A. Schirmer, G. Wen, M. Platzer, S. Hrvatin, D. Butzke, K. Zimmermann and J. Piel, Appl. Environ. Microbiol., 2007, 73, 2144–2155 CrossRef CAS.
- D. E. Cane, C. T. Walsh and C. Khosla, Science, 1998, 282, 63–68 CrossRef CAS.
- K. J. Weissman and P. F. Leadlay, Nat. Rev. Microbiol., 2005, 3, 925–936 Search PubMed.
- M. Oliynyk, M. Samborskyy, J. B. Lester, T. Mironenko, N. Scott, S. Dickens, S. F. Haydock and P. F. Leadlay, Nat. Biotechnol., 2007, 25, 447–453 CrossRef CAS.
- S. Omura, H. Ikeda, J. Ishikawa, A. Hanamoto, C. Takahashi, M. Shinose, Y. Takahashi, H. Horikawa, H. Nakazawa, T. Osonoe, H. Kikuchi, T. Shiba, Y. Sakaki and M. Hattori, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 12215–12220 CrossRef CAS.
- E. M. Meyerowitz, G. M. Guild, L. S. Prestidge and D. S. Hogness, Gene, 1980, 11, 271–282 CrossRef CAS.
- U. J. Kim, H. Shizuya, P. J. de Jong, B. Birren and M. I. Simon, Nucleic Acids Res., 1992, 20, 1083–1085 CrossRef CAS.
- Q. Tao and H. B. Zhang, Nucleic Acids Res., 1998, 26, 4901–4909 CrossRef CAS.
- J. C. Pierce, B. Sauer and N. Sternberg, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 2056–2060 CrossRef CAS.
- H. Shizuya, B. Birren, U. J. Kim, V. Mancino, T. Slepak, Y. Tachiiri and M. Simon, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 8794–8797 CrossRef CAS.
- D. T. Burke, G. F. Carle and M. V. Olson, Science, 1987, 236, 806–812 CAS.
- A. P. Monaco and Z. Larin, Trends Biotechnol., 1994, 12, 280–286 CrossRef CAS.
- C. M. Hamilton, M. Aldea, B. K. Washburn, P. Babitzke and S. R. Kushner, J. Bacteriol., 1989, 171, 4617–4622 CAS.
- C. J. Chiang, P. T. Chen and Y. P. Chao, Biotechnol. Bioeng., 2008, 101, 985–995 CrossRef CAS.
- R. Rong, M. M. Slupska, J. H. Chiang and J. H. Miller, Gene, 2004, 336, 73–80 CrossRef CAS.
- H. Liu, H. Jiang, B. Haltli, K. Kulowski, E. Muszynska, X. Feng, M. Summers, M. Young, E. Graziani, F. Koehn, G. T. Carter and M. He, J. Nat. Prod., 2009, 72, 389–395 CrossRef CAS.
- D. G. Gibson, J. I. Glass, C. Lartigue, V. N. Noskov, R. Y. Chuang, M. A. Algire, G. A. Benders, M. G. Montague, L. Ma, M. M. Moodie, C. Merryman, S. Vashee, R. Krishnakumar, N. Assad-Garcia, C. Andrews-Pfannkoch, E. A. Denisova, L. Young, Z. Q. Qi, T. H. Segall-Shapiro, C. H. Calvey, P. P. Parmar, C. A. Hutchison, 3rd, H. O. Smith and J. C. Venter, Science, 2010, 329, 52–56 CrossRef CAS.
- L. Thomason, D. L. Court, M. Bubunenko, N. Costantino, H. Wilson, S. Datta and A. Oppenheim, Curr. Protoc. Mol. Biol., 2007, ch. 1, unit 1 16 Search PubMed.
- J. P. Muyrers, Y. Zhang and A. F. Stewart, Trends Biochem. Sci., 2001, 26, 325–331 CrossRef CAS.
- D. A. Hopwood, Chem. Rev., 1997, 97, 2465–2498 CrossRef CAS.
- D. A. Hopwood, M. J. Bibb, K. F. Chater, T. Kieser, C. J. Bruton, H. M. Kieser, D. J. Lydiate, C. P. Smith, J. M. Ward, and H. Schrempf, Genetic manipulation of Streptomyces: a laboratory manual, John Innes Foundation, Norwich, UK, 1985 Search PubMed.
- B. M, T. Kieser, M. J. Buttner, K. F. Chater, D. A. Hopwood, Practical Streptomyces genetics, John Innes Centre, Norwich, UK., 2000 Search PubMed.
- K. F. Chater, Bio/Technology, 1990, 8, 115–121 CrossRef CAS.
- C. R. Hutchinson et al., Ann. N. Y. Acad. Sci., 1991, 646, 78–93 CrossRef.
- P. Mazodier, R. Petter and C. Thompson, J. Bacteriol., 1989, 171, 3583–3585 CAS.
- M. Bierman, R. Logan, K. O'Brien, E. T. Seno, R. N. Rao and B. E. Schoner, Gene, 1992, 116, 43–49 CrossRef CAS.
- F. Flett, V. Mersinias and C. P. Smith, FEMS Microbiol. Lett., 1997, 155, 223–229 CrossRef CAS.
- M. Bibb, J. L. Schottel and S. N. Cohen, Nature, 1980, 284, 526–531 CrossRef CAS.
- D. J. Lydiate, F. Malpartida and D. A. Hopwood, Gene, 1985, 35, 223–235 CrossRef CAS.
- D. A. Hopwood, M. J. Bibb, K. F. Chater and T. Kieser, Methods Enzymol., 1987, 153, 116–166 Search PubMed.
- S. C. Wenzel, H. B. Bode, I. Kochems and R. Muller, ChemBioChem, 2008, 9, 2711–2721 CrossRef CAS.
- E. Takano, J. White, C. J. Thompson and M. J. Bibb, Gene, 1995, 166, 133–137 CrossRef CAS.
- C. J. Thompson and G. S. Gray, Proc. Natl. Acad. Sci. U. S. A., 1983, 80, 5190–5194 CAS.
- G. R. Janssen, J. M. Ward and M. J. Bibb, Genes Dev., 1989, 3, 415–429 CrossRef CAS.
- E. Katz, C. J. Thompson and D. A. Hopwood, J. Gen. Microbiol., 1983, 129, 2703–2714 CAS.
- C. J. Rowe, J. Cortes, S. Gaisser, J. Staunton and P. F. Leadlay, Gene, 1998, 216, 215–223 CrossRef CAS.
- J. Vara, M. Lewandowska-Skarbek, Y. G. Wang, S. Donadio and C. R. Hutchinson, J. Bacteriol., 1989, 171, 5872–5881 CAS.
- B. Sthapit, T. J. Oh, R. Lamichhane, K. Liou, H. C. Lee, C. G. Kim and J. K. Sohng, FEBS Lett., 2004, 566, 201–206 CrossRef CAS.
- T. Murakami, T. G. Holt and C. J. Thompson, J. Bacteriol., 1989, 171, 1459–1466 CAS.
- M. J. Bibb, J. White, J. M. Ward and G. R. Janssen, Mol. Microbiol., 1994, 14, 533–545 CrossRef CAS.
- M. Doumith, P. Weingarten, U. F. Wehmeier, K. Salah-Bey, B. Benhamou, C. Capdevila, J. M. Michel, W. Piepersberg and M. C. Raynal, Mol. Gen. Genet., 2000, 264, 477–485 CrossRef CAS.
- M. J. Bibb, G. R. Janssen and J. M. Ward, Gene, 1985, 38, 215–226 CrossRef CAS.
- S. Herai, Y. Hashimoto, H. Higashibata, H. Maseda, H. Ikeda, S. Omura and M. Kobayashi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 14031–14035 CrossRef CAS.
- Z. Hindle and C. P. Smith, Mol. Microbiol., 1994, 12, 737–745 CrossRef CAS.
- A. Rodriguez-Garcia, P. Combes, R. Perez-Redondo and M. C. Smith, Nucleic Acids Res., 2005, 33, e87 CrossRef.
- V. Dangel, L. Westrich, M. C. Smith, L. Heide and B. Gust, Appl. Microbiol. Biotechnol., 2010, 87, 261–269 CrossRef CAS.
- T. Schmitt-John and J. W. Engels, Appl. Microbiol. Biotechnol., 1992, 36, 493–498 CAS.
- C. J. Wilkinson, Z. A. Hughes-Thomas, C. J. Martin, I. Bohm, T. Mironenko, M. Deacon, M. Wheatcroft, G. Wirtz, J. Staunton and P. F. Leadlay, J. Mol. Microbiol. Biotechnol., 2002, 4, 417–426 CAS.
- R. H. Baltz, Bioprocess. Technol., 1995, 22, 309–381 Search PubMed.
- Z. Hu, R. P. Desai, Y. Volchegursky, T. Leaf, E. Woo, P. Licari, D. V. Santi, C. R. Hutchinson and R. McDaniel, J. Ind. Microbiol. Biotechnol., 2003, 30, 161–167 CAS.
- T. Kieser and D. A. Hopwood, Methods Enzymol., 1991, 204, 430–458 CAS.
- Z. Hu, D. A. Hopwood and C. R. Hutchinson, J. Ind. Microbiol. Biotechnol., 2003, 30, 516–522 CrossRef CAS.
- R. Fong, J. A. Vroom, Z. Hu, C. R. Hutchinson, J. Huang, S. N. Cohen and C. M. Kao, Appl. Environ. Microbiol., 2007, 73, 1296–1307 CrossRef CAS.
- A. S. Eustaquio, B. Gust, S. M. Li, S. Pelzer, W. Wohlleben, K. F. Chater and L. Heide, Chem. Biol., 2004, 11, 1561–1572 CrossRef CAS.
- R. Zirkle, J. M. Ligon and I. Molnar, Microbiology, 2004, 150, 2761–2774 CrossRef CAS.
- R. H. Baltz and T. J. Hosted, Trends Biotechnol., 1996, 14, 245–250 CrossRef CAS.
- M. Sosio, F. Giusino, C. Cappellano, E. Bossi, A. M. Puglia and S. Donadio, Nat. Biotechnol., 2000, 18, 343–345 CrossRef CAS.
- A. Martinez, S. J. Kolvek, C. L. Yip, J. Hopke, K. A. Brown, I. A. MacNeil and M. S. Osburne, Appl. Environ. Microbiol., 2004, 70, 2452–2463 CrossRef CAS.
- V. Miao, M. F. Coeffet-Legal, P. Brian, R. Brost, J. Penn, A. Whiting, S. Martin, R. Ford, I. Parr, M. Bouchard, C. J. Silva, S. K. Wrigley and R. H. Baltz, Microbiology, 2005, 151, 1507–1523 CrossRef CAS.
- Z. Feng, L. Wang, S. R. Rajski, Z. Xu, M. F. Coeffet-LeGal and B. Shen, Bioorg. Med. Chem., 2009, 17, 2147–2153 CrossRef CAS.
- J. Wild, Z. Hradecna and W. Szybalski, Genome Res., 2002, 12, 1434–1444 CrossRef CAS.
- M. A. Gregory, R. Till and M. C. Smith, J. Bacteriol., 2003, 185, 5320–5323 CrossRef CAS.
- C. M. Kao, L. Katz and C. Khosla, Science, 1994, 265, 509–512 CrossRef CAS.
- K. Ichinose, D. J. Bedford, D. Tornus, A. Bechthold, M. J. Bibb, W. P. Revill, H. G. Floss and D. A. Hopwood, Chem. Biol., 1998, 5, 647–659 CrossRef CAS.
- K. Ichinose, M. Ozawa, K. Itou, K. Kunieda and Y. Ebizuka, Microbiology, 2003, 149, 1633–1645 CrossRef CAS.
- L. Tang, S. Shah, L. Chung, J. Carney, L. Katz, C. Khosla and B. Julien, Science, 2000, 287, 640–642 CrossRef CAS.
- D. J. Bedford, E. Schweizer, D. A. Hopwood and C. Khosla, J. Bacteriol., 1995, 177, 4544–4548 CAS.
- A. S. Eustaquio, B. Gust, U. Galm, S. M. Li, K. F. Chater and L. Heide, Appl. Environ. Microbiol., 2005, 71, 2452–2459 CrossRef CAS.
- R. McDaniel, S. Ebert-Khosla, D. A. Hopwood and C. Khosla, Science, 1993, 262, 1546–1550 CAS.
- W. Zhang, B. D. Ames, S. C. Tsai and Y. Tang, Appl. Environ. Microbiol., 2006, 72, 2573–2580 CrossRef CAS.
- W. Zhang, K. Watanabe, C. C. Wang and Y. Tang, J. Biol. Chem., 2007, 282, 25717–25725 CrossRef CAS.
- R. H. Baltz, Nat. Biotechnol., 2006, 24, 1533–1540 CrossRef CAS.
- S. Kuhstoss, M. A. Richardson and R. N. Rao, Gene, 1991, 97, 143–146 CrossRef CAS.
- Q. Xue, G. Ashley, C. R. Hutchinson and D. V. Santi, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 11740–11745 CrossRef CAS.
- T. Smokvina, P. Mazodier, F. Boccard, C. J. Thompson and M. Guerineau, Gene, 1990, 94, 53–59 CrossRef CAS.
- R. Ziermann and M. C. Betlach, Biotechniques, 1999, 26, 106–110 CAS.
- F. W. Studier and B. A. Moffatt, J. Mol. Biol., 1986, 189, 113–130 CrossRef CAS.
- F. X. Lussier, F. Denis and F. Shareck, Appl. Environ. Microbiol., 2010, 76, 967–970 CrossRef CAS.
- T. Ito, N. Roongsawang, N. Shirasaka, W. Lu, P. M. Flatt, N. Kasanah, C. Miranda and T. Mahmud, ChemBioChem, 2009, 10, 2253–2265 CrossRef CAS.
- Z. Hu, D. A. Hopwood and C. Khosla, Appl. Environ. Microbiol., 2000, 66, 2274–2277 CrossRef CAS.
- J. T. Kealey, L. Liu, D. V. Santi, M. C. Betlach and P. J. Barr, Proc. Natl. Acad. Sci. U. S. A., 1998, 95, 505–509 CrossRef CAS.
- R. H. Lambalot, A. M. Gehring, R. S. Flugel, P. Zuber, M. LaCelle, M. A. Marahiel, R. Reid, C. Khosla and C. T. Walsh, Chem. Biol., 1996, 3, 923–936 CAS.
- L. E. Quadri, P. H. Weinreb, M. Lei, M. M. Nakano, P. Zuber and C. T. Walsh, Biochemistry, 1998, 37, 1585–1595 CrossRef CAS.
- B. A. Pfeifer, S. J. Admiraal, H. Gramajo, D. E. Cane and C. Khosla, Science, 2001, 291, 1790–1792 CrossRef CAS.
- K. K. Lee, N. A. Da Silva and J. T. Kealey, Anal. Biochem., 2009, 394, 75–80 CrossRef CAS.
- J. F. Sanchez, Y. M. Chiang and C. C. Wang, Mol. Pharmaceutics, 2008, 5, 226–233 CrossRef CAS.
- J. Schumann and C. Hertweck, J. Biotechnol., 2006, 124, 690–703 CrossRef.
- P. Schneider, M. Misiek and D. Hoffmeister, Mol. Pharmaceutics, 2008, 5, 234–242 CrossRef CAS.
- V. Meyer, Biotechnol. Adv., 2008, 26, 177–185 CrossRef CAS.
- S. Bergmann, J. Schumann, K. Scherlach, C. Lange, A. A. Brakhage and C. Hertweck, Nat. Chem. Biol., 2007, 3, 213–217 CrossRef CAS.
- S. Wattanachaisaereekul, A. E. Lantz, M. L. Nielsen, O. S. Andresson and J. Nielsen, Biotechnol. Bioeng., 2007, 97, 893–900 CrossRef.
- S. C. Mutka, S. M. Bondi, J. R. Carney, N. A. Da Silva and J. T. Kealey, FEMS Yeast Res., 2006, 6, 40–47 CrossRef CAS.
- J. R. Fincham, Microbiol. Rev., 1989, 53, 148–170 CAS.
- B. Ruiz-Diez, J. Appl. Microbiol., 2002, 92, 189–195 CrossRef CAS.
- I. Fujii, Y. Ono, H. Tada, K. Gomi, Y. Ebizuka and U. Sankawa, Mol. Gen. Genet., 1996, 253, 1–10 CrossRef CAS.
- P. Lu, A. Zhang, L. M. Dennis, A. M. Dahl-Roshak, Y. Q. Xia, B. Arison, Z. An and J. S. Tkacz, Mol. Genet. Genomics, 2005, 273, 207–216 CrossRef CAS.
- R. B. Waring, G. S. May and N. R. Morris, Gene, 1989, 79, 119–130 CrossRef CAS.
- I. Fujii, Y. Mori, A. Watanabe, Y. Kubo, G. Tsuji and Y. Ebizuka, Biosci., Biotechnol., Biochem., 1999, 63, 1445–1452 CrossRef CAS.
- R. J. Cox, F. Glod, D. Hurley, C. M. Lazarus, T. P. Nicholson, B. A. Rudd, T. J. Simpson, B. Wilkinson and Y. Zhang, Chem. Commun., 2004, 2260–2261 RSC.
- A. M. Bailey, R. J. Cox, K. Harley, C. M. Lazarus, T. J. Simpson and E. Skellam, Chem. Commun., 2007, 4053–4055 RSC.
- J. Kennedy, K. Auclair, S. G. Kendrew, C. Park, J. C. Vederas and C. R. Hutchinson, Science, 1999, 284, 1368–1372 CrossRef CAS.
- H. J. Pel, J. H. de Winde, D. B. Archer, P. S. Dyer, G. Hofmann, P. J. Schaap, G. Turner, R. P. de Vries, R. Albang, K. Albermann, M. R. Andersen, J. D. Bendtsen, J. A. Benen, M. van den Berg, S. Breestraat, M. X. Caddick, R. Contreras, M. Cornell, P. M. Coutinho, E. G. Danchin, A. J. Debets, P. Dekker, P. W. van Dijck, A. van Dijk, L. Dijkhuizen, A. J. Driessen, C. d'Enfert, S. Geysens, C. Goosen, G. S. Groot, P. W. de Groot, T. Guillemette, B. Henrissat, M. Herweijer, J. P. van den Hombergh, C. A. van den Hondel, R. T. van der Heijden, R. M. van der Kaaij, F. M. Klis, H. J. Kools, C. P. Kubicek, P. A. van Kuyk, J. Lauber, X. Lu, M. J. van der Maarel, R. Meulenberg, H. Menke, M. A. Mortimer, J. Nielsen, S. G. Oliver, M. Olsthoorn, K. Pal, N. N. van Peij, A. F. Ram, U. Rinas, J. A. Roubos, C. M. Sagt, M. Schmoll, J. Sun, D. Ussery, J. Varga, W. Vervecken, P. J. van de Vondervoort, H. Wedler, H. A. Wosten, A. P. Zeng, A. J. van Ooyen, J. Visser and H. Stam, Nat. Biotechnol., 2007, 25, 221–231 CrossRef.
- W. Minas, P. Brunker, P. T. Kallio and J. E. Bailey, Biotechnol. Prog., 1998, 14, 561–566 CrossRef CAS.
- J. L. Adrio and A. L. Demain, FEMS Microbiol. Rev., 2006, 30, 187–214 CrossRef CAS.
- J. Cortes, S. F. Haydock, G. A. Roberts, D. J. Bevitt and P. F. Leadlay, Nature, 1990, 348, 176–178 CrossRef CAS.
- S. Donadio, M. J. Staver, J. B. McAlpine, S. J. Swanson and L. Katz, Science, 1991, 252, 675–679 CrossRef CAS.
- R. G. Summers, S. Donadio, M. J. Staver, E. Wendt-Pienkowski, C. R. Hutchinson and L. Katz, Microbiology, 1997, 143(10), 3251–3262 CrossRef CAS.
- H. C. Gramajo, E. Takano and M. J. Bibb, Mol. Microbiol., 1993, 7, 837–845 CrossRef CAS.
- E. Wendt-Pienkowski, Y. Huang, J. Zhang, B. Li, H. Jiang, H. Kwon, C. R. Hutchinson and B. Shen, J. Am. Chem. Soc., 2005, 127, 16442–16452 CrossRef CAS.
- S. D. Bentley, K. F. Chater, A. M. Cerdeno-Tarraga, G. L. Challis, N. R. Thomson, K. D. James, D. E. Harris, M. A. Quail, H. Kieser, D. Harper, A. Bateman, S. Brown, G. Chandra, C. W. Chen, M. Collins, A. Cronin, A. Fraser, A. Goble, J. Hidalgo, T. Hornsby, S. Howarth, C. H. Huang, T. Kieser, L. Larke, L. Murphy, K. Oliver, S. O'Neil, E. Rabbinowitsch, M. A. Rajandream, K. Rutherford, S. Rutter, K. Seeger, D. Saunders, S. Sharp, R. Squares, S. Squares, K. Taylor, T. Warren, A. Wietzorrek, J. Woodward, B. G. Barrell, J. Parkhill and D. A. Hopwood, Nature, 2002, 417, 141–147 CrossRef.
- J. R. Jacobsen, C. R. Hutchinson, D. E. Cane and C. Khosla, Science, 1997, 277, 367–369 CrossRef CAS.
- C. Carreras, S. Frykman, S. Ou, L. Cadapan, S. Zavala, E. Woo, T. Leaf, J. Carney, M. Burlingame, S. Patel, G. Ashley and P. Licari, J. Biotechnol., 2002, 92, 217–228 CrossRef CAS.
- G. A. Roberts, J. Staunton and P. F. Leadlay, Eur. J. Biochem., 1993, 214, 305–311 CAS.
- R. S. Gokhale, S. Y. Tsuji, D. E. Cane and C. Khosla, Science, 1999, 284, 482–485 CrossRef CAS.
- M. Golomb and M. Chamberlin, J. Biol. Chem., 1974, 249, 2858–2863 CAS.
- S. Textor, V. F. Wendisch, A. A. De Graaf, U. Muller, M. I. Linder, D. Linder and W. Buckel, Arch. Microbiol., 1997, 168, 428–436 CrossRef CAS.
- A. R. Horswill and J. C. Escalante-Semerena, J. Bacteriol., 1999, 181, 5615–5623 CAS.
- E. Rodriguez and H. Gramajo, Microbiology, 1999, 145(Pt 11), 3109–3119 CAS.
- Y. Wang and B. A. Pfeifer, Metab. Eng., 2008, 10, 33–38 CrossRef CAS.
- S. Peiru, H. G. Menzella, E. Rodriguez, J. Carney and H. Gramajo, Appl. Environ. Microbiol., 2005, 71, 2539–2547 CrossRef CAS.
- H. Y. Lee and C. Khosla, PLoS Biol., 2007, 5, e45 CrossRef.
- K. Watanabe, M. A. Rude, C. T. Walsh and C. Khosla, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9774–9778 CrossRef CAS.
- H. Zhang, Y. Wang, J. Wu, K. Skalina and B. A. Pfeifer, Chem. Biol., 2010 Search PubMed , in press.
- H. G. Menzella, S. J. Reisinger, M. Welch, J. T. Kealey, J. Kennedy, R. Reid, C. Q. Tran and D. V. Santi, J. Ind. Microbiol. Biotechnol., 2006, 33, 22–28 CrossRef CAS.
- R. H. Baltz, Curr. Opin. Chem. Biol., 2009, 13, 144–151 CrossRef CAS.
- R. H. Baltz, Curr. Top. Med. Chem., 2008, 8, 618–638 CrossRef CAS.
- F. Lombo, A. Velasco, A. Castro, F. de la Calle, A. F. Brana, J. M. Sanchez-Puelles, C. Mendez and J. A. Salas, ChemBioChem, 2006, 7, 366–376 CrossRef CAS.
- J. Penn, X. Li, A. Whiting, M. Latif, T. Gibson, C. J. Silva, P. Brian, J. Davies, V. Miao, S. K. Wrigley and R. H. Baltz, J. Ind. Microbiol. Biotechnol., 2006, 33, 121–128 CrossRef.
- E. A. Felnagle, M. R. Rondon, A. D. Berti, H. A. Crosby and M. G. Thomas, Appl. Environ. Microbiol., 2007, 73, 4162–4170 CrossRef CAS.
- F. Rusnak, M. Sakaitani, D. Drueckhammer, J. Reichert and C. T. Walsh, Biochemistry, 1991, 30, 2916–2927 CrossRef CAS.
- H. von Dohren, de Gruyter, Berlin, New York, 1990, pp. 411–506.
- B. A. Pfeifer, C. C. Wang, C. T. Walsh and C. Khosla, Appl. Environ. Microbiol., 2003, 69, 6698–6702 CrossRef CAS.
- S. C. Mutka, J. R. Carney, Y. Liu and J. Kennedy, Biochemistry, 2006, 45, 1321–1330 CrossRef CAS.
- S. Gruenewald, H. D. Mootz, P. Stehmeier and T. Stachelhaus, Appl. Environ. Microbiol., 2004, 70, 3282–3291 CrossRef CAS.
- K. Watanabe, K. Hotta, A. P. Praseuth, K. Koketsu, A. Migita, C. N. Boddy, C. C. Wang, H. Oguri and H. Oikawa, Nat. Chem. Biol., 2006, 2, 423–428 CrossRef CAS.
- C. N. Boddy, K. Hotta, M. L. Tse, R. E. Watts and C. Khosla, J. Am. Chem. Soc., 2004, 126, 7436–7437 CrossRef CAS.
- K. Watanabe, K. Hotta, M. Nakaya, A. P. Praseuth, C. C. Wang, D. Inada, K. Takahashi, E. Fukushi, H. Oguri and H. Oikawa, J. Am. Chem. Soc., 2009, 131, 9347–9353 CrossRef CAS.
- D. A. Hopwood and M. J. Merrick, Bacteriol Rev, 1977, 41, 595–635 CAS.
- H. Kleinkauf and H. von Dohren, Antonie van Leeuwenhoek, 1995, 67, 229–242 CrossRef CAS.
- S. Bron, W. Meijer, S. Holsappel and P. Haima, Res. Microbiol., 1991, 142, 875–883 CrossRef CAS.
- D. R. Zeigler, Bacillus Genetic Stock Center Catalog of Strains (7th edn), Volume 4: Integration Vectors for Gram-Positive Organisms, http://www.bgsc.org/Catpart4.pdf Search PubMed.
- K. Eppelmann, S. Doekel and M. A. Marahiel, J. Biol. Chem., 2001, 276, 34824–34831 CrossRef CAS.
- Y. Saito, H. Taguchi and T. Akamatsu, J. Biosci. Bioeng., 2006, 101, 257–262 CrossRef CAS.
- M. Itaya and S. Kaneko, Nucleic Acids Res., 2010, 38, 2551–2557 CrossRef CAS.
- B. A. Boghigian and B. A. Pfeifer, Biotechnol. Lett., 2008, 30, 1323–1330 CrossRef CAS.
- S. Doekel, K. Eppelmann and M. A. Marahiel, FEMS Microbiol. Lett., 2002, 216, 185–191 CrossRef CAS.
- S. C. Wenzel, F. Gross, Y. Zhang, J. Fu, A. F. Stewart and R. Muller, Chem. Biol., 2005, 12, 349–356 CrossRef CAS.
- F. Gross, M. W. Ring, O. Perlova, J. Fu, S. Schneider, K. Gerth, S. Kuhlmann, A. F. Stewart, Y. Zhang and R. Muller, Chem. Biol., 2006, 13, 1253–1264 CrossRef CAS.
- J. Fu, S. C. Wenzel, O. Perlova, J. Wang, F. Gross, Z. Tang, Y. Yin, A. F. Stewart, R. Muller and Y. Zhang, Nucleic Acids Res., 2008, 36, e113 CrossRef.
- K. A. Datsenko and B. L. Wanner, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 6640–6645 CrossRef CAS.
- B. Gust, G. L. Challis, K. Fowler, T. Kieser and K. F. Chater, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1541–1546 CrossRef CAS.
- D. R. Hahn, P. J. Solenberg, M. A. McHenney and R. H. Baltz, J. Ind. Microbiol., 1991, 7, 229–234 CrossRef CAS.
- S. J. Kodumal, K. G. Patel, R. Reid, H. G. Menzella, M. Welch and D. V. Santi, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15573–15578 CrossRef CAS.
- R. S. Gokhale and C. Khosla, Curr. Opin. Chem. Biol., 2000, 4, 22–27 CrossRef CAS.
- L. Katz, Chem. Rev., 1997, 97, 2557–2576 CrossRef CAS.
- P. R. Arsenault, K. K. Wobbe and P. J. Weathers, Curr. Med. Chem., 2008, 15, 2886–2896 CrossRef CAS.
- V. J. Martin, D. J. Pitera, S. T. Withers, J. D. Newman and J. D. Keasling, Nat. Biotechnol., 2003, 21, 796–802 CrossRef CAS.
- M. C. Chang, R. A. Eachus, W. Trieu, D. K. Ro and J. D. Keasling, Nat. Chem. Biol., 2007, 3, 274–277 CrossRef CAS.
- D. K. Ro, E. M. Paradise, M. Ouellet, K. J. Fisher, K. L. Newman, J. M. Ndungu, K. A. Ho, R. A. Eachus, T. S. Ham, J. Kirby, M. C. Chang, S. T. Withers, Y. Shiba, R. Sarpong and J. D. Keasling, Nature, 2006, 440, 940–943 CrossRef CAS.
- M. Hezari, N. G. Lewis and R. Croteau, Arch. Biochem. Biophys., 1995, 322, 437–444 CrossRef CAS.
- M. R. Wildung and R. Croteau, J. Biol. Chem., 1996, 271, 9201–9204 CrossRef CAS.
- J. Hefner, S. M. Rubenstein, R. E. Ketchum, D. M. Gibson, R. M. Williams and R. Croteau, Chem. Biol., 1996, 3, 479–489 CAS.
- K. Walker, R. E. Ketchum, M. Hezari, D. Gatfield, M. Goleniowski, A. Barthol and R. Croteau, Arch. Biochem. Biophys., 1999, 364, 273–279 CrossRef CAS.
- K. Walker and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 583–587 CrossRef CAS.
- K. Walker and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2000, 97, 13591–13596 CrossRef CAS.
- K. Walker, A. Schoendorf and R. Croteau, Arch. Biochem. Biophys., 2000, 374, 371–380 CrossRef CAS.
- A. Schoendorf, C. D. Rithner, R. M. Williams and R. B. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 1501–1506 CrossRef CAS.
- S. Jennewein, C. D. Rithner, R. M. Williams and R. B. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2001, 98, 13595–13600 CrossRef CAS.
- K. Walker, S. Fujisaki, R. Long and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 12715–12720 CrossRef CAS.
- K. Walker, R. Long and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2002, 99, 9166–9171 CrossRef CAS.
- M. Chau and R. Croteau, Arch. Biochem. Biophys., 2004, 427, 48–57 CrossRef CAS.
- M. Chau, S. Jennewein, K. Walker and R. Croteau, Chem. Biol., 2004, 11, 663–672 CAS.
- S. Jennewein, M. R. Wildung, M. Chau, K. Walker and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 9149–9154 CrossRef CAS.
- K. X. Huang, Q. L. Huang, M. R. Wildung, R. Croteau and A. I. Scott, Protein Expression Purif., 1998, 13, 90–96 CrossRef CAS.
- K. C. Nicolaou et al., Nature, 1994, 367, 630–634 CrossRef CAS.
- J. N. Denis, A. E. Greene, D. Guenard, F. Guerittevoegelein, L. Mangatal and P. Potier, J. Am. Chem. Soc., 1988, 110, 5917–5919 CrossRef CAS.
- D. Frense, Appl. Microbiol. Biotechnol., 2007, 73, 1233–1240 CrossRef CAS.
- Y. Yukimune, H. Tabata, Y. Higashi and Y. Hara, Nat. Biotechnol., 1996, 14, 1129–1132 CrossRef CAS.
- C. De Dobbeleer, M. Cloutier, M. Fouilland, R. Legros and M. Jolicoeur, Biotechnol. Bioeng., 2006, 95, 1126–1137 CrossRef CAS.
- S. Bentebibel, E. Moyano, J. Palazon, R. M. Cusido, M. Bonfill, R. Eibl and M. T. Pinol, Biotechnol. Bioeng., 2005, 89, 647–655 CrossRef CAS.
- O. Besumbes, S. Sauret-Gueto, M. A. Phillips, S. Imperial, M. Rodriguez-Concepcion and A. Boronat, Biotechnol. Bioeng., 2004, 88, 168–175 CrossRef CAS.
- K. Kovacs, L. Zhang, R. S. Linforth, B. Whittaker, C. J. Hayes and R. G. Fray, Transgenic Res., 2007, 16, 121–126 CrossRef CAS.
- A. Stierle, G. Strobel and D. Stierle, Science, 1993, 260, 214–216 CrossRef CAS.
- D. Zhou and X. W. Ping, J. Microbiol., 2001, 21, 18–20 Search PubMed.
- Z. K, D. P. Zhou, W. X. Ping, J. P. Ge, X. Ma and L. Jun, J. Am. Sci., 2005, 1, 55–62 Search PubMed.
- P. W, K. Zhao, X. Ma, J. Liu and D. P. Zhou, Acta Microbiol. Sinica, 2005, 45, 355–358 Search PubMed.
- D. C. Williams, M. R. Wildung, A. Q. Jin, D. Dalal, J. S. Oliver, R. M. Coates and R. Croteau, Arch. Biochem. Biophys., 2000, 379, 137–146 CrossRef CAS.
- Q. Huang, C. A. Roessner, R. Croteau and A. I. Scott, Bioorg. Med. Chem., 2001, 9, 2237–2242 CrossRef CAS.
- L. Z. Yuan, P. E. Rouviere, R. A. Larossa and W. Suh, Metab. Eng., 2006, 8, 79–90 CrossRef CAS.
- J. M. Dejong, Y. Liu, A. P. Bollon, R. M. Long, S. Jennewein, D. Williams and R. B. Croteau, Biotechnol. Bioeng., 2006, 93, 212–224 CrossRef CAS.
- B. Engels, P. Dahm and S. Jennewein, Metab. Eng., 2008, 10, 201–206 CrossRef CAS.
- P. K. Ajikumar, W.-H. Xiao, K. E. J. Tyo, Y. Wang, F. Simeon, E. Leonard, O. Mucha, T. H. Phon, B. Pfeifer and G. Stephanopoulos, Science, 2010, 330, 70–74 CrossRef CAS.
- L. Bai, L. Li, H. Xu, K. Minagawa, Y. Yu, Y. Zhang, X. Zhou, H. G. Floss, T. Mahmud and Z. Deng, Chem. Biol., 2006, 13, 387–397 CrossRef CAS.
- L. M. Halo, J. W. Marshall, A. A. Yakasai, Z. Song, C. P. Butts, M. P. Crump, M. Heneghan, A. M. Bailey, T. J. Simpson, C. M. Lazarus and R. J. Cox, ChemBioChem, 2008, 9, 585–594 CrossRef CAS.
- L. Kaysser, E. Wemakor, S. Siebenberg, J. A. Salas, J. K. Sohng, B. Kammerer and B. Gust, Appl. Environ. Microbiol., 2010, 76, 4008–4018 CrossRef CAS.
- S. M. Ma, J. W. Li, J. W. Choi, H. Zhou, K. K. Lee, V. A. Moorthie, X. Xie, J. T. Kealey, N. A. Da Silva, J. C. Vederas and Y. Tang, Science, 2009, 326, 589–592 CrossRef CAS.
- V. Siewers, X. Chen, L. Huang, J. Zhang and J. Nielsen, Metab. Eng., 2009, 11, 391–397 CrossRef CAS.
- R. I. Amann, W. Ludwig and K. H. Schleifer, Microbiol. Rev., 1995, 59, 143–169 CAS.
- C. T. Walsh and M. A. Fischbach, J. Am. Chem. Soc., 2010, 132, 2469–2493 CrossRef CAS.
- E. Zazopoulos, K. Huang, A. Staffa, W. Liu, B. O. Bachmann, K. Nonaka, J. Ahlert, J. S. Thorson, B. Shen and C. M. Farnet, Nat. Biotechnol., 2003, 21, 187–190 CrossRef CAS.
- K. Lewis, S. Epstein, A. D'Onofrio and L. L. Ling, J. Antibiot., 2010 Search PubMed.
- K. E. Davis, S. J. Joseph and P. H. Janssen, Appl. Environ. Microbiol., 2005, 71, 826–834 CrossRef CAS.
- P. D. Schloss and J. Handelsman, GenomeBiology, 2005, 6, 229 CrossRef.
- H. L. Steele, K. E. Jaeger, R. Daniel and W. R. Streit, J. Mol. Microbiol. Biotechnol., 2009, 16, 25–37 CrossRef CAS.
- H. B. Bode and R. Muller, Angew. Chem., Int. Ed., 2005, 44, 6828–6846 CrossRef CAS.
- B. Wilkinson and J. Micklefield, Nat. Chem. Biol., 2007, 3, 379–386 CrossRef CAS.
- C. Corre and G. L. Challis, Nat. Prod. Rep., 2009, 26, 977–986 RSC.
- M. Nett, H. Ikeda and B. S. Moore, Nat. Prod. Rep., 2009, 26, 1362–1384 RSC.
- J. B. McAlpine, J. Nat. Prod., 2009, 72, 566–572 CrossRef CAS.
- E. Rodriguez, Z. Hu, S. Ou, Y. Volchegursky, C. R. Hutchinson and R. McDaniel, J. Ind. Microbiol. Biotechnol., 2003, 30, 480–488 CrossRef CAS.
- A. M. Lum, J. Huang, C. R. Hutchinson and C. M. Kao, Metab. Eng., 2004, 6, 186–196 CrossRef CAS.
- G. Stephanopoulos, Metab. Eng., 1999, 1, 1–11 CrossRef CAS.
- B. A. Boghigian, G. Seth, R. Kiss and B. A. Pfeifer, Metab. Eng., 2010, 12, 81–95 CrossRef CAS.
- P. Brunke, O. Sterner, J. E. Bailey and W. Minas, Antonie van Leeuwenhoek, 2001, 79, 235–245 CrossRef CAS.
- L. Xiang and B. S. Moore, J. Biol. Chem., 2002, 277, 32505–32509 CrossRef CAS.
- M. Otsuka, K. Ichinose, I. Fujii and Y. Ebizuka, Antimicrob. Agents Chemother., 2004, 48, 3468–3476 CrossRef CAS.
- W. Zhang, K. Watanabe, C. C. Wang and Y. Tang, J. Nat. Prod., 2006, 69, 1633–1636 CrossRef CAS.
- C. Khosla, R. McDaniel, S. Ebert-Khosla, R. Torres, D. H. Sherman, M. J. Bibb and D. A. Hopwood, J. Bacteriol., 1993, 175, 2197–2204 CAS.
- D. E. Cane, G. Luo, C. Khosla, C. M. Kao and L. Katz, J. Antibiot., 1995, 48, 647–651 CAS.
- G. Blanco, H. Fu, C. Mendez, C. Khosla and J. A. Salas, Chem. Biol., 1996, 3, 193–196 CrossRef CAS.
- P. Brunker, K. McKinney, O. Sterner, W. Minas and J. E. Bailey, Gene, 1999, 227, 125–135 CrossRef CAS.
- C. Olano, S. J. Moss, A. F. Brana, R. M. Sheridan, V. Math, A. J. Weston, C. Mendez, P. F. Leadlay, B. Wilkinson and J. A. Salas, Mol. Microbiol., 2004, 52, 1745–1756 CrossRef CAS.
- Z. Xu, K. Jakobi, K. Welzel and C. Hertweck, Chem. Biol., 2005, 12, 579–588 CrossRef CAS.
- S. Gullon, C. Olano, M. S. Abdelfattah, A. F. Brana, J. Rohr, C. Mendez and J. A. Salas, Appl. Environ. Microbiol., 2006, 72, 4172–4183 CrossRef CAS.
- P. Lopez, A. Hornung, K. Welzel, C. Unsin, W. Wohlleben, T. Weber and S. Pelzer, Gene, 2010, 461, 5–14 CrossRef CAS.
- J. M. Winter, M. C. Moffitt, E. Zazopoulos, J. B. McAlpine, P. C. Dorrestein and B. S. Moore, J. Biol. Chem., 2007, 282, 16362–16368 CrossRef CAS.
- S. Y. Kim, P. Zhao, M. Igarashi, R. Sawa, T. Tomita, M. Nishiyama and T. Kuzuyama, Chem. Biol., 2009, 16, 736–743 CrossRef CAS.
- S. L. Ward, Z. Hu, A. Schirmer, R. Reid, W. P. Revill, C. D. Reeves, O. V. Petrakovsky, S. D. Dong and L. Katz, Antimicrob. Agents Chemother., 2004, 48, 4703–4712 CrossRef CAS.
- E. Rodriguez, S. Ward, H. Fu, W. P. Revill, R. McDaniel and L. Katz, Appl. Microbiol. Biotechnol., 2004, 66, 85–91 CrossRef CAS.
- U. von Mulert, A. Luzhetskyy, C. Hofmann, A. Mayer and A. Bechthold, FEMS Microbiol. Lett., 2004, 230, 91–97 CrossRef CAS.
- F. Malpartida and D. A. Hopwood, Nature, 1984, 309, 462–464 CrossRef CAS.
- Y. J. Yoon, B. J. Beck, B. S. Kim, H. Y. Kang, K. A. Reynolds and D. H. Sherman, Chem. Biol., 2002, 9, 203–214 CrossRef CAS.
- W. S. Jung, S. K. Lee, J. S. Hong, S. R. Park, S. J. Jeong, A. R. Han, J. K. Sohng, B. G. Kim, C. Y. Choi, D. H. Sherman and Y. J. Yoon, Appl. Microbiol. Biotechnol., 2006, 72, 763–769 CrossRef CAS.
- N. P. Kurumbang, T. J. Oh, K. Liou and J. K. Sohng, J. Microbiol. Biotechnol., 2008, 18, 866–873 CAS.
- J. W. Park, J. S. Hong, N. Parajuli, W. S. Jung, S. R. Park, S. K. Lim, J. K. Sohng and Y. J. Yoon, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 8399–8404 CrossRef CAS.
- B. B. Pageni, T. J. Oh, H. C. Lee and J. K. Sohng, Biotechnol. Lett., 2008, 30, 1609–1615 CrossRef CAS.
- S. Maharjan, T. J. Oh, H. C. Lee and J. K. Sohng, Biotechnol. Lett., 2008, 30, 1621–1626 CrossRef CAS.
- L. P. Thapa, T. J. Oh, K. Liou and J. K. Sohng, J. Appl. Microbiol., 2008, 105, 300–308 CrossRef CAS.
- L. P. Thapa, T. J. Oh, H. C. Lee, K. Liou, J. W. Park, Y. J. Yoon and J. K. Sohng, Appl. Microbiol. Biotechnol., 2007, 76, 1357–1364 CrossRef CAS.
- S. R. Park, J. A. Yoon, J. H. Paik, J. W. Park, W. S. Jung, Y. H. Ban, E. J. Kim, Y. J. Yoo, A. R. Han and Y. J. Yoon, J. Biotechnol., 2009, 141, 181–188 CrossRef CAS.
- C. J. Martin, M. C. Timoney, R. M. Sheridan, S. G. Kendrew, B. Wilkinson, J. C. Staunton and P. F. Leadlay, Org. Biomol. Chem., 2003, 1, 4144–4147 RSC.
- T. A. Cropp, D. J. Wilson and K. A. Reynolds, Nat. Biotechnol., 2000, 18, 980–983 CrossRef CAS.
- M. Komatsu, T. Uchiyama, S. Omura, D. E. Cane and H. Ikeda, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 2646–2651 CrossRef CAS.
- A. Hautala, S. Torkkell, K. Raty, T. Kunnari, J. Kantola, P. Mantsala, J. Hakala and K. Ylihonko, J. Antibiot., 2003, 56, 143–153 CAS.
- Y. Volchegursky, Z. Hu, L. Katz and R. McDaniel, Mol. Microbiol., 2000, 37, 752–762 CrossRef CAS.
- S. Shah, Q. Xue, L. Tang, J. R. Carney, M. Betlach and R. McDaniel, J. Antibiot., 2000, 53, 502–508 CAS.
- J. Kantola, T. Kunnari, A. Hautala, J. Hakala, K. Ylihonko and P. Mantsala, Microbiology, 2000, 146(Pt 1), 155–163 CAS.
- S. Wohlert, N. Lomovskaya, K. Kulowski, L. Fonstein, J. L. Occi, K. M. Gewain, D. J. MacNeil and C. R. Hutchinson, Chem. Biol., 2001, 8, 681–700 CrossRef CAS.
- L. Xiang, J. A. Kalaitzis and B. S. Moore, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 15609–15614 CrossRef CAS.
- J. A. Kalaitzis and B. S. Moore, J. Nat. Prod., 2004, 67, 1419–1422 CrossRef CAS.
- Z. Hu, R. Reid and H. Gramajo, J. Antibiot., 2005, 58, 625–633 CrossRef CAS.
- S. Tatsuno, K. Arakawa and H. Kinashi, J. Antibiot., 2007, 60, 700–708 CrossRef CAS.
- L. B. Pickens, W. Kim, P. Wang, H. Zhou, K. Watanabe, S. Gomi and Y. Tang, J. Am. Chem. Soc., 2009, 131, 17677–17689 CrossRef CAS.
- K. Zaleta-Rivera, L. K. Charkoudian, C. P. Ridley and C. Khosla, J. Am. Chem. Soc., 2010, 132, 9122–9128 CrossRef CAS.
- M. K. Kharel, S. E. Nybo, M. D. Shepherd and J. Rohr, ChemBioChem, 2010, 11, 523–532 CrossRef CAS.
- K. Ylihonko, J. Hakala, T. Kunnari and P. Mantsala, Microbiology, 1996, 142(8), 1965–1972 CrossRef CAS.
- S. T. Hong, J. R. Carney and S. J. Gould, J. Bacteriol., 1997, 179, 470–476 CAS.
- S. J. Gould, S. T. Hong and J. R. Carney, J. Antibiot., 1998, 51, 50–57 CAS.
- L. Tang, H. Fu, M. C. Betlach and R. McDaniel, Chem. Biol., 1999, 6, 553–558 CrossRef CAS.
- J. J. Barkei, B. M. Kevany, E. A. Felnagle and M. G. Thomas, ChemBioChem, 2009, 10, 366–376 CrossRef CAS.
- S. I. Patzer and V. Braun, J. Bacteriol., 2010, 192, 426–435 CrossRef CAS.
- M. A. Rude and C. Khosla, J. Antibiot., 2006, 59, 464–470 CrossRef CAS.
- W. Zhang, Y. Li and Y. Tang, Proc. Natl. Acad. Sci. U. S. A., 2008, 105, 20683–20688 CrossRef CAS.
- Y. Xu, R. Orozco, E. M. Wijeratne, A. A. Gunatilaka, S. P. Stock and I. Molnar, Chem. Biol., 2008, 15, 898–907 CrossRef CAS.
- H. J. Bouwmeester, J. Kodde, F. W. Verstappen, I. G. Altug, J. W. de Kraker and T. E. Wallaart, Plant Physiol., 2002, 129, 134–144 CrossRef CAS.
- S. Steiger, S. Takaichi and G. Sandmann, J. Biotechnol., 2002, 97, 51–58 CrossRef CAS.
- S. Steiger and G. Sandmann, Biotechnol. Lett., 2004, 26, 813–817 CrossRef CAS.
- S. K. Choi, S. Matsuda, T. Hoshino, X. Peng and N. Misawa, Appl. Microbiol. Biotechnol., 2006, 72, 1238–1246 CrossRef CAS.
- L. Tao, H. Yao, H. Kasai, N. Misawa and Q. Cheng, Mol. Genet. Genomics, 2006, 276, 79–86 CrossRef CAS.
- G. Sandmann, C. Zhu, P. Krubasik and P. D. Fraser, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2006, 1761, 1085–1092 CrossRef CAS.
- C. Ramel, M. Tobler, M. Meyer, L. Bigler, M. O. Ebert, B. Schellenberg and R. Dudler, BMC Biochem., 2009, 10, 26 CrossRef.
- R. Verwaal, J. Wang, J. P. Meijnen, H. Visser, G. Sandmann, J. A. van den Berg and A. J. van Ooyen, Appl. Environ. Microbiol., 2007, 73, 4342–4350 CrossRef CAS.
- M. A. Asadollahi, J. Maury, K. Moller, K. F. Nielsen, M. Schalk, A. Clark and J. Nielsen, Biotechnol. Bioeng., 2008, 99, 666–677 CrossRef CAS.
- Y. Seshime, P. R. Juvvadi, M. Tokuoka, Y. Koyama, K. Kitamoto, Y. Ebizuka and I. Fujii, Bioorg. Med. Chem. Lett., 2009, 19, 3288–3292 CrossRef CAS.
- R. L. Arslanian, L. Tang, S. Blough, W. Ma, R. G. Qiu, L. Katz and J. R. Carney, J. Nat. Prod., 2002, 65, 1061–1064 CrossRef CAS.
- O. Perlova, J. Fu, S. Kuhlmann, D. Krug, A. F. Stewart, Y. Zhang and R. Muller, Appl. Environ. Microbiol., 2006, 72, 7485–7494 CrossRef CAS.
- D. C. Stevens, M. R. Henry, K. A. Murphy and C. N. Boddy, Appl. Environ. Microbiol., 2010, 76, 2681–2683 CrossRef CAS.
- H. Decker and S. Haag, J. Bacteriol., 1995, 177, 6126–6136 CAS.
- B. Julien and S. Shah, Antimicrob. Agents Chemother., 2002, 46, 2772–2778 CrossRef CAS.
- P. L. Bartel, C. B. Zhu, J. S. Lampel, D. C. Dosch, N. C. Connors, W. R. Strohl, J. M. Beale, Jr. and H. G. Floss, J. Bacteriol., 1990, 172, 4816–4826 CAS.
- C. Binnie, M. Warren and M. J. Butler, J. Bacteriol., 1989, 171, 887–895 CAS.
- J. K. Epp, M. L. Huber, J. R. Turner, T. Goodson and B. E. Schoner, Gene, 1989, 85, 293–301 CrossRef CAS.
- D. A. Hopwood, F. Malpartida, H. M. Kieser, H. Ikeda, J. Duncan, I. Fujii, B. A. Rudd, H. G. Floss and S. Omura, Nature, 1985, 314, 642–644 CrossRef.
- H. Motamedi and C. R. Hutchinson, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 4445–4449 CAS.
- S. L. Otten, K. J. Stutzman-Engwall and C. R. Hutchinson, J. Bacteriol., 1990, 172, 3427–3434 CAS.
- L. Tang, L. Chung, J. R. Carney, C. M. Starks, P. Licari and L. Katz, J. Antibiot., 2005, 58, 178–184 CrossRef CAS.
- Y. Tang, T. S. Lee and C. Khosla, PLoS Biol., 2004, 2, E31 CrossRef.
- T. Marti, Z. Hu, N. L. Pohl, A. N. Shah and C. Khosla, J. Biol. Chem., 2000, 275, 33443–33448 CrossRef CAS.
- H. Fu, M. A. Alvarez, C. Khosla and J. E. Bailey, Biochemistry, 1996, 35, 6527–6532 CrossRef CAS.
- M. A. Alvarez, H. Fu, C. Khosla, D. A. Hopwood and J. E. Bailey, Nat. Biotechnol., 1996, 14, 335–338 CrossRef CAS.
- C. D. Reeves, S. L. Ward, W. P. Revill, H. Suzuki, M. Marcus, O. V. Petrakovsky, S. Marquez, H. Fu, S. D. Dong and L. Katz, Chem. Biol., 2004, 11, 1465–1472 CrossRef CAS.
- L. Tang, H. Fu and R. McDaniel, Chem. Biol., 2000, 7, 77–84 CrossRef CAS.
- Y. Chen, E. Wendt-Pienkowski and B. Shen, J. Bacteriol., 2008, 190, 5587–5596 CrossRef CAS.
- S. Kuhstoss, M. Huber, J. R. Turner, J. W. Paschal and R. N. Rao, Gene, 1996, 183, 231–236 CrossRef CAS.
- C. Bormann, V. Mohrle and C. Bruntner, J. Bacteriol., 1996, 178, 1216–1218 CAS.
- H. J. Kwon, W. C. Smith, L. Xiang and B. Shen, J. Am. Chem. Soc., 2001, 123, 3385–3386 CrossRef CAS.
- R. A. Lacalle, J. A. Tercero and A. Jimenez, EMBO J., 1992, 11, 785–792 CAS.
- P. J. Solenberg, P. Matsushima, D. R. Stack, S. C. Wilkie, R. C. Thompson and R. H. Baltz, Chem. Biol., 1997, 4, 195–202 CrossRef CAS.
- C. N. Tetzlaff, Z. You, D. E. Cane, S. Takamatsu, S. Omura and H. Ikeda, Biochemistry, 2006, 45, 6179–6186 CrossRef CAS.
- L. Tang, Y. G. Wang and X. W. Zhu, Chin. J. Biotechnol., 1991, 7, 33–42 Search PubMed.
- K. T. Nguyen, X. He, D. C. Alexander, C. Li, J. Q. Gu, C. Mascio, A. Van Praagh, L. Mortin, M. Chu, J. A. Silverman, P. Brian and R. H. Baltz, Antimicrob. Agents Chemother., 2010, 54, 1404–1413 CrossRef CAS.
- E. S. Kim, K. D. Cramer, A. L. Shreve and D. H. Sherman, J. Bacteriol., 1995, 177, 1202–1207 CAS.
- R. McDaniel, S. Ebert-Khosla, D. A. Hopwood and C. Khosla, Nature, 1995, 375, 549–554 CrossRef CAS.
- K. T. Seow, G. Meurer, M. Gerlitz, E. Wendt-Pienkowski, C. R. Hutchinson and J. Davies, J. Bacteriol., 1997, 179, 7360–7368 CAS.
- A. Li and J. Piel, Chem. Biol., 2002, 9, 1017–1026 CrossRef CAS.
- J. He and C. Hertweck, ChemBioChem, 2005, 6, 908–912 CrossRef CAS.
- S. Gaisser, A. Trefzer, S. Stockert, A. Kirschning and A. Bechthold, J. Bacteriol., 1997, 179, 6271–6278 CAS.
- J. He and C. Hertweck, Chem. Biol., 2003, 10, 1225–1232 CrossRef CAS.
- M. Albrecht, S. Takaichi, N. Misawa, G. Schnurr, P. Boger and G. Sandmann, J. Biotechnol., 1997, 58, 177–185 CrossRef CAS.
- J. Crock, M. Wildung and R. Croteau, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 12833–12838 CrossRef CAS.
- V. J. Martin, Y. Yoshikuni and J. D. Keasling, Biotechnol. Bioeng., 2001, 75, 497–503 CrossRef CAS.
- K. K. Reiling, Y. Yoshikuni, V. J. Martin, J. Newman, J. Bohlmann and J. D. Keasling, Biotechnol. Bioeng., 2004, 87, 200–212 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2011 |