Binding region and interaction properties of sulfoquinovosylacylglycerol (SQAG) with human vascular endothelial growth factor 165 revealed by biosensor-based assays†
Yoichi
Takakusagi‡
abc,
Kaori
Takakusagi
abc,
Noriko
Ida
c,
Mihoko
Takami
c,
Yuki
Matsumoto
c,
Tomoe
Kusayanagi
c,
Tadashi
Nakabayashi
c,
Satoko
Aoki
c,
Hiroshi
Murata
c,
Keisuke
Ohta
c,
Fumio
Sugawara
*abc and
Kengo
Sakaguchi
*abc
aDivision of Social Collaboration, Research Institute for Science and Technology (RIST), University of Science, TokyoJapan
bDivision of Chemical Biology, Research Institute for Science and Technology (RIST), University of Science, TokyoJapan
cDepartment of Applied Biological Science, Faculty of Science and Technology, University of Science, Tokyo. Japan. E-mail: sugawara@rs.noda.tus.ac.jp; Fax: +81 4 7123 9767; Tel: +81 4 7124 1501 ext. 3400 5009kengo@rs.noda.tus.ac.jp
First published on 5th October 2011
Abstract
Sulfoquinovosylacylglycerol (SQAG) is a sulfoglycolipid showing anti-angiogenic and radiosensitizing effects for treatment of solid tumors both in vitro and in vivo. Here we elucidated the interaction of SQAG with various growth factors and their cognate receptors for vascular formation using biosensor-based assays. The structure–binding relationship was also determined. Our results show that βSQDG selectively recognizes heparin binding domain (HBD) in human vascular endothelial growth factor 165 (hVEGF165) with an affinity in the order of 10−11 M. The presence of both a sulfate moiety and at least one C18 length fatty acid chain is essential for binding. Conversion of anomeric configurations in SQAG did not alter the affinity with hVEGF165. This SQAG association inhibited T7 phage-displayed HBD binding to neuropilin-1 (NRP1), a VEGF receptor on the endothelial cell surface of blood vessels that specifically recognizes HBD in hVEGF165.
In a broad sense, neovascularization is primarily classified into two groups, vasculogenesis and angiogenesis. Vasculogenesis is the formation of primitive vascular networks through differentiation of endothelial progenitor cells (EPC) into endothelial cells in the developmental process,1 while angiogenesis is the growth and sprouting of additional blood vessels from pre-existing blood vessels in postnatal vascular formation.2 As well as ontogenesis or homeostasis, angiogenesis is also associated with various diseases,3 such as rheumatic inflammation,4 retinopathy in diabetes mellitus5 and tumor progression.6 At each stage in these vascular formation processes, growth factors and cognate receptors regulate blood endothelial and smooth muscle cells differentiation and/or growth to determine subsequent blood vessel formation.2,7,8 These include vascular endothelial growth factor (VEGF),2,7–9 transforming growth factor (TGF),10insulin-like growth factor (IGF),11fibroblast growth factor (FGF),12 angiopoietin (Ang),8,13ephrin (Eph),8epidermal growth factor (EGF),14platelet-derived growth factor (PDGF),2 bone morphogenetic protein (BMP)10 and others, as well as their cognate receptors on cell surfaces.
Sulfoquinovosylacylglycerol (SQAG) is a sulfoglycolipid that was isolated from natural resources, such as sea algae, and shown to exhibit various biological effects including inhibition of HIV reverse transcriptase or DNA polymerase (Fig. 1).15–17 The SQAG molecule is made up of a core glycerol unit bonded with one or two long acyl chains at position 1 and/or 2. At the 3-hydroxyl group, a sulfoquinovose unit that has a sulfonate group at position 6′ is bonded via an anomeric position (Fig. 1). Several SQAG analogues were synthesized, each with a different stereochemistry at the anomeric carbon and acylation (with varying lengths of fatty acid) at the 1 and/or 2 hydroxyl group in glycerol.18 Some recent studies have demonstrated that SQAG shows growth suppression of certain solid tumors,19,20 and displays anti-angiogenic21,22 and strong radiosensitizing effects both in vitro and in vivo.22,23 CG-0321, a synthetic analogue of SQAG, is a lead compound and is currently being scheduled to begin phase I clinical tests as an anti-angiogenic type of radiosensitizer for the treatment of solid tumors. In a pre-clinical test, this compound displayed very few side effects and gave a one-year extension to a prognosis of dogs with primary sarcoma by combination therapy with X-irradiation, abolishing the tumor angiogenesis (unpublished data). However, apart from the downregulation of Tie-2 upon treatment with SQAG,21 the molecular mechanisms for the anti-angiogenic and radio-sensitizing effects of this compound have not been elucidated. Indeed, it is not even known whether there is direct interaction of SQAG with growth factors or their receptors for angiogenesis.
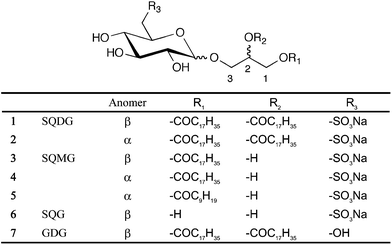 | ||
| Fig. 1 Structure of SQAGs used in this study. Sulfoquinovosyldiacylglycerol (SQDG): C45H85NaO12S, Mw 873.2. Sulfoquinovosylmonoacylglycerol (SQMG) C18: C27H51NaO11S, Mw 606.74, C10: C19H35NaO11S, Mw 494.53. Sulfoquinovosylglycerol (SQG): C9H17NaO10S, Mw 340.04. Glucosyldiacylglycerol (GDG): C45H86O10, Mw 787.16. | ||
The aim of this study was to elucidate the interaction between SQAG and growth factors or their receptors on vascular formation by making use of biosensor-based assays. Here, we employed two distinctive biosensors by taking advantage of each characteristic. One is a flow injection-type surface plasmon resonance (SPR)24 apparatus (Biacore® 3000, GE Healthcare, Piscataway, NJ) and the other is a cuvette-type 27-MHz quartz-crystal microbalance (QCM)25 device (AffinixQ, Initium, Tokyo). This SPR biosensor enables elucidation of the interactions with reversibility and is dependent on the refractive index of an analyte, which occurs by surface plasmon phenomena at a gold–liquid interface and increases proportionally to the mass on the gold. Non-linear fitting of the resulting sensorgram obtained by initial sub-minutes of association and subsequent dissociation analysis using various concentrations of analyte gives the kinetic parameters and the potential binding models.26 In addition, successive injections of label-free analyte are feasible in this flow-injection system fitted with an autosampler. This biosensor is, therefore, adaptive for studies investigating the interaction with dose-dependency or structure–binding relationships for a target of interest. QCM is a biosensor whose shear modulus of quartz (AT-cut) decreases in proportion to the increase of mass on the gold electrode attached on the quartz by vapor deposition.25 The interaction on the gold electrode (mass increase) is, therefore, detected as a decrease of oscillatory resonance frequency in real time. This QCM device comprises a buffer-filled cuvette, which is constantly stirred, where the sensor chip is immersed. The guest molecule is then injected into the cuvette to analyze the interaction on the gold electrode.27 In addition, the sensor chip is readily detached from the device to facilitate recovery of the bound guest molecule. The multiple binding molecules can be simultaneously identified by a single affinity selection.28,29 Thus, this biosensor is suitable for use in combination with other molecular biological techniques, such as T7 phage display.28–31
Using the SPR biosensor, we screened the binding of 1 to growth factors or their associated receptors that are involved in vascular formation. Each protein was immobilized on a sensor chip CM5 (GE Healthcare) by an amine coupling reaction and interacted with various concentrations of 1. Of the 25 of molecules tested (Table 1), only hVEGF165, which plays the central role in angiogenic activity, showed a response after injection of 1 ranging in concentration from 0.125 to 1 μM (Fig. 2A). The kinetic parameters were obtained from global fitting of the resulting sensorgram using BIAevaluation 3.2 software (Table 1). The SPR profile by injecting 1 showed slow association (ka: 3.0 × 103 M−1s−1) with irreversibility (kd: 1.4 × 10−7s−1) in this concentration range. The dissociation constant (KD) between 1 and hVEGF165, which was calculated assuming a stoichiometry of interaction of a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, was estimated to be 4.6 × 10−11 M, suggesting a very high level of affinity. By contrast, no response was observed with regard to other growth factors or their receptors even at 2 μM of 1 (Table 1 and Fig. S1†). In particular, hVEGF121, a splicing variant that lacks N115-C160 (exon7) in hVEGF165 (Fig. 3), did not show any response against 1 at the concentration range tested (Fig. 2B).
:1, was estimated to be 4.6 × 10−11 M, suggesting a very high level of affinity. By contrast, no response was observed with regard to other growth factors or their receptors even at 2 μM of 1 (Table 1 and Fig. S1†). In particular, hVEGF121, a splicing variant that lacks N115-C160 (exon7) in hVEGF165 (Fig. 3), did not show any response against 1 at the concentration range tested (Fig. 2B).
| Protein (human) | M w/kDa | Amino acid | K D/Ma |
|---|---|---|---|
| a The KD value was obtained from the resulting sensorgram using BIAevaluation 3.2 software (GE Healthcare). b No response by injecting compound 1 (0.125–2 μM). c The mouse PDGFRB was used. d This Mw includes the fusion GST tag. | |||
| VEGF165 | 19.1 | A1-R165 | 4.6 × 10−11 |
| VEGF121 | 14 | A1-R121 | NR b |
| VEGFR1 | 60.7 | S27-I328 | NR |
| VEGFR2 | 116 | A20-E764 | NR |
| NRP1 | 90 | M1-K644 | NR |
| TGFβ2 | 25 | A1-S112 | NR |
| TGFβ RII | 18 | M1-D159 | NR |
| IGF-I | 7.6 | G49-A118 | NR |
| IGF-I R | 102.9 | E31-N932 | NR |
| FGF1 | 15.8 | M1-D141 | NR |
| FGF2 | 17.2 | A1-S154 | NR |
| FGF R2α | 66 | R22-E378 | NR |
| Ang-1 | 66 | N21-F496 | NR |
| Tie-2 | 100 | A23-K745 | NR |
| EphB2c | 85.3 | V27-K458 | NR |
| EphB4 | 57.9 | L16-A539 | NR |
| EGF | 6.2 | N1-R53 | NR |
| EGFR | 68.6 | L25-S645 | NR |
| PDGF-BB | 12 | S1-T110 | NR |
| PDGFRB | 36.7d | L33-E133 | NR |
| BMP-2 | 26 | M1-R115 | NR |
| BMP RII | 41.5 | A26-I151 | NR |
| sDLL-4 | 54 | S1-P498 | NR |
| Jagged-1 | 180 | S32-S1046 | NR |
| Notch-1 | 80.1 | A19-Q526 | NR |
| DNA pol λ (cont.) | 14.3 | M1-R95 | 9.9 × 10−9 |
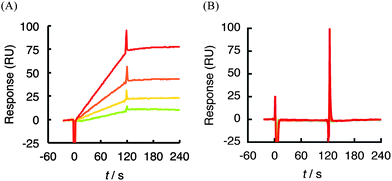 | ||
| Fig. 2 SPR sensorgram between 1 (0.125–1 μM) and hVEGF165 (A) or 1 (0.125–2 μM) and hVEGF121 (B). Each protein was immobilized on a sensor chip CM5 by an amine coupling reaction and then various concentrations of 1 were injected. Association: 120 s, dissociation: 120 s. Response curves were generated by subtraction of the background signals generated simultaneously on the control flow cell (protein-non-immobilized cell), the injection of vehicle, and bulk response by DMSO. RU: resonance unit. 1 RU = 1 pg mm−2. | ||
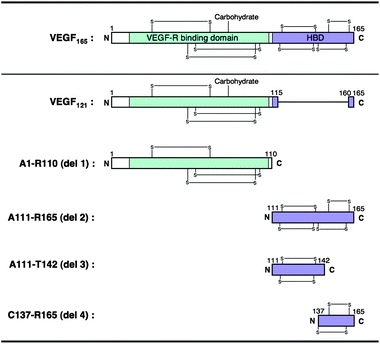 | ||
| Fig. 3 Schematic representations of hVEGF165 monomer and its truncated version designed in this study. | ||
Recent studies toward the development of anti-angiogenic agents or radiosensitizers have demonstrated hVEGF165 to be a promising therapeutic target of angiogenesis-related diseases or tumors.2,32,33 In particular, Bevacizumab (Avastin™), a VEGF-specific antibody,34–36 and Pegaptanib (Macugen®), a 28-nucleotide RNA aptamer,37–41 have been approved by the Food and Drug Administration (FDA) in the United States for treatment of colorectal cancer42 and age-related macular degeneration (AMD),37–39 respectively. Intriguingly, despite the strong affinity of bevacizumab and pegaptanib for hVEGF165 and their efficient anti-angiogenic activity both in vitro and in vivo, the site of interaction of each molecule within hVEGF165 is discrete. Specifically, bevacizumab recognizes the VEGFR-binding region (A1-R110),43 while pegaptanib binds to HBD (A111-R165).44 In order to refine the recognition site in the case of βSQDG, we used the QCM-based affinity selection of a library of T7 phage-displayed polypeptides designated as del 1–4 (Fig. 3). Compound 1 was immobilized on the gold electrode of a sensor chip and then inserted into the buffer-filled cuvette. After stabilizing the QCM sensor, the T7 phage library was injected into the cuvette. Fig. 4A shows the representative sensorgram that was obtained by injection of the T7 phage library. The resulting frequency decrease indicated binding of the T7 phage to compound 1 on the gold electrode. After monitoring the frequency decrease for 10 min, the sensor chip was dislodged from the device. The hostEscherichia coli (BLT5615) culture was then dropped onto the electrode and further incubated for 30 min at 37 °C to recover the bound T7 phage DNA directly.28–31 Twenty-four phage particles were randomly extracted from the resulting solution and part of the phage DNA encoding capsid protein was amplified by PCR. The products were then subjected to DNA sequencing to define the population of recovered T7 phages. Fig. 4B shows the emergence ratio obtained by a comparison between the content of the respective T7 phage particles in the resulting solution to that of the unscreened parent phage library, which was detected by an agarose gel electrophoresis. The emergence ratio of T7 phage particles displaying HBD (A111-R165; del 2 phage) and C-terminal fragment of HBD (C137-R165; del 4 phage) in hVEGF165 increased following selection (Table S5†). This result suggests that 1 recognizes the HBD polypeptide, especially the C137-R165 region (Fig. 3).
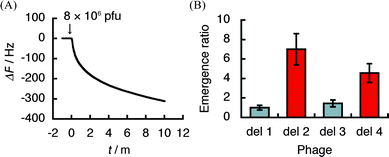 | ||
| Fig. 4 The scanning of βSQDG (1)-binding region by T7 phage display selection using a quartz-crystal microbalance (QCM). (A) A representative QCM sensorgram obtained by the injection of a T7 phage library that displays defined segments (del 1–4) of hVEGF165. (B) Emergence of T7 phage particles after the selection by 1. The data are displayed in terms of the emergence ratio of each T7 phage of 24 arbitrarily selected T7 phage particles from the recovered solution with 1 on the gold electrode of the sensor chip and that from the unscreened parent library. The data show means ± SE of three individual experiments. 1 Hz = 0.62 pg mm−2. | ||
Further evidence for the specific interaction of 1 was obtained by SPR analysis using a truncated version of hVEGF165 engineered into an E. coli expression system. The respective fragment was expressed as a His-tagged recombinant polypeptide and purified by affinity chromatography. The sensorgrams obtained from each analysis are shown in Fig. 5. Compound 1 (0.125–1 μM) showed a dose-dependent response against HBD (A111-R165) in hVEGF165 (Fig. 5B), although the response against immobilzed del 2 was slightly decreased due to the decreased mass by deletion of A1-R110 region (i.e., partial loss of piggyback or mass enhancer effect).45,46 Nonetheless, the experiment gave a similar profile of sensorgram and kinetic parameters to that against full length hVEGF165 (Fig. 2A and Table 2). A response was also detected using the C-terminal fragment of HBD (C137-R165), though the strength of affinity was reduced by approximately 4 × 104-fold due to the faster dissociation profile (kd: 5.9 × 10−3s−1) compared with full-length hVEGF165 (kd: 1.4 × 10−7s−1) (Fig. 2A and 5D, Table 2). By contrast, the A1-R110 protein that corresponds to the VEGFR-binding region, and the A111-T142 fragment of HBD did not show any binding with 1 at the concentration range tested (Fig. 5A and C). These data are well correlated with the result of T7 phage display analysis and strongly suggest that 1 recognizes HBD in hVEGF165 as a ‘hot spot’ of binding.
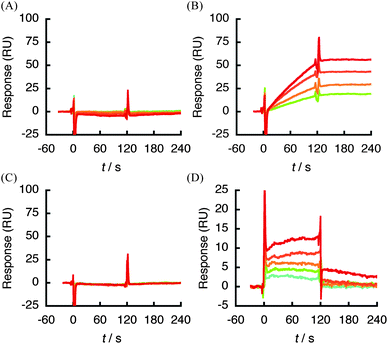 | ||
| Fig. 5 SPR sensorgram of analysis between 1 (0.125–2 μM) and truncated hVEGF165protein immobilized on a sensor chip CM5. (A) A1-R110 (del 1) in hVEGF165. (B) 1 (0.125–1 μM) and A111-R165 (del 2). (C) A111-T142 (del 3). (D) C137-R165 (del 4). | ||
| k a/M−1s−1 | k d/s−1 | K D (kd/ka)/M | R max/RU | χ 2 | |
|---|---|---|---|---|---|
|
a
Analytical conditions: HBS-EP (10 mM HEPES pH 7.4, 150 mM NaCl, 3 mM EDTA, 0.005% surfactant P20) −8% DMSO, 25 °C. ka: association rate constant, kd: dissociation rate constant, KD: dissociation constant, Rmax: maximum binding amount, χ2: fitting to a 1:1 drifted baseline model using BIAevaluation 3.2 software (complete fitting; χ2 = 0).
b No response (NR) at each concentration range tested.
|
|||||
| 1–hVEGF165 | 3.0 × 103 | 1.4 × 10−7 | 4.6 × 10−11 | 248 | 0.345 |
| 1–hVEGF121 | NR b | NR | NR | — | — |
| 1–del 1 | NR | NR | NR | — | — |
| 1–del 2 | 8.1 × 103 | 2.1 × 10−7 | 2.5 × 10−11 | 99 | 0.392 |
| 1–del 3 | NR | NR | NR | — | — |
| 1–del 4 | 4.3 × 103 | 5.9 × 10−3 | 1.4 × 10−6 | 7.6 | 0.082 |
| 2–hVEGF165 | 4.4 × 103 | 3.6 × 10−7 | 2.5 × 10−11 | 284 | 0.266 |
| 3–hVEGF165 | 1.7 × 103 | 2.6 × 10−2 | 1.6 × 10−5 | 37 | 0.23 |
| 4–hVEGF165 | 1.8 × 103 | 3.2 × 10−2 | 1.8 × 10−5 | 35.7 | 0.288 |
| 5–hVEGF165 | NR | NR | NR | — | — |
| 6–hVEGF165 | NR | NR | NR | — | — |
| 7–hVEGF165 | NR | NR | NR | — | — |
To define the structural features of SQAG that is responsible for binding to hVEGF165, βSQDG (1) analogues 2–7 (Fig. 1) were prepared and subjected to SPR analysis. Of the SQAG derivatives tested in this study, both 1 and 2 displayed the strongest affinity for hVEGF165 in the order of 10−11 M (Table 2). Furthermore, configuration of the anomeric center did not appear to affect this interaction (Fig. 2A and 6A). SQMGs (3, 4), which lack acyl group at position 2 in compound 1 (Fig. 1), showed decreased affinity against hVEGF165 (∼2 × 105-fold increase in kd) compared with 1 (Table 2). In addition, the interaction of 3 and 4 with hVEGF165 was found to be reversible (Fig. 6B and C). The reduced length of the fatty acid chain from C18 to C10 (5), and the removal of both fatty acid chains in 1 (βSQG, 6) showed no response against hVEGF165 at the concentration range tested (Fig. 6D and E). Intriguingly, βGDG (7), which lacks only the sulfate moiety in 1 (Fig. 1), did not respond at all (Fig. 6F). Overall, these data suggest that the presence of both a sulfate moiety and C18 length fatty acid, at least in position 1 of SQAG, are essential for interaction with hVEGF165.
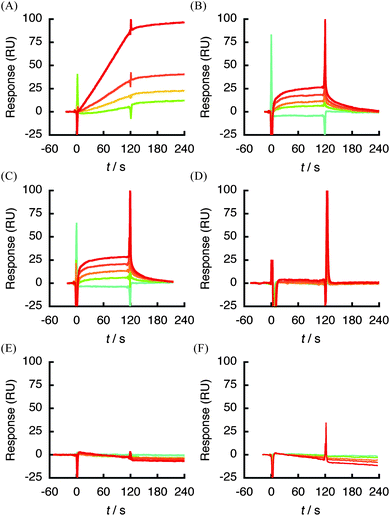 | ||
| Fig. 6 Structure–binding relationship studies using a SPR biosensor. hVEGF165 was immobilized on sensor chip CM5 and various concentrations of SQAG analogues (2; 0.125–1 μM, 3–5; 1.25–20 μM, 6, 7; 0.125–2 μM) were respectively injected. (A) 2 and hVEGF165. (B) 3 and hVEGF165. (C) 4 and hVEGF165. (D) 5 and hVEGF165. (E) 6 and hVEGF165. (F) 7 and hVEGF165. | ||
The hVEGF165 is known to have a cognate protein–tyrosine kinase receptor, VEGFR1 (Flt1)47,48 and VEGFR2 (KDR, flk-1)49 on the endothelial cells of blood vessels. VEGFR2 is the main signaling pathway for angiogenesis, while VEGFR1 plays a regulatory role. Both receptors bind to hVEGF165via the receptor binding domain in A1-R110 region (Fig. 3), which lead to cellular events for new blood vessel formation. By contrast, neuropilin-1 (NRP1), a glycoprotein present on endothelial cells of blood vessels or the surface of hemocytes,50 is an alternative receptor of hVEGF165.51–53 The hVEGF165 binds to NRP1 through HBD54–56 and mediates formation of complexes containing VEGFR2 and NRP1, resulting in an enhancement of hVEGF165-receptor binding.57 Our experiments suggest that SQAG shows a pegaptanib-type of binding to hVEGF165i.e., specific recognition of HBD but no interaction with A1-R110 (del 1) or the cognate VEGFR1/2 and NRP1 (Fig. 3, 4B and 5A and Tables 1 and 2). To ascertain binding of SQAG to HBD and its subsequent influence on NRP1 binding, we carried out a binding experiment using QCM apparatus. T7 phage particles displaying HBD (A111-R165, del 2 phage) were pre-incubated with compound 1 or 4 and injected into buffer-filled cuvette where NRP1 immobilized QCM sensor chip was immersed. Fig. 7 shows the resulting data with or without the pretreatment of each compound. Compared to control (vehicle only), binding of del 2 phage to NRP1 was significantly inhibited by the pretreatment of compound 1 and 4. Thus, SQAG shows the inhibitory effect of HBD–NRP1 interaction by specifically binding to HBD.
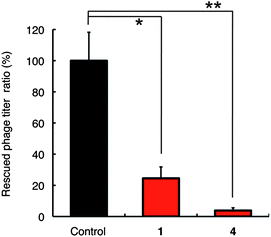 | ||
| Fig. 7 The effect of 1 μM βSQDG (1) or 10 μM αSQMG (4) for NRP1 binding of T7 phage-displayed HBD on a QCM. The data are displayed in terms of the rescued phage titer ratio (vehicle vs. compound) of T7 phage particles displaying HBD (del 2 phage) from the recovered solution on the gold electrode of the sensor chip. The data show means ± SD of three individual experiments. *P < 0.05, **P < 0.01. 1 Hz = 0.62 pg mm−2. | ||
Recent studies have generated inhibitors of hVEGF165–NRP1 interaction. EG3287 is a bicyclic peptideNRP1 antagonist mimicking the C-terminal region of hVEGF165,58 whereas EG00229 is a small-molecule ligand that targets the hVEGF165 binding pocket in NRP1.59 Together with SQAGs reported in this study, these molecules are expected to be valuable therapeutic agents for treatment of solid tumors targeting hVEGF165–NRP1 interaction. Further experiments of SQAG are currently underway.
Conclusions
In this study we have shown that SQAGs selectively recognize hVEGF165 through interaction with HBD. Our results also show that the affinity was strongest for SQDG and the presence of both a sulfate moiety and at least one C18 fatty acid in βSQDG are essential for HBD association. Furthermore, this association specifically inhibits cognate HBD–NRP1 interaction. These data provide a sound platform for further studies concerning anti-angiogenic and radiosensitizing activities as well as the development of SQAG for clinical applications.Acknowledgements
This work was supported by the Program for Promotion of Fundamental Studies in Health Science of the National Institute of Biomedical Innovation (NIBIO), and a research grant from Cango, Co. Ltd.Notes and references
- T. Asahara and A. Kawamoto, Am. J. Physiol.: Cell Physiol., 2004, 287, C572–C579 CrossRef CAS.
- N. Ferrara and R. S. Kerbel, Nature, 2005, 438, 967–974 CrossRef CAS.
- R. F. Gariano and T. W. Gardner, Nature, 2005, 438, 960–966 CrossRef CAS.
- A. E. Koch, Arthritis Rheum., 1998, 41, 951–962 CrossRef CAS.
- B. P. Nicholson and A. P. Schachat, Graefe's Arch. Clin. Exp. Ophthalmol., 2010, 248, 915–930 CrossRef CAS.
- P. Carmeliet and R. K. Jain, Nature, 2000, 407, 249–257 CrossRef CAS.
- N. Ferrara, Nat. Rev. Cancer, 2002, 2, 795–803 CrossRef CAS.
- G. D. Yancopoulos, S. Davis, N. W. Gale, J. S. Rudge, S. J. Wiegand and J. Holash, Nature, 2000, 407, 242–248 CrossRef CAS.
- K. J. Kim, B. Li, J. Winer, M. Armanini, N. Gillett, H. S. Phillips and N. Ferrara, Nature, 1993, 362, 841–844 CrossRef CAS.
- N. M. Shah, A. K. Groves and D. J. Anderson, Cell, 1996, 85, 331–343 CrossRef CAS.
- R. S. Bar, M. Boes, B. L. Dake, B. A. Booth, S. A. Henley and A. Sandra, Am. J. Med., 1988, 85, 59–70 CrossRef CAS.
- M. Presta, P. Dell'Era, S. Mitola, E. Moroni, R. Ronca and M. Rusnati, Cytokine Growth Factor Rev., 2005, 16, 159–178 CrossRef CAS.
- C. Suri, P. F. Jones, S. Patan, S. Bartunkova, P. C. Maisonpierre, S. Davis, T. N. Sato and G. D. Yancopoulos, Cell, 1996, 87, 1171–1180 CrossRef CAS.
- F. Ciardiello and G. Tortora, Clin. Cancer Res., 2001, 7, 2958–2970 CAS.
- Y. Mizushina, I. Watanabe, K. Ohta, M. Takemura, H. Sahara, N. Takahashi, S. Gasa, F. Sugawara, A. Matsukage, S. Yoshida and K. Sakaguchi, Biochem. Pharmacol., 1998, 55, 537–541 CrossRef CAS.
- K. Ohta, Y. Mizushina, N. Hirata, M. Takemura, F. Sugawara, A. Matsukage, S. Yoshida and K. Sakaguchi, Chem. Pharm. Bull. (Tokyo), 1998, 46, 684–686 CAS.
- C. Benning, Annu. Rev. Plant Physiol. Plant Mol. Biol., 1998, 49, 53–75 CrossRef CAS.
- S. Hanashima, Y. Mizushina, T. Yamazaki, K. Ohta, S. Takahashi, H. Sahara, K. Sakaguchi and F. Sugawar, Bioorg. Med. Chem., 2001, 9, 367–376 CrossRef CAS.
- H. Sahara, S. Hanashima, T. Yamazaki, S. Takahashi, F. Sugawara, S. Ohtani, M. Ishikawa, Y. Mizushina, K. Ohta, K. Shimozawa, S. Gasa, K. Jimbow, K. Sakaguchi, N. Sato and N. Takahashi, Jpn. J. Cancer Res., 2002, 93, 85–92 CrossRef CAS.
- K. Ohta, Y. Mizushina, T. Yamazaki, S. Hanashima, F. Sugawara and K. Sakaguchi, Biochem. Biophys. Res. Commun., 2001, 288, 893–900 CrossRef CAS.
- Y. Mori, H. Sahara, K. Matsumoto, N. Takahashi, T. Yamazaki, K. Ohta, S. Aoki, M. Miura, F. Sugawara, K. Sakaguchi and N. Sato, Cancer Sci., 2008, 99, 1063–1070 CrossRef CAS.
- M. Miura, I. Sakimoto, K. Ohta, F. Sugawara and K. Sakaguchi, Anti-Cancer Drugs, 2007, 18, 1–5 CrossRef CAS.
- I. Sakimoto, K. Ohta, T. Yamazaki, S. Ohtani, H. Sahara, F. Sugawara, K. Sakaguchi and M. Miura, Cancer Res., 2006, 66, 2287–2295 CrossRef CAS.
- J. Homola, S. S. Yee and G. Gauglitz, Sens. Actuators, B, 1999, 54, 3–15 CrossRef.
- C. K. O'Sullivan and G. G. Guilbault, Biosens. Bioelectron., 1999, 14, 663–670 CrossRef CAS.
- K. Andersson, D. Areskoug and E. Hardenborg, J. Mol. Recognit., 1999, 12, 310–315 CrossRef CAS.
- H. Matsuno, H. Furusawa and Y. Okahata, Chem.–Eur. J., 2004, 10, 6172–6178 CrossRef CAS.
- Y. Takakusagi, K. Kuramochi, M. Takagi, T. Kusayanagi, D. Manita, H. Ozawa, K. Iwakiri, K. Takakusagi, Y. Miyano, A. Nakazaki, S. Kobayashi, F. Sugawara and K. Sakaguchi, Bioorg. Med. Chem., 2008, 16, 9837–9846 CrossRef CAS.
- Y. Takakusagi, K. Takakusagi, K. Kuramochi, S. Kobayashi, F. Sugawara and K. Sakaguchi, Bioorg. Med. Chem., 2007, 15, 7590–7598 CrossRef CAS.
- Y. Takakusagi, K. Takakusagi, F. Sugawara and K. Sakaguchi, Expert Opin. Drug Discovery, 2010, 5, 361–389 CrossRef CAS.
- Y. Takakusagi, K. Takakusagi, F. Sugawara and K. Sakaguchi, Tanpakushitsu Kakusan Koso, 2009, 54, 1203–1209 CAS.
- J. Brieger, P. Schroeder, J. Gosepath and W. J. Mann, Int. J. Mol. Med., 2005, 15, 145–151 CAS.
- D. H. Gorski, M. A. Beckett, N. T. Jaskowiak, D. P. Calvin, H. J. Mauceri, R. M. Salloum, S. Seetharam, A. Koons, D. M. Hari, D. W. Kufe and R. R. Weichselbaum, Cancer Res., 1999, 59, 3374–3378 CAS.
- N. Ferrara, K. J. Hillan and W. Novotny, Biochem. Biophys. Res. Commun., 2005, 333, 328–335 CrossRef CAS.
- L. G. Presta, H. Chen, S. J. O'Connor, V. Chisholm, Y. G. Meng, L. Krummen, M. Winkler and N. Ferrara, Cancer Res., 1997, 57, 4593–4599 CAS.
- K. J. Kim, B. Li, K. Houck, J. Winer and N. Ferrara, Growth Factors, 1992, 7, 53–64 CrossRef CAS.
- E. W. Ng and A. P. Adamis, Can. J. Ophthalmol., 2005, 40, 352–368 Search PubMed.
- S. L. Fine, D. F. Martin and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2005, 4, 187–188 CrossRef CAS.
- E. S. Gragoudas, A. P. Adamis, E. T. Cunningham, Jr, M. Feinsod and D. R. Guyer, N. Engl. J. Med., 2004, 351, 2805–2816 CrossRef CAS.
- C. Bell, E. Lynam, D. J. Landfair, N. Janjic and M. E. Wiles, In Vitro Cell. Dev. Biol.: Anim., 1999, 35, 533–542 CrossRef CAS.
- J. Ruckman, L. S. Green, J. Beeson, S. Waugh, W. L. Gillette, D. D. Henninger, L. Claesson-Welsh and N. Janjic, J. Biol. Chem., 1998, 273, 20556–20567 CrossRef CAS.
- R. S. Warren, H. Yuan, M. R. Matli, N. A. Gillett and N. Ferrara, J. Clin. Invest., 1995, 95, 1789–1797 CrossRef CAS.
- Y. A. Muller, Y. Chen, H. W. Christinger, B. Li, B. C. Cunningham, H. B. Lowman and A. M. de Vos, Structure, 1998, 6, 1153–1167 CrossRef CAS.
- J. H. Lee, M. D. Canny, A. De Erkenez, D. Krilleke, Y. S. Ng, D. T. Shima, A. Pardi and F. Jucker, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 18902–18907 CrossRef CAS.
- W. A. Henne, D. D. Doorneweerd, J. Lee, P. S. Low and C. Savran, Anal. Chem., 2006, 78, 4880–4884 CrossRef CAS.
- X. Mao, L. Yang, X. L. Su and Y. Li, Biosens. Bioelectron., 2006, 21, 1178–1185 CrossRef CAS.
- M. A. Starovasnik, H. W. Christinger, C. Wiesmann, M. A. Champe, A. M. de Vos and N. J. Skelton, J. Mol. Biol., 1999, 293, 531–544 CrossRef CAS.
- C. Wiesmann, G. Fuh, H. W. Christinger, C. Eigenbrot, J. A. Wells and A. M. de Vos, Cell, 1997, 91, 695–704 CrossRef CAS.
- B. A. Keyt, H. V. Nguyen, L. T. Berleau, C. M. Duarte, J. Park, H. Chen and N. Ferrara, J. Biol. Chem., 1996, 271, 5638–5646 CrossRef CAS.
- Y. Yamada, Y. Oike, H. Ogawa, Y. Ito, H. Fujisawa, T. Suda and N. Takakura, Blood, 2003, 101, 1801–1809 CrossRef CAS.
- L. M. Ellis, Mol. Cancer Ther., 2006, 5, 1099–1107 CrossRef CAS.
- H. Q. Miao, P. Lee, H. Lin, S. Soker and M. Klagsbrun, FASEB J., 2000, 14, 2532–2539 CrossRef CAS.
- S. Soker, S. Takashima, H. Q. Miao, G. Neufeld and M. Klagsbrun, Cell, 1998, 92, 735–745 CrossRef CAS.
- B. A. Appleton, P. Wu, J. Maloney, J. Yin, W. C. Liang, S. Stawicki, K. Mortara, K. K. Bowman, J. M. Elliott, W. Desmarais, J. F. Bazan, A. Bagri, M. Tessier-Lavigne, A. W. Koch, Y. Wu, R. J. Watts and C. Wiesmann, EMBO J., 2007, 26, 4902–4912 CrossRef CAS.
- R. Mamluk, Z. Gechtman, M. E. Kutcher, N. Gasiunas, J. Gallagher and M. Klagsbrun, J. Biol. Chem., 2002, 277, 24818–24825 CrossRef CAS.
- S. Soker, H. Fidder, G. Neufeld and M. Klagsbrun, J. Biol. Chem., 1996, 271, 5761–5767 CrossRef CAS.
- S. Soker, H. Q. Miao, M. Nomi, S. Takashima and M. Klagsbrun, J. Cell. Biochem., 2002, 85, 357–368 CrossRef CAS.
- H. Jia, A. Bagherzadeh, B. Hartzoulakis, A. Jarvis, M. Lohr, S. Shaikh, R. Aqil, L. Cheng, M. Tickner, D. Esposito, R. Harris, P. C. Driscoll, D. L. Selwood and I. C. Zachary, J. Biol. Chem., 2006, 281, 13493–13502 CrossRef CAS.
- A. Jarvis, C. K. Allerston, H. Jia, B. Herzog, A. Garza-Garcia, N. Winfield, K. Ellard, R. Aqil, R. Lynch, C. Chapman, B. Hartzoulakis, J. Nally, M. Stewart, L. Cheng, M. Menon, M. Tickner, S. Djordjevic, P. C. Driscoll, I. Zachary and D. L. Selwood, J. Med. Chem., 2010, 53, 2215–2226 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental procedures, additional SPR sensorgrams, and detailed data of T7 phage display selection. See DOI: 10.1039/c1md00180a |
| ‡ Current affiliation: National Cancer Institute (NCI), National Institutes of Health (NIH), Bethesda, MD, USA. |
| This journal is © The Royal Society of Chemistry 2011 |