Inhibitors of Stat5 protein signalling
Abbarna A.
Cumaraswamy
a,
Aleksandra
Todic
a,
Diana
Resetca
a,
Mark D.
Minden
b and
Patrick T.
Gunning
*a
aDepartment of Chemistry, University of Toronto, 3359 Mississauga Road North, Mississauga, ON L5L 1C6, Canada. E-mail: patrick.gunning@utoronto.ca; Tel: +905-828-5354
bPrincess Margaret Hospital, Ontario Cancer Institute, 610 University Avenue, Toronto, ON M5G 2M9, Canada. E-mail: mark.minden@uhn.on.ca; Fax: +416-946-6546; Tel: +416.946.4501
First published on 3rd October 2011
Abstract
Evidence shows that signal transducer and activator of transcription 5 (Stat5) protein, a member of the STAT family of signalling proteins, plays a pivotal role in the progression of many human cancers including acute myeloid leukemias and prostate cancer. This mini-review outlines progress made towards identifying agents capable of silencing aberrant Stat5 signalling.
Introduction
Signal transducer and activator of transcription 5 (Stat5) protein, a member of the STAT family of signalling proteins, plays a crucial transcriptional role in numerous human cancers and has become a target for therapeutic intervention. Due to growth factor receptor mutations, amplified expression or misdirected kinase activity in the cytoplasm, constitutively activated Stat5 is present in a diverse number of human cancers, including those of the breast, prostate, liver, skin, blood, head and neck.1 The STAT family of proteins is comprised of seven members including Stat1, Stat2, Stat3, Stat4, Stat5A and 5B, as well as Stat6. These proteins perform dual roles as both cytosolic signalling proteins and as nuclear transcription factors that mediate the expression of specific sets of genes. In particular, expression of genes involved in proliferation (Bcl-xl, c-Myc, pim-1), apoptosis (JAB), cell differentiation (p21),2 and inflammation is mediated. In cancer cells, STAT proteins are routinely hyperactivated through either somatic mutation or dysregulated expression of upstream signalling pathways. As a result, several members of the STAT family, including Stat5, can act as oncogenes by facilitating the aberrant expression of key proteins associated with the cancer phenotype. This article will review the progress made towards the development of Stat5-targeting molecular therapeutics.Stat5 proteins are activated/phosphorylated by kinases associated with transmembrane receptors, including IL-2, GM-CSF, erythropoietin, IL-5, IL-7, thrombopoietin, prolactin, and growth hormone receptors.1Extracellular ligand binding to these target receptors induces receptor dimerization and intracellular activation of receptor-associated kinases, such as JAK. JAK-mediated phosphorylation of key tyrosine (Y) residues on the cytoplasmic receptor domain creates docking sites for non-phosphorylated Stat5 proteins. Stat5 binds to the receptor through its phosphotyrosine binding, Src Homology 2 (SH2) domain. Once recruited, Stat5 is phosphorylated by receptor associated JAK kinases, such as Jak2, which phosphorylate Stat5 proteins at specific Y residues in the C-terminus (Y694 for Stat5a and Y699 for Stat5b). Phosphorylated Stat5 protein dissociates from the receptor and forms either homo- or hetero- Stat5–StatX dimers, through reciprocal SH2 domain-pY interactions. Activated Stat5 translocates to the nucleus and binds to Stat5 response elements and induces target gene transcription.
In normal cells, Stat5 proteins are transiently activated by extracellular cytokine or ligand-receptor engagement and rapidly deactivated by a number of intracellular mechanisms, including cytosolic and nuclear phosphatases. For example, deactivation of Stat5 can occur directly viaphosphatases such as protein inhibitors of aberrant STAT proteins (PIAS) or proteintyrosine phosphatases such as SHP-2, or indirectly by down-regulation of cytokine signalling with SOCS1, SOCS3 and CIS proteins.1,3 However, in cancer cells Stat5 is constitutively phosphorylated, leading to elevated expression levels of its target genes, such as the anti-apoptotic proteins Bcl-xl, Bcl-2, Myc and MCL. Aberrant Stat5 activity has been associated with hyperactivated upstream cytosolic tyrosine kinases including TEL-Jak2 and Bcr-Abl, mutated FMS-like tyrosine kinase 3 (FLT3), as well as overactive receptor tyrosine kinases, such as SRC and EGFR.4 Medicinal chemists have attempted to silence aberrant Stat5 activity through a number of direct and indirect molecular approaches. These therapeutic efforts have included the targeting of upstream effector proteins or the direct targeting of Stat5 protein. This review will explore the progress made towards developing effective inhibitors of Stat5 function in human cancers.
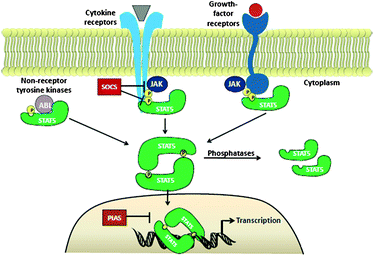 | ||
| Fig. 1 Jak2/Stat5 signalling pathway. | ||
Indirect inhibitors of Stat5 signalling
Stat5's crucial role in cellular signalling and gene transcription is mediated by an intricate cascade of protein–protein interactions. Consequently, there are several junctures at which inhibitors may disrupt the Stat5 signalling pathway. Notably, most inhibitors of Stat5 function have not directly targeted Stat5 protein complexation events, but rather focussed on inhibiting upstream tyrosine kinases including Bcr/Abl, FLT3, and Jak. In addition to indirect kinase inhibitors, Stat5 function has been inhibited by suppression of proteases responsible for protein terminal truncation.Suppressing Stat5 function through Bcr/Abl inhibitors
Bcr/Abl is a novel oncogenic protein that arises in cells as a result of a chromosomal translocation event. This chimeric protein drives constitutive Abl kinase signalling within the cytoplasm of the cell. Aberrant Bcr/Abl signalling leads to constitutive Stat5 activation and subsequent dysregulation of downstream targets.5 Bcr/Abl is the primary genetic abnormality leading to Chronic Myelogenous Leukemia (CML).6 The breakthrough small molecule inhibitor of Bcr/Abl, imatinib or Gleevec (1, Fig. 2), was first approved by the FDA in 2001 as a highly selective inhibitor that provided rapid reduction in the CML clone, recovery of normal hematopoiesis, and improved survival. Consequently the drug quickly became the primary agent for treatment of CML. Imatinib is a highly specific inhibitor of the Abl tyrosine kinase (Abl, IC50 = 25 nM; cf. c-Src, IC5 > 100 μM),7 and potently inhibits downstream phosphorylation of Stat5.8 However, acquired imatinib resistance through development of novel Bcr/Abl mutations has highlighted the need for next-generation inhibitors with activity against imatinib resistant cancers. A promising candidate, PD180970 (2, Fig. 2), containing a pyridopyrimidinone core is a highly potent and selective Abl kinase inhibitor that induces apoptosis in imatinib-resistant leukemia cells. PD180970 has been shown to potently inhibit Stat5–Stat5 DNA-binding activity with an IC50 of 5 nM.9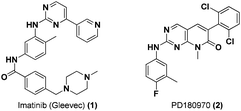 | ||
| Fig. 2 Chemical structures of Bcr/Abl inhibitors. | ||
Suppressing Stat5 function through FLT3 inhibitors
In addition to Bcr/Abl targeting, Stat5 signalling has been inhibited through inhibition of the FLT3 receptortyrosine kinase. FLT3 is highly expressed in lymphoid and myeloid progenitor cells.10 Approximately one third of all AML patients harbour constitutive FLT3 activity due to the development of internal tandem duplication (ITD) mutations. This dysregulation of FLT3 leads to aberrant activation of numerous signalling cascades, including Jak2/Stat5, PI3K/PTEN/Akt/mTOR, and RAS/Raf/MEK/ERK resulting in the up-regulation of Stat5 transcriptional targets such as Bcl-xl, c-Myc, pim-1, JAB and p-21.11 Thus, constitutively activated FLT3 increases cancer cell survival and apoptotic resistance.12 Gain-of-function point mutations, which are associated with hyperactivation of Stat5 and increased growth of lethal myeloproliferative disease cell clones, have been identified in different FLT3 receptor domains.13To deal with this, a number of small-molecule FLT3 inhibitors have been developed and advanced to clinical trials as potential treatments for AML patients. For example, the indolocarbazole alkaloid, lestaurtinib (3, Fig. 3), also known to be a potent Jak2 inhibitor, is an orally bioavailable inhibitor of FLT3 phosphorylation (IC50 ∼ 1.5–3.0 nM). As a result, lestaurtinib has been shown to inhibit FLT3 proliferation in MV4-11 leukemia cells and in turn inhibit Stat5 phosphorylation (IC50 = 2.5–5 nM).14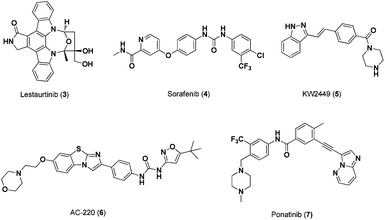 | ||
| Fig. 3 Chemical structures of FLT3 Inhibitors. | ||
Multi-kinase inhibitor, sorafenib (4, Fig. 3), has also been shown to indirectly inhibit Stat5 function. This biaryl urea-based compound inhibited both FLT3-ITD and wild-type FLT3 phosphorylation in MV4-11 and EOL-1 leukemia cell lines and, in turn, potently inhibited cell proliferation with IC50 values of 0.87 nM and 2.3 nM, respectively.15 Moreover, sorafenib produced complete remissions in AML patients, alone or in combination with other drugs, and is currently being evaluated for safety and efficacy.16 In addition to sorafenib, multi-kinase inhibitor, KW-2449 (5, Fig. 3), suppressed FLT3 phosphorylation and subsequent Stat5 phosphorylation in MolM-14 cell lines (IC50 values of 13.1 nM and 14.8 nM, respectively).
AC220 (6, Fig. 3), a second-generation FLT3 inhibitor, has been shown to more potently and selectively inhibit cell proliferation in MV4-11 cells (IC50 = 0.56 nM) than first-generation FLT3 inhibitors, such as 3 and 4.17 In addition, ponatinib (7, Fig. 3), a potent Bcr-Abl inhibitor from ARIAD Pharmaceuticals (IC50 = 0.5–36 nM), has demonstrated comparable inhibition against FLT3 in MV4-11 cells (IC50 = 0.3–17 nM) and subsequent inhibition of Stat5 phosphorylation.18
Despite the identification of chemical compounds with excellent FLT3 inhibitory potency in vitro, clinical trials of these inhibitors demonstrated only moderate inhibition and therefore have not yet resulted in a commercially available FLT3 inhibitor drug. Three possible explanations have been proposed. Firstly, it has been shown that FLT3 mutations are not fundamentally important to the development of AML. Thus, inhibition of FLT3-ITD mutations are simply selecting for AML subclones that are less dependent on complex signallingvia the bone marrow stroma.17 Secondly, Sato et al. have recently suggested that the significant up-regulation of the FLT3 ligand, observed in patients previously treated with chemotherapy, could drastically impair the efficacy of FLT3 inhibitorsin vivo.19 Finally, limitations in vivo may be due to poor pharmacokinetic profiles of these drugs. Despite major successes in effectively inhibiting FLT3, the rapid evolution of resistance to this class of drugs in AML patients remains an ongoing concern.
Suppressing Stat5 function through Jak2 inhibitors
In addition to FLT3 and Bcr/Abl, Jak2 kinase activates Stat5 protein and has been shown to be the most frequently activated oncogenic protein in myeloproliferative neoplasms.20 Somatic mutations in the JAK2 allele can render Jak2 constitutively active and contribute to a number of myeloproliferative disorders. Critically, up-regulation of Jak2 results in constitutively active Stat5 signalling, leading to increased Stat5 transcriptional activity and over-expression of different pro-survival factors.A commonly observed somatic mutation in the JAK2 loci in cancer cells results from a G to T transversion at position 1849, substituting Phe617 with a Val (Jak2-V617F).21 Jak2 has a multi-domain structure containing two Jak homology domains (JH), JH1 and JH2. The C-terminal JH1 domain is a highly conserved kinase domain and is responsible for Jak2 kinase activity. JH2 is a pseudokinase domain lacking ATP binding activity and is responsible for inhibition of the JH1 domain. Gnanasambandan et al. hypothesised that the V617F mutation resulted in a more energetically favourable structural conformation, mediated by a π–π stacking interaction between Phe617 and Phe595. Thus, constitutive Jak2-V617F activity has been attributed to a stabilized aberrant structure, which reduces the auto-inhibitory function of the JH2 domain on the JH1 kinase domain.
A number of small-molecule inhibitors have been identified that selectively modulate the activity of receptor-associated kinases, like Jak2, and thus inhibit Stat5 protein function indirectly. Compounds identified to date have exhibited encouraging oral bioavailability and have progressed to various advanced stages of preclinical and clinical development. Quintas-Cardama and coworkers identified compound 8 (Fig. 4) as a potent and orally bioavailable Jak1/2 inhibitor with increased apoptosis in patients with the Jak2-V617F mutation. Preclinical evaluation in Jak2-V615F+Ba/F3 cells showed inhibition of Jak1 and Jak2 phosphorylation (IC50 = 126 nM).22 This effective pan-Jak kinase activity resulted in the inhibition of Stat5 phosphorylation (IC50 > 100 nM) and, subsequently, Stat5's downstream transcriptional activity.
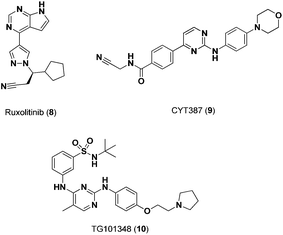 | ||
| Fig. 4 Chemical structures of Jak2 inhibitors. | ||
CYT387 (9, Fig. 4), an ATP mimetic, has exhibited promising activity in preclinical studies, inhibiting Jak2 activity in Jak2V615F+Ba/F3 cells (IC50 = 1.5 μM).23,24 In addition to CYT387, TG101348 (10, Fig. 4) has been shown to be a selective Jak2 inhibitor (IC50 = 3 nM) with a 334 fold selectivity over Jak3.25 As mentioned previously, compound 3, an ATP mimetic, and inhibitor of FLT3, potently inhibits both Jak2 and Jak3 in AML cells thereby silencing Stat5 activity.14
However, while potent small molecule inhibitors of upstream activators of Stat5 have been forthcoming, such inhibitors have a number of disadvantages as indirect inhibitors of Stat5 activity. First, kinase inhibitors often lack target specificity. For example, compounds targeting Jak2 often exhibit pan-JAK activity. Jak2 inhibitors 8 and 9, both potently inhibit Jak1 kinase activity with IC50 values of 3.3 nM and 11 nM, respectively. In addition to pan-JAK kinase activity, JAK inhibitors have exhibited significant off-target effects against other kinase families. As previously mentioned, compound 3 also acts upon FLT3 (IC50 = 2–3 nM). Off-target reactivity and kinase promiscuity remains a significant limitation, often resulting in variable clinical outcomes. Moreover, the majority of small-molecule Jak2 inhibitors are not selective for Jak2 mutants (Jak2-V617F). However, evidence suggests that malignant cells have increased sensitivity to Jak2 inhibitors, thus deriving a therapeutic window to target cancer cells selectively. Nevertheless, the lack of selectivity makes these molecules potent immune-modulators that are generally both pro-inflammatory and immunosuppressive.26 While Stat5 and other STAT family proteins are principle targets of Jak2, activated Jak2 phosphorylates other non-STAT targets feeding into additional pathways.27 These arguments suggest that achieving selectivity for Stat5 over other targets is difficult when using small molecule-based inhibitors to indirectly inhibit Stat5 downstream signalling. Furthermore, Stat5 mutations have been identified in leukemia Balb/c mouse models that render Stat5 constitutively active, independent of the upstream Jak2 signalling pathway.28 Indirect inhibition of Stat5 by targeting Jak2 accelerates the evolution of mutations and drug resistance, maintaining the activity of other Jak2-regulated signalling pathways.
Suppressing Stat5 function through truncation inhibitors
In both healthy and cancerous cells, there naturally exists a proportion of terminally truncated Stat5 proteins. Although the concentration of these variants is not sufficient in healthy cells to exhibit a dominant negative effect over wildtype Stat5, dominant negative effects have been observed in various cancers, including prostate cancer cells (CWR22Rv1, DU145, PC-3, LNCaP)29 and AML patient samples.30One of the most commonly over-expressed Stat5 variants, particularly in prostate cancers, is the N-terminally truncated Stat5 protein.29 In healthy cells, PIAS3 protein serves to downregulate wildtype Stat5 by interacting with the N-terminal domain within the nucleus and thereby inhibiting DNA binding (Fig. 1). N-terminal truncation therefore provides a mechanism by which Stat5 evades PIAS3 interaction and continues to bind DNA, leading to metastasis, tumour growth, and an increased histological grade of the cancer. Inhibiting the generation of these variants could therefore restore PIAS3 inhibition and provide a potential therapy for these dominant negative effects. Presently, the proteases responsible for N-terminal truncation have not been effectively identified and purified in order for the development of small-molecule inhibitors. However, these proteases responsible for truncation could provide a novel and selective therapeutic target for Stat5.
In addition to N-terminal truncation, the C-terminal truncated Stat5 protein is a commonly occurring variant of Stat5. These 77 or 80 kDa variants typically lack their transactivation domain (TAD, aa750–772), which is necessary for interaction with co-activators and gene transcription. The heterodimer formed between these variants and wildtype Stat5 binds DNA with more stability than a wildtype homodimer and thereby suppresses the expression of genes normally regulated by Stat5.31 In healthy mammary cells, this truncation has been shown to reduce cell proliferation rates, increase apoptosis by three-fold, and decrease β-lactoglobulin/luciferase activity.32 Presently, cathepsin G has been identified as being responsible for this truncation in myeloid cells (32D, FDC-P1).33 As with the N-terminally truncated Stat5 variants, targeting this protease may provide a mechanism for inhibiting variant activity in cancerous cells as well as reducing dominant negative effects in healthy cells. However, recent evidence suggests that these C-terminally truncated proteins are in fact artificially generated during sample preparation and do not occur naturally in vivo.34 The physiological significance of C-terminal truncation is therefore brought into question. Generation of N-terminally truncated Stat5, on the other hand, was shown to occur prior to sample preparation and therefore these variants do occur in nature.29 Thus, Stat5 truncation at the N-terminus seems to be a more promising therapeutic target than C-terminal truncated isoforms, and may provide a more selective indirect inhibitory method than kinase targeting.
Direct inhibitors of Stat5 signalling
A significant drawback associated with indirect STAT inhibition strategies has been the large potential for off-target effects and subsequent increases in toxicity. This lack of selectivity is often responsible for poor clinical development of indirect inhibitors, such as kinase inhibitors. Therefore, it is therapeutically appealing to target Stat5 protein directly using small-molecule inhibitors. Presently, two different strategies have been employed to inhibit Stat5 directly: (A) direct inhibition of Stat5 binding to DNA and (B) inhibiting Stat5–Stat5 protein–protein interactions via disruption of the reciprocal phosphotyrosine-SH2 domain interaction.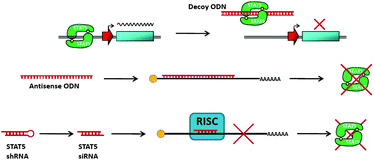 | ||
| Fig. 5 Schematic of oligonucleotide inhibitors of Stat5. | ||
An alternative strategy for inhibiting Stat5 activity, which has been successfully implemented with many other transcription factors,40,41 is based on delivering decoy ODNs that mimic Stat5's cis-elements on DNA. Decoy ODNs occupy the DNA binding site and sequester activated Stat5 proteins in the cytoplasm, thereby reducing their ability to bind to nuclear transcriptional targets and ultimately inhibiting transcription of target genes. This approach holds promise for faster kinetics of Stat5 inhibition than strategies targeting Stat5 isoforms, which rely on the cellular turn-over rates of the Stat5a and Stat5b proteins. According to Behbod et al., these turnover rates could be as little as 6 h, for Stat5a, or as much as 48 h, for Stat5b. While decoy ODNs provide an effective approach for the inhibition of transcriptional activity, the delivery of these ODNs by transfection or other methods limits their sub-cellular compartmentalization, potentially restricting the inhibition of activated Stat5 in the cytoplasm.42
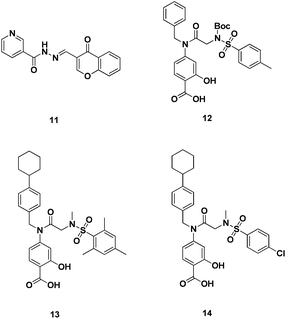 | ||
| Fig. 6 Direct inhibitors of Stat5 dimerization. | ||
Similarly, Gunning and co-workers conducted an in vitro screen of a focussed library of SH2 domain binding salicylic acid-containing inhibitors against Stat5, Stat3 and Stat1 for SH2 domain selectivity. Several potent leads were identified with Ki < 10 μM. Most notably, 12 (Fig. 6), 13 (Fig. 6) and 14 (Fig. 6) (Ki = 7.9 μM, Ki = 2.8 μM and Ki = 8.3μM, respectively) exhibited a 3-fold selectivity for Stat5 over Stat3 and Stat1. Of these Stat5 inhibitors, 12 exhibited the highest Stat5 selectivity (Stat5 Ki = 7.9 μM cf. Stat1, >25 μM and Stat3 >25 μM). When evaluated in K562 and MV4-11 leukemia cells, these lead agents demonstrated potent and selective suppression of Stat5 phosphorylation, inhibition of Stat5's target genes and induction of apoptosis (IC50 ∼ 20μM). Compound 12, in particular, showed negligible cytotoxic effects against healthy bone marrow cells, which do not harbour constitutively activated Stat5. The compounds identified in this study represent the most potent, non-phosphorylated direct small molecule inhibitors of Stat5 to date.44
Finally, a function-based screening approach was used to discover the drug pimozide (15, Fig. 7), which was found to significantly inhibit Stat5 gene expression. Further studies in KU812 and K562 CML cells revealed that both cell lines exhibited dose-dependent decreases in Stat5 tyrosine phosphorylation at 3–10μM doses of 15. Moreover, treatment of CML cells with 15 (10 μM) resulted in decreased viability and induction of apoptosis. Encouragingly, 15 showed no toxicity against healthy cells at similar concentrations. Notably, Nelson and co-workers demonstrated that 15 did not directly inhibit kinases such as Bcr-Abl or its downstream pathway (MAPK) or Jak2 kinase. Rather, 15 is proposed to interact with Stat5 protein directly. Nelson and co-workers showed that 15, in combination with imatinib (1), is effective against Bcr-Abl mutant cancer cells.49
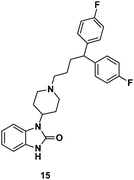 | ||
| Fig. 7 Chemical structure of pimozide. | ||
Conclusions
Significant effort has been exerted for the targeting of upstream effectors of Stat5 function, including, Bcr-Abl, Jak2 and FLT-3. Most notably, imatinib, the potent Bcr/Abl targeting drug, has achieved considerable success in the clinic. In general, however, kinase inhibitors upstream of Stat5 have suffered from the development of resistance, toxicity as a result of poor kinase selectivity, as well as cardiovascular toxicity. In an effort to overcome these issues, downstream targeting of Stat5 protein and Stat5-DNA binding has been attempted through application of oligonucleotide-based therapeutics, various truncated Stat5 proteins and small molecule inhibitors. However, to date, the number of direct inhibitors of Stat5 protein has been limited to only a handful of examples. Given the recent successes of small molecule therapeutic approaches against Stat3 complexation events,45–48 there is much scope for developing improved agents against Stat5 directly. The prospect for a Stat5-selective therapeutic for treatment of leukemias and prostate cancer is highly appealing.References
- T. Shyh-Han and M. T. Nevaleinen, Endocr. Relat. Cancer, 2008, 15, 367 CrossRef.
- T. Nosaka, T. Kawashima and K. Misawa, EMBO J., 1999, 18, 4754 CrossRef CAS.
- K. J. Peltola and K. Paukku, et al. , Blood, 2004, 103, 3744 CrossRef CAS.
- H. Yu and R. Jove, Nat. Rev. Cancer, 2004, 4, 97 CrossRef CAS.
- K. Shuai and J. Halpern, et al. , Oncogene, 1996, 13, 247 CAS.
- J. D. Rowley, Nature, 1973, 243, 290 CrossRef CAS.
- B. J. Druker and S. Tamura, et al. , Nat. Med., 1996, 2, 561 CrossRef CAS.
- B. M. Mow and J. Chandra, et al. , Blood, 2002, 99, 664 CrossRef CAS.
- M. Huang and J. F. Dorsey, et al. , Oncogene, 2002, 21, 8804 CrossRef CAS.
- E. Weisberg and M. Sattler, et al. , Oncogene, 2010, 29, 5120 CrossRef CAS.
- J. L. Rocnik and R. Okabe, et al. , Blood, 2006, 108, 1339 CrossRef CAS.
- S. M. Kornblau and M. Womble, et al. , Blood, 2006, 108, 2358 CrossRef CAS.
- L. M. Kelly and Q. Liu, et al. , Blood, 2002, 99, 310 CrossRef CAS.
- M. Levis and J. Allebach, et al. , Blood, 2002, 99, 3885 CrossRef CAS.
- D. Auclair and D. Miller, et al. , Leukemia, 2007, 21, 439 CrossRef CAS.
- A. T. Fathi, S. Grant and J. E. Karp, Cancer Treat. Rev., 2010, 36, 142 CrossRef CAS.
- Q. Chao and K. G. Sprankle, et al. , J. Med. Chem., 2009, 52, 7808 CrossRef CAS.
- T. Sato and X. Yang, et al. , Blood, 2011, 117, 3286 CrossRef CAS.
- J. M. Gozgit and M. J. Wong, et al. , Blood, 2011, 10, 1 Search PubMed.
- A. Pardanani and A. M. Vannucchi, et al. , Leukemia, 2011, 25, 218 CrossRef CAS.
- K. Gnanasambandan and A. Magis, et al. , Biochemistry, 2010, 49, 9972 CrossRef CAS.
- A. Quintas-Cardama and K. Vaddi, et al. , Blood, 2010, 115, 3109 CrossRef CAS.
- A. Pardanani and T. Lasho, et al. , Leukemia, 2009, 23, 1441 CrossRef CAS.
- F. P. Santos and H. M. Kantarjian, et al. , Blood, 2010, 115, 1131 CrossRef CAS.
- G. Wernig and M. G. Kharas, et al. , Cancer Cell, 2008, 13, 311 CrossRef CAS.
- T. Ikezoe and J. Yang, et al. , Int. J. Cancer, 2011, 128, 2317 CrossRef CAS.
- C. Carter-Su, L. Rui and M. R. Stofega, Recent Prog Horm Res, 2000, 55, 293 CAS.
- R. L. Ilaria and R. G. Hawley, et al. , Blood, 1999, 93, 4154 CAS.
- A. Dagvadorj and S. Tan, et al. , Int. J. Biochem. Cell Biol., 2010, 42, 2037 CrossRef CAS.
- Z. Xia and N. J. Sheila, et al. , Leuk. Res., 2001, 25, 473 CrossRef CAS.
- D. Wang and D. Stravopodis, et al. , Mol. Cell Biol., 1996, 16, 6141 CAS.
- E. Iavnilovitch and T. Eilon, et al. , Mol. Reprod. Dev., 2006, 73, 841 CrossRef CAS.
- B. Schuster and L. Hendry, et al. , Biochem. J., 2007, 404, 81 CrossRef CAS.
- H. L. Ramos, J. J. O'Shea and W. T. Watford, Biochem. J., 2007, 404, e1 CrossRef CAS.
- F. Behbod and Z. S. Nagy, et al. , J Immunol, 2003, 171, 3919 CAS.
- S. Cao and C. Wang, et al. , Neurosci. Lett., 2011, 487, 228 CrossRef CAS.
- X. Liu and G. W. Robinson, et al. , Proc. Natl. Acad. Sci. U. S. A., 1995, 92, 8831 CrossRef CAS.
- C. A. Sledz and B. R. Williams, Biochem. Soc. Trans., 2004, 32, 952 CrossRef CAS.
- S. K. Klosek and K. Nakashiro, et al. , Oncol Rep, 2008, 20, 873 CAS.
- X. Wang and J. Zeng, et al. , DNA Cell Biol., 2011, 30, 71 CrossRef CAS.
- J. Shen and R. Li, et al. , In Vivo, 2009, 23, 237 CAS.
- D. W. Kim and J. H. Kim, et al. , Biomaterials, 2011, 32, 2593 CrossRef CAS.
- J. Muller and T. Berg, et al. , ChemBioChem, 2008, 9, 723 CrossRef.
- B. D. Page and P. T. Gunning, et al. , J Med Chem, 2011 DOI:jm-2011-00720n , submitted.
- M. Avadisian, S. Haftchenary and P. T. Gunning, Anticancer Drugs, 2010, 22, 115 Search PubMed.
- S. Fletcher and J. A. Drewry, et al. , Biochem. Cell Biol., 2009, 87, 825 CrossRef CAS.
- S. Fletcher, J. Turkson and P. T. Gunning, ChemMedChem, 2008, 3, 1159 CrossRef CAS.
- B. D. Page, D. P. Ball and P. T. Gunning, Expert Opin. Ther. Pat., 2011, 21, 65 CrossRef CAS.
- E. A. Nelson and S. R. Walker, et al. , Blood, 2011, 117, 3421 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2012 |