From the protein's perspective: the benefits and challenges of protein structure-based pharmacophore modeling
Marijn P. A.
Sanders
a,
Ross
McGuire
b,
Luc
Roumen
c,
Iwan J. P.
de Esch
c,
Jacob
de Vlieg
ad,
Jan P. G.
Klomp
e and
Chris
de Graaf
*c
aComputational Drug Discovery Group, CMBI, Radboud University Nijmegen, Geert Grooteplein Zuid 26-28, 6525 GA, Nijmegen, The Netherlands. E-mail: M.Sanders@cmbi.ru.nl
bBioAxis Research BV, Bergse Heihoek 56, 5351 SL, Berghem, The Netherlands. E-mail: ross.mcguire@bioaxisresearch.com
cDivision of Medicinal Chemistry, LACDR, VU University Amsterdam, de Boelelaan 1083, 1081 HV, Amsterdam, The Netherlands. E-mail: C.de.Graaf@vu.nl
dLead Pharma Medicine, Kapittelweg 29, 6525 EN, Nijmegen, The Netherlands. E-mail: jan.klomp@leadpharma.com
eNetherlands eScience Center, Science Park 140, 1098XG, Amsterdam, The Netherlands. E-mail: J.devlieg@esciencecenter.nl
First published on 12th December 2011
Abstract
A pharmacophore describes the arrangement of molecular features a ligand must contain to efficaciously bind a receptor. Pharmacophore models are developed to improve molecular understanding of ligand–protein interactions, and can be used as a tool to identify novel compounds that fulfil the pharmacophore requirements and have a high probability of being biologically active. Protein structure-based pharmacophores (SBPs) derive these molecular features by conversion of protein properties to reciprocal ligand space. Unlike ligand-based pharmacophore models, which require templates of ligands in their bioactive conformation, SBPs do not depend on ligand information. The current review describes the different steps in the construction of SBPs: (i) protein structure preparation, (ii) binding site detection, (iii) pharmacophore feature definition, and (iv) pharmacophore feature selection. We show that the SBP modeling workflow poses different challenges than ligand-based pharmacophore modeling, including the definition of protein pharmacophore features essential for ligand binding. A comprehensive overview of different SBP modeling and screening methods and applications is provided to illustrate that SBPs can be efficiently used for virtual screening, ligand binding mode prediction, and binding site similarity detection. Our review demonstrates that SBPs are valuable tools for hit and lead optimization, compound library design and target hopping, especially in cases where ligand information is scarce.
1. Introduction
The pharmacophore concept was first introduced in 1909 by Ehrlich who defined a pharmacophore as ‘a molecular framework that carries (phoros) the essential features responsible for a drug's (pharmacon) biological activity’.1 Later, the pharmacophore concept was more precisely described by Kier2 who stated that a drug must possess ‘(a) those atomic features suitable for the requisite drug–receptor interaction phenomena and (b) the appropriate spatial disposition of these features necessary to bring about the required simultaneous or required sequential interaction events with the receptor’.3 Gund further updated Kier's definition and described a pharmacophore in 1977 as “a set of structural features in a molecule that is recognized at a receptor site and is responsible for that molecule's biological activity”.4 Pharmacophores have proven to be extremely effective in silico filters in the search for bioactive molecules on several targets. Their use reduces the number of compounds and costs which have to be considered in large biophysical screenings.5–8 In contrast to e.g. topological/2D ligand similarity searches which take a whole-structure, ‘global,’ view on the activity of molecules, pharmacophores focus on ‘local’ similarity and study the molecular determinants and their specific arrangement required for biological activity.9 As a result they provide an explanation for the predicted activity of molecules. Ligand-based pharmacophores (LBPs), i.e., pharmacophore models derived from one or multiple active ligand(s), have been extensively used in the discovery and design of biologically active molecules.6Protein structure-based pharmacophores (SBPs), derived from the three-dimensional (3D) structure(s) of one or more protein target(s), are receiving more and more attention in the past few years.6,10 One of the reasons for the rising interest in SBPs is the significant increase in high resolution protein structures. Currently more than 75![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 three-dimensional structures of biological macromolecules (mostly proteins) are deposited in the Protein Databank (9th August 2011),11 leading to unprecedented understanding of these molecular drug targets. SBPs can be used as a tool to give insights into ligand–protein interactions and to enable large scale structural chemogenomics studies to identify new ligands for specific proteins (ligand profiling), or find new targets for specific ligands (target fishing).12–16 As such, there are three benefits of SBPs over LBPs: (i) SBPs allow the identification of novel scaffolds, as they are less biased towards existing ligand chemotypes. (ii) SBPs can be used to elucidate protein–ligand binding mode hypotheses within the protein structural framework,17–20 making SBPs suitable tools for structure-based ligand optimization. (iii) SBPs lead to better understanding of ligand binding sites. These insights can for example be used to find ligands for orphan receptors21 or to study ligand binding site similarities between different proteins to address cross-pharmacology22 and suggest new targets for existing drugs.23
000 three-dimensional structures of biological macromolecules (mostly proteins) are deposited in the Protein Databank (9th August 2011),11 leading to unprecedented understanding of these molecular drug targets. SBPs can be used as a tool to give insights into ligand–protein interactions and to enable large scale structural chemogenomics studies to identify new ligands for specific proteins (ligand profiling), or find new targets for specific ligands (target fishing).12–16 As such, there are three benefits of SBPs over LBPs: (i) SBPs allow the identification of novel scaffolds, as they are less biased towards existing ligand chemotypes. (ii) SBPs can be used to elucidate protein–ligand binding mode hypotheses within the protein structural framework,17–20 making SBPs suitable tools for structure-based ligand optimization. (iii) SBPs lead to better understanding of ligand binding sites. These insights can for example be used to find ligands for orphan receptors21 or to study ligand binding site similarities between different proteins to address cross-pharmacology22 and suggest new targets for existing drugs.23
Generally four different steps in the construction of SBPs can be distinguished: (i) protein structure preparation, (ii) binding site detection, (iii) pharmacophore feature definition, and (iv) pharmacophore feature selection (Fig. 1). Protein structure preparation and binding site detection are obviously specific aspects of SBP modeling. The protonation states of functional groups and conformation of the pharmacophore modeling template are clearly equally important determinants of LBPs and SBPs. For SBPs however, the possible variation in protonation states and conformations (at the protein backbone, sidechain, or polar hydrogen atoms level) is in principle larger, simply because ligand binding sites (in most cases) contain more atoms than ligands do. Whereas the features included in SBPs are generally the same as in LBPs, the initial number of features in SBPs is generally higher. As a result, pharmacophore feature selection (an important step in both LBP and SBP modeling workflows) requires a different approach and poses different challenges in LBP and SBP modeling protocols. Structural alignment of different ligands can be used to identify essential features in LBPs, and experimental data such as structure–activity relationships (SAR) can be used to further emphasize specific features in LBPs. Selection of essential features in SBPs is however not straightforward, even when guided directly by experimental data such as site-directed mutagenesis studies or indirectly by including amino acid sequence based knowledge or ligand information. The larger number of conformational and spatial possibilities in SBPs make the selection of pharmacophore features more complex than in LBPs.
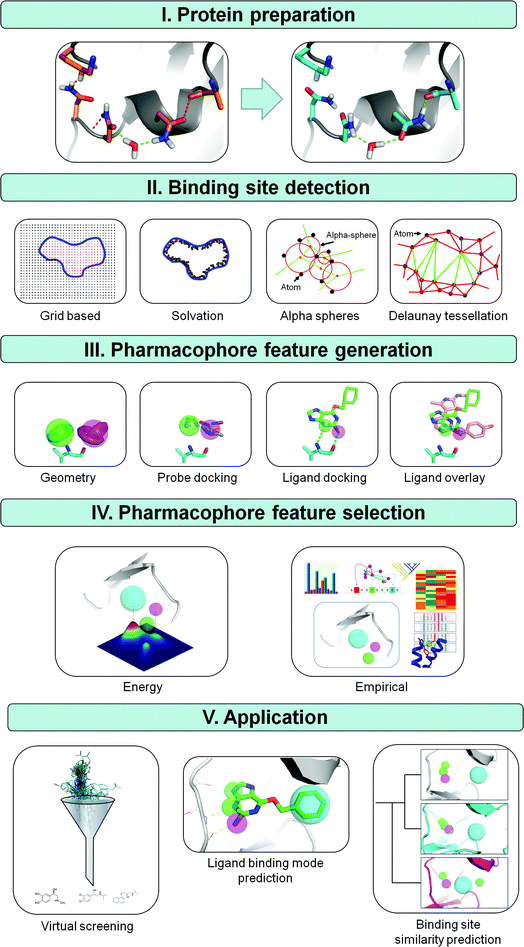 | ||
| Fig. 1 Methods for deriving structure based pharmacophores (SBPs). (I) The first step in the derivation of a SBP is the preparation of the protein structure. An example of PDB file 2BNU is shown with favorable interactions in green and unfavorable interactions in red. Three unfavorable interactions are observed in the original input file; an asparagine bumps into a lysine, a hydrogen of a neighboring asparagine clashes with the backbone and two partial negatively charged oxygens of an asparagine and backbone alanine are in close proximity. Flipping these asparagines removes all unfavorable interactions and gains one additional favorable interaction.65 (II) Next, a cavity is defined using a grid-based, solvation, alpha sphere or the Delaunay tessellation based approach. (III) Pharmacophore features are subsequently derived by calculation of spatially favorable positions for functional groups by using known interaction geometries, probe docking, or by docking one or more ligands and extracting this information from ligand poses. (IV) To increase pharmacophore specificity an energy or statistics based method is finally applied to reduce the number of pharmacophore features in the final pharmacophore hypothesis. (V) Three possible applications for pharmacophores are virtual screening, ligand binding mode prediction and binding site similarity prediction. | ||
The current review gives a comprehensive overview of different methodologies to construct SBPs and describes the specific benefits and challenges (Section 2). Furthermore, representative applications of SBPs are provided to illustrate that SBPs can be efficiently used for virtual screening, ligand binding mode prediction, and binding site similarity detection (Section 3). Our review demonstrates that SBPs are valuable tools for hit and lead optimization, compound library design and target hopping, especially in cases where ligand information is scarce.
2. Protein structure based pharmacophore modeling methodology
In principle, the availability of a three dimensional protein structure or model is the only absolute pre-requisite for SBP methods. If the target protein structure is not known a protein structure model may be generated by homology modeling with the use of the sequence and the three dimensional structure of a close homolog.24 The subsequent steps in the SBP modeling workflow, protein preparation (2.1), binding site detection (2.2), pharmacophore feature definition (2.3), and pharmacophore feature selection (2.4) are outlined in Fig. 1 and discussed in this section. An overview and classification of the different SBP methods for binding site detection, feature definition and selection is presented in Table 1.| Method/software | Protein preparation | Binding site detection | Feature definition | Feature selection | Resolutiona | Automationb | Reference |
|---|---|---|---|---|---|---|---|
| a High resolution methods require one high resolution structure from which all information is deduced. Medium resolution methods are able to combine different high resolution structures. Low resolution models do not require a high resolution structure, instead typically use interaction geometries, rotamer ensembles or simulations to cover a range of probable conformations. b Highly automated methods can produce pharmacophores by the push of a button and have a fully integrated workflow. Medium automation requires a call to separate procedures. Low automation methods require the user to specify at which location or residue a feature should be positioned without giving much guidance except possibly the coloring of surfaces according to calculated properties. c The method uses a fixed position for pharmacophore features and positions them solely based on sequence information. d The method requires a ligand. | |||||||
| Catalyst/Discovery Studio | Yes | Grid | Geometry | Energy | Medium | Medium | 50 |
| Chemogenomics (Klabunde) | No | Fixedc | Fixedc | — | Low | High | 28 |
| FLAP | Yes | Grid | Geometry | Energy | Medium | Medium | 66 and 67 |
| GBPM | Yes | Grid | Geometry | Energyd | Medium | Medium | 68 |
| HS-Pharm | Yes | Grid | Geometry | Empirical | Medium | Medium | 69 |
| LigandScout | Yes | Ligand | Energy | High | High | 70 | |
| MOE | Yes | Alpha spheres | Ligand | Energy/manual | High | High | 52 |
| MUSIC | Yes | Solvation | Probe | Energy | Low | Medium | 47 |
| Pocket v2 | Yes | Grid | Geometry | Energy | Medium | High | 71 |
| Schrodinger | Yes | Grid | Probe | Energy | High | Medium | 72 |
| Ligand | Energy | High | |||||
| Snooker | No | Delaunay | Geometry | Empirical | Low | High | 73 |
| Sybyl | Yes | Grid | Geometry | Knowledge | Medium | Low | 49 |
| Solvation | Ligand | Energy/manual |
2.1 Protein structure preparation
2.2 Binding site detection
The second step in the SBP modeling workflow (Fig. 1) is to define the location of the ligand binding site by the application of binding site detection algorithms. Retrospective studies show that these algorithms perform very well in the case of ligand bound crystal structures, correctly detecting up to 95% of the ligand binding pockets, but are less accurate for apo-structures.44 Structure based binding site detection can be divided into energy based methods (including solvation methods) and geometry based methods (including grid based, alpha sphere, Delaunay tessellation) as summarized in Fig. 1 and Table 1. Energy-based algorithms, like PocketFinder,45 SuperStar,46 and MUSIC,47 the solvation method of SiteID (Tripos) and SiteMap (Schrödinger) try to describe the local surface properties of a cavity by the simulation of solvent molecule interactions with the protein surface. SiteMap48 uses a grid to sample binding site properties but is nevertheless fully based on interaction energy calculation. MUSIC47 floods the pocket with probe fragments instead of solvent molecules and simulates the interactions of those fragments in short MD-simulations. Geometry based methods can be divided into two categories: discrete grid-based sampling and analytic methods like Delaunay tessellation and alpha sphere based methods. Grid-based methods available in SiteID (Tripos49), GRID40 and in the binding site analysis module of InsightII (Accelrys50) typically select grid points close to but not overlapping with protein atoms, and define the pocket after a flood filling algorithm of grid points located in a cavity, where cavity grid points are defined as grid points with a substantial number of contacts with the protein. Snooker identifies the protein pocket by a Delaunay tessellation51 of Cα-atoms and a calculated mean side chain atom, followed by the indexing of tetrahedra with at least one ‘long’ edge (Fig. 1). Delaunay tessellation implies the generation of an aggregate of space-filling irregular tetrahedra. These tetrahedra are deduced from a set of coordinates such that for each tetrahedron, the four vertices are on the circumspheres while no other vertices are inside the circumsphere. It is used to identify all tetrahedra which span large distances and cover potential ligand binding pockets. Finally, all indexed tetrahedra having a triangle in common are merged and the largest merged volume is defined as the pocket, while all non-indexed tetrahedra form the protein volume. SiteFinder (MOE52), PASS,53SURFNET,54 LIGSITE,55 APROPOS,56 and CAST57,58 use alpha complexes to detect pockets. Alpha shapes are an extension of the convex hulls proposed by Edelsbrunner and Mucke.59 Alpha spheres associated with 4 atoms can be generated from the simplices of a Delaunay tessellation (Fig. 1). Solvent exposed alpha spheres and those corresponding to inaccessible areas (small spheres) are removed and the pocket is detected by aggregation of remaining nearby alpha spheres. Two reviews summarizing and explaining ligand-binding site detection have been written by Prymula et al.60 and Henrich et al.612.3 Pharmacophore feature definition
In the third step in the SBP flow scheme (Fig. 1), pharmacophore features are derived from the co-crystallized ligand or from the ligand binding site (determined in the previous step) itself (Table 1). Protein structure based pharmacophore methods typically use geometric entities, such as spheres, vectors and planes with given attributes to characterize favorable interactions. The commonly used interaction types include H-bond acceptors, H-bond donors, positive and negative ionizable groups, lipophilic regions and aromatic rings. Their positions are set according to either positions of co-crystallized ligands or basic interaction geometry rules.Derivation of pharmacophores from ligands is ostensibly straightforward with features positioned at functional groups of the ligand. Ligand based pharmacophore modeling tools are generally similar although they may produce slightly different pharmacophores due to differences in feature definitions and algorithmic search strategies.62,63 An excellent review providing a detailed explanation of the available molecular alignment techniques has been written by Lemmen and Lengauer.64 As each software package has differences in feature definition criteria, it is appropriate to use the same algorithm for both pharmacophore elucidation and pharmacophore searching, in order to ensure compatibility. If the structure of a protein–ligand complex is available, a pharmacophore can be constructed by positioning features at the functional groups of ligands (Table 1).
Structure based pharmacophore derivation from apo structures is in contrast more challenging. The number of potential interacting residues in a ligand binding site is typically larger than the number of observed interactions in protein–ligand complexes. Furthermore it is not straightforward to determine the optimal interaction geometry and there is no guarantee that ligands will interact at the predicted favorable sites of interaction. Some targets even have diverse ligand-binding modes and require a set of different pharmacophores to cover the interaction observed for all ligands.74–80 Pharmacophore feature placement is therefore less accurate in structure based pharmacophore methods and tolerances of pharmacophore features are typically less strict. FLAP,66,67GBPM,68 Pocket V2,71 Discovery Studio (Accelrys50), Sybyl (Tripos49) and Snooker29 can create SBPs without the use of any ligand information and apply knowledge from residue based interaction geometries to predict likely interactions and their locations. MUSIC47 and Schrodinger37,38 also do not require information about known actives and use a multiple copy simultaneous search (MCSS) method81 to identify energetically favorable positions and properties of probes to generate pharmacophore features.38 In the MCSS method a protein's active site is filled with thousands of copies of organic functional groups which are allowed to energy-minimize onto the protein surface. Groups that minimize at the same location and bind most tightly are subsequently converted into pharmacophore features. MOE52 and LigandScout70 at least require one known active ligand and a protein structure. Both can be used to generate a binding mode hypothesis and extract pharmacophore features from the modelled interactions. An improved pharmacophore can potentially be defined if multiple ligands are docked into the receptor active site and if only the conserved interactions are translated into pharmacophore features. Two recent comparative reviews on pharmacophore elucidation methods have recently been published by Luu et al. and Wallach.82,83
2.4 Selection of essential pharmacophore features
To obtain valid binding mode hypotheses and subsequent compound library enrichment it is important to select only those features that correlate with biological activity (step 4 in the SBP modeling workflow (Fig. 1)). Three different approaches can be defined to select essential pharmacophore features (Fig. 1 and Table 1): (i) using interaction energy calculations (2.4.1), (ii) using protein–ligand interaction information (2.4.2), and (iii) based on analysis of amino acid sequence variation (2.4.3). Finally, SBPs can be refined by training pharmacophore models with known active compounds (2.4.4) and by complementing SBPs with shape restraints (2.4.5).000 compounds was achieved. Filtering of this set with 25 excluded volume spheres placed at positions occupied by the receptor gave 56000 compounds while only 38000 compounds were selected after the reduction in HBA tolerances to 1.3 Å. A final shape filter of 110% and 100% of the reference structure volume reduced the selection to 35000 and 16665 compounds, respectively. Seventeen compounds were selected based on high fit values as well as diverse structures and subjected to experimental validation in a bioassay. All these compounds showed an inhibitory effect on ACE2 activity. Some virtual screening methodologies are even entirely based on shape, like the ROCS method from OpenEye and Shape4 developed by Ebalunode et al.92 Both have been reported to perform well in terms of virtual screening.93,94 However to derive specific shape restraints the conformation and binding mode of at least one known active molecule are required and these methods have therefore limited compatibility with structure based pharmacophores.
3. Applications of SBPs
SBPs are developed to improve molecular understanding of ligand–protein interactions (3.1). As already mentioned in Section 1, SBPs can be used for virtual screening (3.2), ligand binding mode prediction (3.3), and binding site similarity detection (3.4). The lower part of Fig. 1 gives a pictorial description of three different uses of SBPs and an overview of relevant articles concerning the application of structure-based pharmacophores is presented in Table 2. The application papers discussed in this review are diverse with respect to the generation method, target family, available data and used software and will be discussed in more detail in this section. The SBP models described in the current section furthermore exemplify: (1) some of the potential advantages of SBPs over LBPs; (2) the discovery of novel scaffolds different from known chemotypes; (3) the elucidation of protein–ligand binding modes, and (4) better understanding of ligand binding sites.| Target | Family | Input | Method | Predictionc | Result | Reference |
|---|---|---|---|---|---|---|
| a The chemogenomics approach has fixed positions for features and positions features depending on the protein sequence. b Features are positioned on the protein (do not describe desired ligand features). c VS: virtual screening, BM: binding mode hypothesis generation, BS similarity: binding site similarity prediction. | ||||||
| AlaR | Racemase | X-Ray | Overlay | VS | 19 Compounds selected for testing | 121 |
| C3AR1 | GPCR | — | Chemoprintsa | VS | 4 New ligands found | 28 |
| Various | Me–Lys binders | X-Ray | Protein b | BS similarity | Similar sites identified | 139 |
| CDK2 | Transferase | X-Ray | Overlay | Model validation | Recognition known ligands | 140 |
| CHK1 | Transferase | X-Ray | Overlay | VS | Enrichment > 9-fold | 122 |
| DHFR-TS | Synthase | X-Ray | Complex | BM + affinity | Correct prediction 6 ligands | 141 |
| DNMT1 | Transferase | Model | Probe | BM + VS | Explanatory pharmacophore model derived | 142 |
| HIV-1 RT | Transcriptase | X-Ray-NMR | Geometry | BM | HIV-1RT superligand generated | 138 |
| HtrA | Protease | Model | Geometry | VS | 6 New ligands found | 120 |
| Kinase | Transferase | X-Ray | Geometry | BS similarity | Relevant clustering of kinase families | 90 |
| Kinase | Transferase | X-Ray | Geometry | BS similarity | Bio-isosters proposed for multiple targets | 143 |
| Thrombin | Protease | |||||
| Kv1.5 | Potassium channel | Model | Geometry | VS | 19 New ligands found | 124 |
| NK1R | GPCR | Model | Overlay | VS | 1 New ligands found | 123 |
| NS3 | Protease/helicase/NTPase | X-Ray | Overlay | BM + VS | 15 Compounds selected for testing, ligand interacting residues identified | 135 |
| Renin | Angiotensinogenase | X-Ray | Interaction | BM + VS | 2 New ligands found | 136 |
| RSK2 | Transferase | Model | Overlay | VS + BM | 2 New ligands found | 144 |
| Thrombin | Protease | X-Ray | Geometry | VS + scaffold hopping | Enrichment > 15-fold, successful scaffold replacement | 137 |
| TrXr, HIV-1N | Reductase–Integrase | X-Ray | Geometry | VS | 1 New ligand found | 119 |
| Various | — | X-Ray | Complex | VS | Average 40-fold enrichment | 38 |
| Various | — | X-Ray | Geometry | VS | Average 17-fold enrichment | 67 |
3.1 SBPs versus LBPs
While comparativeligand- and structure-based pharmacophore modelling studies are relatively scarce,95 more and more protocols are reported in which LBP and SBPs are combined.96–99 This is in line with recent comparative virtual screening studies which show that ligand- and protein structure-based methods are complementary approaches in identifying different ligand chemotypes.99–101Thangapandian et al.95 report a comparative study of ligand- and structure-based pharmacophores for the design of novel histone deacetylase 8 inhibitors. The LBP comprised 4 features and retrieved 117 compounds of which 87 were active (corresponding to an ∼8-fold enrichment over random picking), while the SBP contained 6 features and retrieved 74 compounds of which 63 were active (corresponding to a comparable enrichment value of ∼10). Kumar et al.96 describe a method which combines LBPs and SBPs in order to identify additional interaction sites with c-Jun N-terminal kinase-3 that cannot be derived by ligand-based approaches alone. Using a training set of 21 c-Jun N-terminal kinase-3 inhibitors a LBP of 4 features was derived and used to construct a quantitative pharmacophore model to predict the affinities of 85 inhibitors with a correlation coefficient r2 of 0.846. Conserved hydrogen bond interactions with the hinge region were subsequently identified by docking of the 85 inhibitors into the kinase binding site and a SBP was derived which contained three additional features (2 donors and 1 acceptor) compared to the previously constructed LBP. Griffith et al.97 and Singh et al.102 have described protocols that combine the speed of LBPs with the ability of SBPs to predict ligand binding modes and to discover novel molecules.
Comparative retrospective virtual screening studies report similar99 or somewhat higher100,101 overall enrichment for ligand-based methods compared to structure-based virtual screening approaches.101 The performance of different ligand-based103,104 and docking-based methods105 as well as the relative performance of ligand- vs. structure-based methods99–101 can however be highly target dependent, justifying the use of both ligand-based and structure-based drug design techniques as complementary ligand discovery tools. Evers et al.100 for example performed a retrospective virtual screening study on four biogenic amine-binding G-protein coupled receptors (GPCRs) in which three ligand-based methods (LBPs,106,107 2D,108 and 3D similarity searches109) were compared with docking110–112 in homology models. They showed that ligand-based methods outperform structure based methods, although structure-based methods still have satisfying enrichment factors (up to 60% of actives in the top-ranked 1% of the screened database). Krüger and Evers99 compared docking,113–116 3D similarity searches117 and 2D similarity searches118 and reported almost equal enrichments. Hit lists obtained from different algorithms were however complementary and the combination of different approaches is likely to result in more (and more diverse) actives.
3.2 Virtual screening (VS) for new ligands
Structure based pharmacophores (SBPs) are very well suited to combine the efficient screening methodologies of pharmacophores with structural information obtained from X-ray crystallization and NMR spectroscopy efforts. In this section 7 virtual screening studies are described28,38,67,119–123 which used different methods (geometry (3.2.1), probe (3.2.2), complex (3.2.3), overlay (3.2.4), and chemogenomics (3.2.5) based) to derive and select pharmacophore features (Fig. 1).3.3 Ligand binding mode prediction
In contrast to ligand-based pharmacophores, structure-based pharmacophores add to the understanding of the interaction of a (set of) ligand(s) with the protein. This is beneficial for affinity and selectivity prediction, defining Structure–Activity Relationships (SAR), and hit optimization. SBPs can also be used for a complete exploration of the binding pocket and enable the targeting of residues that have not previously been utilized in interactions with ligands. A prerequisite for all these studies is the correct prediction of the ligand binding. So far only a few studies have shown that SBPs are capable to reproduce ligand binding modes of experimentally determined protein–ligand complexes. Loving et al.37 showed that they could reproduce the binding modes observed in 12 different protein–ligand complexes, while Sanders et al. used SBPs to correctly predict the binding modes of a set of β-2-adrenergic agonists and antagonists/inverse agonists. SBPs can also be used in pharmacophore constrained docking and several studies have shown that pharmacophore constraints can significantly improve binding mode predictions and virtual screening enrichment.39,132–134 In this section several different methods to derive and use SBPs in binding mode prediction will be discussed.3.4 Ligand binding site comparison
Many different methods to compare ligand binding sites based on pharmacophore or pharmacophore related properties have been developed in the past decade (for extensive reviews see Henrich et al.61 and Kellenberger et al.146). Most methods, like FuzCav,147 CavBase148 and PocketMatch,149 compare binding sites by assigning pharmacophore features directly to the protein. Campagna-Slater identified ligand binding sites that are chemically similar to known methyl-lysine binding domains using such SBP models.139 The KRIPO method, developed by Ritschel et al., uses protein–ligand interaction features derived from the ligand binding site to create 3D-pharmacophore fingerprints. This method has been successfully used to identify similar binding pockets and suggest structural modifications to ligands based on presumed bioisosteres.143 Sciabola et al. used FLAP to compare protein binding sites and were able to cluster kinase protein families in a relevant manner, predict ligand activity across related targets and perform protein–protein virtual screening.90 These methods are suitable for the identification of proteins which can be selectively targeted and compounds which might have activity on proteins with pharmacophorically similar binding sites.4. Conclusion
The current review describes the different steps in the construction of SBPs: (i) protein structure preparation, (ii) binding site detection, (iii) pharmacophore feature definition, and (iv) pharmacophore feature selection. SBP generation typically starts with a protein preparation step to correct and optimize the starting structure. Subsequently the ligand binding pocket can be defined by a pocket detection algorithm and several strategies can be used to convert the protein properties to ligand. A choice of method can be made depending on the resolution of the available protein structure or model and the availability of known active ligands and corresponding binding mode hypotheses. Geometric methods to derive SBPs are typically the least restrictive and are together with the probe based method the only approaches which can be applied in the absence of known ligands. A real challenge in SBP design is the reduction of the typically high number of features in a structure based pharmacophore to only those features which are related to biological activity. Energy based methods to construct SBPs in these cases typically depend on the accuracy of the input structure and the binding mode hypothesis generated, while statistics based measures, such as those relying on the protein–ligand complex or protein variance information, are generally more robust with a greater capability to deal with low resolution or low quality structures. The relatively simple concept of a pharmacophore makes it an attractive tool for various research applications. Several studies have described the successful use of SBPs for binding mode hypotheses generation, virtual screening and binding site similarity calculations, demonstrating that SBPs are valuable tools for hit and lead optimization, compound library design and target hopping, especially in cases where ligand information is scarce.Abbreviations
| LBP | ligand based pharmacophore; |
| SBP | structure based pharmacophore; |
| QSAR | quantitative structure–activity relationship; |
| SAR | structure–activity relationship; |
| HBD | hydrogen bond donor; |
| HBA | hydrogen bond acceptor; |
| MD | molecular dynamics; |
| MIFs | molecular interaction fields; |
| MCSS | multiple copy simultaneous search; |
| XED | extended election distribution; |
| SNPs | single nucleotide polymorphisms; |
| MSA | multiple sequence alignment |
Acknowledgements
The authors would like to thank Tina Ritschel and Sander Nabuurs for critical reading of the manuscript. This work is supported by the Top Institute Pharma [project number D1.105: the GPCR Forum]. CdG is supported by The Netherlands Organization for Scientific Research (NWO) through VENI grant 700.59.408.References
- P. Ehrlich, Ber. Dtsch. Chem. Ges., 1909, 42, 17–47 Search PubMed.
- L. B. Kier, Mol. Pharmacol., 1967, 3, 487–494 Search PubMed.
- L. B. Kier, Pure Appl. Chem., 1973, 35, 509–520 Search PubMed.
- P. Gund, Prog. Mol. Subcell. Biol., 1977, 11, 117–143 Search PubMed.
- T. Langer, Mol. Inf., 2010, 29, 470–475 Search PubMed.
- A. R. Leach, V. J. Gillet, R. A. Lewis and R. Taylor, J. Med. Chem., 2010, 53, 539–558 CrossRef CAS.
- H. Sun, Curr. Med. Chem., 2008, 15, 1018–1024 Search PubMed.
- Success Stories of Computer-Aided Design, ed. H. Kubinyi, Wiley-Interscience, New York, 2006 Search PubMed.
- H. Eckert and J. Bajorath, Drug Discovery Today, 2007, 12, 225–233 CrossRef CAS.
- T. Langer, Mol. Inf., 2010, 29, 470–475 Search PubMed.
- F. C. Bernstein, T. F. Koetzle, G. J. Williams, E. F. Meyer, Jr, M. D. Brice, J. R. Rodgers, O. Kennard, T. Shimanouchi and M. Tasumi, J. Mol. Biol., 1977, 112, 535–542 CAS.
- D. Rognan, Mol. Inf., 2009, 29, 176–187 Search PubMed.
- D. Rognan, Br. J. Pharmacol., 2007, 152, 38–52 CrossRef CAS.
- S. Ekins, J. Mestres and B. Testa, Br. J. Pharmacol., 2007, 152, 9–20 CrossRef CAS.
- S. Ekins, J. Mestres and B. Testa, Br. J. Pharmacol., 2007, 152, 21–37 Search PubMed.
- J. Meslamani and D. Rognan, J. Chem. Inf. Model., 2011, 51, 1593–1603 Search PubMed.
- A. C. Pike, A. M. Brzozowski, J. Walton, R. E. Hubbard, T. Bonn, J. A. Gustafsson and M. Carlquist, Biochem. Soc. Trans., 2000, 28, 396–400 CrossRef CAS.
- A. C. Pike, A. M. Brzozowski and R. E. Hubbard, J. Steroid Biochem. Mol. Biol., 2000, 74, 261–268 CrossRef CAS.
- C. de Graaf and D. Rognan, J. Med. Chem., 2008, 51, 4978–4985 Search PubMed.
- D. Moras and H. Gronemeyer, Curr. Opin. Cell Biol., 1998, 10, 384–391 Search PubMed.
- A. Evers, G. Hessler, H. Matter and T. Klabunde, J. Med. Chem., 2005, 48, 5448–5465 CrossRef.
- F. Milletti and A. Vulpetti, J. Chem. Inf. Model., 2010, 50, 1418–1431 Search PubMed.
- V. J. Haupt and M. Schroeder, Briefings Bioinf., 2011, 12, 312–326 Search PubMed.
- C. N. Cavasotto and S. S. Phatak, Drug Discovery Today, 2009, 14, 676–683 Search PubMed.
- R. W. Hooft, C. Sander and G. Vriend, Proteins, 1996, 26, 363–376 Search PubMed.
- L. R. Forrest and B. Honig, Proteins, 2005, 61, 296–309 Search PubMed.
- R. P. Joosten, T. A. te Beek, E. Krieger, M. L. Hekkelman, R. W. Hooft, R. Schneider, C. Sander and G. Vriend, Nucleic Acids Res., 2011, 39, D411–419 Search PubMed.
- T. Klabunde, C. Giegerich and A. Evers, J. Med. Chem., 2009, 52, 2923–2932 Search PubMed.
- M. P. Sanders, S. Verhoeven, C. De Graaf, L. Roumen, B. Vroling, S. Nabuurs, J. D. Vlieg and J. P. Klomp, J. Chem. Inf. Model., 2011, 51, 2277–2292 Search PubMed.
- L. B. Kier, Fundamental Concepts in Drug–Receptor Interactions, Academic Press, London, New York, 1970 Search PubMed.
- L. B. Kier and L. H. Hall, Molecular Orbital Theory in Drug Research, Academic Press, New York, 1971 Search PubMed.
- E. Perola and P. S. Charifson, J. Med. Chem., 2004, 47, 2499–2510 CrossRef CAS.
- H. Alonso, A. A. Bliznyuk and J. E. Gready, Med. Res. Rev., 2006, 26, 531–568 CrossRef CAS.
- H. A. Carlson, Curr. Opin. Chem. Biol., 2002, 6, 447–452 Search PubMed.
- M. L. Teodoro and L. E. Kavraki, Curr. Pharm. Des., 2003, 9, 1635–1648 Search PubMed.
- K. L. Meagher and H. A. Carlson, J. Am. Chem. Soc., 2004, 126, 13276–13281 Search PubMed.
- K. Loving, N. K. Salam and W. Sherman, J. Comput.-Aided Mol. Des., 2009, 23, 541–554 Search PubMed.
- N. K. Salam, R. Nuti and W. Sherman, J. Chem. Inf. Model., 2009, 49, 2356–2368 Search PubMed.
- S. L. Dixon, A. M. Smondyrev, E. H. Knoll, S. N. Rao, D. E. Shaw and R. A. Friesner, J. Comput.-Aided Mol. Des., 2006, 20, 647–671 Search PubMed.
- P. J. Goodford, J. Med. Chem., 1985, 28, 849–857 CrossRef CAS.
- H. J. Bohm, J. Comput.-Aided Mol. Des., 1994, 8, 623–632 CrossRef CAS.
- H. J. Bohm, J. Comput.-Aided Mol. Des., 1992, 6, 61–78 CAS.
- S. C. Lovell, J. M. Word, J. S. Richardson and D. C. Richardson, Proteins, 2000, 40, 389–408 CrossRef CAS.
- P. Schmidtke, C. Souaille, F. Estienne, N. Baurin and R. T. Kroemer, J. Chem. Inf. Model., 2010, 50, 2191–2200 Search PubMed.
- J. An, M. Totrov and R. Abagyan, Mol. Cell. Proteomics, 2005, 4, 752–761 CrossRef CAS.
- M. L. Verdonk, J. C. Cole and R. Taylor, J. Mol. Biol., 1999, 289, 1093–1108 CrossRef CAS.
- H. A. Carlson, K. M. Masukawa, K. Rubins, F. D. Bushman, W. L. Jorgensen, R. D. Lins, J. M. Briggs and J. A. McCammon, J. Med. Chem., 2000, 43, 2100–2114 Search PubMed.
- T. Halgren, Chem. Biol. Drug Des., 2007, 69, 146–148 Search PubMed.
- Tripos, http://tripos.com/, 2011.
- Accelrys, http://accelrys.com/products/discovery-studio/, 2011.
- B. Delaunay, Izv. Akad. Nauk SSSR, Otd. Mat. Estestv. Nauk, 1934, 7, 793–800 Search PubMed.
- C. C. Group, http://www.chemcomp.com/software.htm, 2011.
- G. P. Brady, Jr and P. F. Stouten, J. Comput.-Aided Mol. Des., 2000, 14, 383–401 Search PubMed.
- R. A. Laskowski, J. Mol. Graphics, 1995, 13, 323–330 CrossRef CAS , 307–328.
- M. Hendlich, F. Rippmann and G. Barnickel, J. Mol. Graphics Modell., 1997, 15, 359–363 CrossRef CAS , 389.
- K. P. Peters, J. Fauck and C. Frommel, J. Mol. Biol., 1996, 256, 201–213 Search PubMed.
- J. Liang, H. Edelsbrunner and C. Woodward, Protein Sci., 1998, 7, 1884–1897 Search PubMed.
- T. A. Binkowski, S. Naghibzadeh and J. Liang, Nucleic Acids Res., 2003, 31, 3352–3355 Search PubMed.
- H. Edelsbrunner and L. Mucke, ACM Trans. Graphics, 1994, 13, 43–72 Search PubMed.
- K. Prymula, T. Jadczyk and I. Roterman, J. Comput.-Aided Mol. Des., 2011, 25, 117–133 Search PubMed.
- S. Henrich, O. M. Salo-Ahen, B. Huang, F. F. Rippmann, G. Cruciani and R. C. Wade, J. Mol. Recognit., 2010, 23, 209–219 Search PubMed.
- G. Wolber, T. Seidel, F. Bendix and T. Langer, Drug Discovery Today, 2008, 13, 23–29 CrossRef CAS.
- G. M. Spitzer, M. Heiss, M. Mangold, P. Markt, J. Kirchmair, G. Wolber and K. R. Liedl, J. Chem. Inf. Model., 2010, 50, 1241–1247 Search PubMed.
- C. Lemmen and T. Lengauer, J. Comput.-Aided Mol. Des., 2000, 14, 215–232 CrossRef CAS.
- J. M. Word, S. C. Lovell, J. S. Richardson and D. C. Richardson, J. Mol. Biol., 1999, 285, 1735–1747 CrossRef CAS.
- M. Baroni, G. Cruciani, S. Sciabola, F. Perruccio and J. S. Mason, J. Chem. Inf. Model., 2007, 47, 279–294 Search PubMed.
- S. Cross, M. Baroni, E. Carosati, P. Benedetti and S. Clementi, J. Chem. Inf. Model., 2010, 50, 1442–1450 Search PubMed.
- F. Ortuso, T. Langer and S. Alcaro, Bioinformatics, 2006, 22, 1449–1455 Search PubMed.
- C. Barillari, G. Marcou and D. Rognan, J. Chem. Inf. Model., 2008, 48, 1396–1410 Search PubMed.
- Inteligand, http://www.inteligand.com/ligandscout/, 2011.
- J. Chen and L. Lai, J. Chem. Inf. Model., 2006, 46, 2684–2691 Search PubMed.
- Schrodinger, http://www.schrodinger.com/, 2011.
- Z. Chen, G. Tian, Z. Wang, H. Jiang and J. Shen, J. Chem. Inf. Model., 2010, 50, 615–625 Search PubMed.
- A. Al-Nadaf, G. Abu Sheikha and M. O. Taha, Bioorg. Med. Chem., 2010, 18, 3088–3115 Search PubMed.
- M. O. Taha, A. G. Al-Bakri and W. A. Zalloum, Bioorg. Med. Chem. Lett., 2006, 16, 5902–5906 CrossRef CAS.
- M. O. Taha, Y. Bustanji, A. G. Al-Bakri, A. M. Yousef, W. A. Zalloum, I. M. Al-Masri and N. Atallah, J. Mol. Graphics Modell., 2007, 25, 870–884 Search PubMed.
- M. O. Taha, Y. Bustanji, M. A. Al-Ghussein, M. Mohammad, H. Zalloum, I. M. Al-Masri and N. Atallah, J. Med. Chem., 2008, 51, 2062–2077 CrossRef CAS.
- M. O. Taha, L. A. Dahabiyeh, Y. Bustanji, H. Zalloum and S. Saleh, J. Med. Chem., 2008, 51, 6478–6494 Search PubMed.
- M. O. Taha, M. Tarairah, H. Zalloum and G. Abu-Sheikha, J. Mol. Graphics Modell., 2010, 28, 383–400 Search PubMed.
- I. Wallach and R. Lilien, J. Chem. Inf. Model., 2009, 49, 2116–2128 Search PubMed.
- A. Miranker and M. Karplus, Proteins, 1991, 11, 29–34 CAS.
- T. T. Luu, N. O. Malcolm and K. Nadassy, Comb. Chem. High Throughput Screening, 2011, 14, 488–489 Search PubMed.
- I. Wallach, Drug Dev. Res., 2011, 72, 17–25 Search PubMed.
- T. Cheeseright, M. Mackey, S. Rose and A. Vinter, Expert Opin. Drug Discovery, 2007, 2, 131–144 Search PubMed.
- M. J. McGregor, J. Chem. Inf. Model., 2007, 47, 2374–2382 Search PubMed.
- Z. Deng, C. Chuaqui and J. Singh, J. Med. Chem., 2004, 47, 337–344 CrossRef CAS.
- S. Renner and G. Schneider, J. Med. Chem., 2004, 47, 4653–4664 CrossRef CAS.
- A. Nicholls, G. B. McGaughey, R. P. Sheridan, A. C. Good, G. Warren, M. Mathieu, S. W. Muchmore, S. P. Brown, J. A. Grant, J. A. Haigh, N. Nevins, A. N. Jain and B. Kelley, J. Med. Chem., 2010, 53, 3862–3886 CrossRef CAS.
- P. A. Greenidge, B. Carlsson, L. G. Bladh and M. Gillner, J. Med. Chem., 1998, 41, 2503–2512 Search PubMed.
- S. Sciabola, R. V. Stanton, J. E. Mills, M. M. Flocco, M. Baroni, G. Cruciani, F. Perruccio and J. S. Mason, J. Chem. Inf. Model., 2010, 50, 155–169 Search PubMed.
- M. Rella, C. A. Rushworth, J. L. Guy, A. J. Turner, T. Langer and R. M. Jackson, J. Chem. Inf. Model., 2006, 46, 708–716 Search PubMed.
- J. O. Ebalunode, Z. Ouyang, J. Liang and W. Zheng, J. Chem. Inf. Model., 2008, 48, 889–901 Search PubMed.
- P. C. Hawkins, A. G. Skillman and A. Nicholls, J. Med. Chem., 2007, 50, 74–82 CrossRef CAS.
- A. Nicholls, G. B. McGaughey, R. P. Sheridan, A. C. Good, G. Warren, M. Mathieu, S. W. Muchmore, S. P. Brown, J. A. Grant, J. A. Haigh, N. Nevins, A. N. Jain and B. Kelley, J. Med. Chem., 2010, 53, 3862–3886 CrossRef CAS.
- S. Thangapandian, S. John, S. Sakkiah and K. W. Lee, Eur. J. Med. Chem., 2010, 45, 4409–4417 Search PubMed.
- B. V. Kumar, R. Kotla, R. Buddiga, J. Roy, S. S. Singh, R. Gundla, M. Ravikumar and J. A. Sarma, J. Mol. Model., 2011, 17, 151–163 Search PubMed.
- R. Griffith, T. T. Luu, J. Garner and P. A. Keller, J. Mol. Graphics Modell., 2005, 23, 439–446 Search PubMed.
- L. Tan, H. Geppert, M. T. Sisay, M. Gutschow and J. Bajorath, ChemMedChem, 2008, 3, 1566–1571 Search PubMed.
- D. M. Kruger and A. Evers, ChemMedChem, 2010, 5, 148–158 CrossRef.
- A. Evers, G. Hessler, H. Matter and T. Klabunde, J. Med. Chem., 2005, 48, 5448–5465 CrossRef.
- M. von Korff, J. Freyss and T. Sander, J. Chem. Inf. Model., 2009, 49, 209–231 Search PubMed.
- N. Singh, G. Cheve, D. M. Ferguson and C. R. McCurdy, J. Comput.-Aided Mol. Des., 2006, 20, 471–493 Search PubMed.
- J. Kirchmair, S. Distinto, P. Markt, D. Schuster, G. M. Spitzer, K. R. Liedl and G. Wolber, J. Chem. Inf. Model., 2009, 49, 678–692 Search PubMed.
- A. J. Kooistra, T. W. Binsl, J. H. van Beek, C. de Graaf and J. Heringa, J. Chem. Inf. Model., 2010, 50, 1772–1780 Search PubMed.
- N. Moitessier, P. Englebienne, D. Lee, J. Lawandi and C. R. Corbeil, Br. J. Pharmacol., 2008, 153(Suppl. 1), S7–26 CrossRef CAS.
- O. Guner, O. Clement and Y. Kurogi, Curr. Med. Chem., 2004, 11, 2991–3005 Search PubMed.
- O. F. Guner, Curr. Comput.-Aided Drug Des., 2011, 7, 158 Search PubMed.
- K. Roy, Mol. Diversity, 2004, 8, 321–323 Search PubMed.
- C. Lemmen, T. Lengauer and G. Klebe, J. Med. Chem., 1998, 41, 4502–4520 CrossRef CAS.
- I. Halperin, B. Ma, H. Wolfson and R. Nussinov, Proteins, 2002, 47, 409–443 CrossRef CAS.
- D. B. Kitchen, H. Decornez, J. R. Furr and J. Bajorath, Nat. Rev. Drug Discovery, 2004, 3, 935–949 CrossRef CAS.
- C. Sotriffer and G. Klebe, Farmaco, 2002, 57, 243–251 Search PubMed.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, M. Shelley, J. K. Perry, D. E. Shaw, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739–1749 CrossRef CAS.
- G. Jones, P. Willett, R. C. Glen, A. R. Leach and R. Taylor, J. Mol. Biol., 1997, 267, 727–748 CrossRef CAS.
- A. N. Jain, J. Med. Chem., 2003, 46, 499–511 CrossRef CAS.
- B. Kramer, M. Rarey and T. Lengauer, Proteins, 1999, 37, 228–241 CrossRef CAS.
- T. S. Rush, 3rd, J. A. Grant, L. Mosyak and A. Nicholls, J. Med. Chem., 2005, 48, 1489–1495 CrossRef.
- G. Hessler, M. Zimmermann, H. Matter, A. Evers, T. Naumann, T. Lengauer and M. Rarey, J. Med. Chem., 2005, 48, 6575–6584 Search PubMed.
- C. Tintori, V. Corradi, M. Magnani, F. Manetti and M. Botta, J. Chem. Inf. Model., 2008, 48, 2166–2179 Search PubMed.
- M. Lower, T. Geppert, P. Schneider, B. Hoy, S. Wessler and G. Schneider, PLoS One, 2011, 6, e17986 Search PubMed.
- G. I. Mustata and J. M. Briggs, J. Comput.-Aided Mol. Des., 2002, 16, 935–953 Search PubMed.
- X. M. Chen, T. Lu, S. Lu, H. F. Li, H. L. Yuan, T. Ran, H. C. Liu and Y. D. Chen, J. Mol. Model., 2010, 16, 1195–1204 Search PubMed.
- A. Evers and G. Klebe, J. Med. Chem., 2004, 47, 5381–5392 Search PubMed.
- B. Pirard, J. Brendel and S. Peukert, J. Chem. Inf. Model., 2005, 45, 477–485 Search PubMed.
- S. Cross and G. Cruciani, Drug Discovery Today, 2010, 7, 213–219 Search PubMed.
- M. Weisel, E. Proschak and G. Schneider, Chem. Cent. J., 2007, 1, 7 Search PubMed.
- M. Weisel, E. Proschak, J. M. Kriegl and G. Schneider, Proteomics, 2009, 9, 451–459 Search PubMed.
- C. Bissantz, B. Kuhn and M. Stahl, J. Med. Chem., 2010, 53, 5061–5084 CrossRef CAS.
- Y. Tanrikulu, M. Nietert, U. Scheffer, E. Proschak, K. Grabowski, P. Schneider, M. Weidlich, M. Karas, M. Gobel and G. Schneider, ChemBioChem, 2007, 8, 1932–1936 Search PubMed.
- R. Wang, Y. Gao and L. Lai, J. Mol. Model., 2000, 6, 498–516 CrossRef CAS.
- A. Evers, H. Gohlke and G. Klebe, J. Mol. Biol., 2003, 334, 327–345 Search PubMed.
- H. Claussen, M. Gastreich, V. Apelt, J. Greene, S. A. Hindle and C. Lemmen, Curr. Drug Discovery Technol., 2004, 1, 49–60 Search PubMed.
- S. A. Hindle, M. Rarey, C. Buning and T. Lengaue, J. Comput.-Aided Mol. Des., 2002, 16, 129–149 Search PubMed.
- D. Joseph-McCarthy, B. E. T. Thomas, M. Belmarsh, D. Moustakas and J. C. Alvarez, Proteins, 2003, 51, 172–188 Search PubMed.
- A. Kaczor and D. Matosiuk, FEMS Immunol. Med. Microbiol., 2010, 58, 91–101 Search PubMed.
- S. Thangapandian, S. John, S. Sakkiah and K. W. Lee, Eur. J. Med. Chem., 2011, 46, 2469–2476 Search PubMed.
- M. M. Ahlstrom, M. Ridderstrom, K. Luthman and I. Zamora, J. Chem. Inf. Model., 2005, 45, 1313–1323 Search PubMed.
- R. Griffith, T. T. Luu, J. Garner and P. A. Keller, J. Mol. Graphics Modell., 2005, 23, 439–446 Search PubMed.
- V. Campagna-Slater, A. G. Arrowsmith, Y. Zhao and M. Schapira, J. Chem. Inf. Model., 2010, 50, 358–367 Search PubMed.
- J. Zou, H. Z. Xie, S. Y. Yang, J. J. Chen, J. X. Ren and Y. Q. Wei, J. Mol. Graphics Modell., 2008, 27, 430–438 Search PubMed.
- N. Schormann, O. Senkovich, K. Walker, D. L. Wright, A. C. Anderson, A. Rosowsky, S. Ananthan, B. Shinkre, S. Velu and D. Chattopadhyay, Proteins, 2008, 73, 889–901 Search PubMed.
- J. Yoo and J. L. Medina-Franco, J. Comput.-Aided Mol. Des., 2011, 25, 555–567 Search PubMed.
- T. Ritschel, D. J. Wood, J. de Vlieg and M. Wagener, J. Cheminf., 2011, 3, 37 Search PubMed.
- T. L. Nguyen, R. Gussio, J. A. Smith, D. A. Lannigan, S. M. Hecht, D. A. Scudiero, R. H. Shoemaker and D. W. Zaharevitz, Bioorg. Med. Chem., 2006, 14, 6097–6105 Search PubMed.
- A. M. Doweyko, J. Med. Chem., 1994, 37, 1769–1778 Search PubMed.
- E. Kellenberger, C. Schalon and D. Rognan, Curr. Comput.-Aided Drug Des., 2008, 4, 209–220 Search PubMed.
- N. Weill and D. Rognan, J. Chem. Inf. Model., 2010, 50, 123–135 Search PubMed.
- S. Schmitt, D. Kuhn and G. Klebe, J. Mol. Biol., 2002, 323, 387–406 Search PubMed.
- K. Yeturu and N. Chandra, BMC Bioinf., 2008, 9, 543 Search PubMed.
| This journal is © The Royal Society of Chemistry 2012 |