“Artificial mitotic spindle” generated by dielectrophoresis and protein micropatterning supports bidirectional transport of kinesin-coated beads†
Maruti
Uppalapati
a,
Ying-Ming
Huang
b,
Vidhya
Aravamuthan
a,
Thomas N.
Jackson
b and
William O.
Hancock
*a
aDepartment of Bioengineering, The Pennsylvania State University, 229 Hallowell Bldg., University Park, PA 16802. E-mail: wohbio@engr.psu.edu; Fax: +1 (814) 863-0490; Tel: +1 (814) 863-0492
bDepartment of Electrical Engineering, The Pennsylvania State University, 216 Electrical Engineering West, University Park, PA 16802
First published on 29th October 2010
Abstract
The mitotic spindle is a dynamic assembly of microtubules and microtubule-associated proteins that controls the directed movement of chromosomes during cell division. Because proper segregation of the duplicated genome requires that each daughter cell receives precisely one copy of each chromosome, numerous overlapping mechanisms have evolved to ensure that every chromosome is transported to the cell equator during metaphase. However, due to the inherent redundancy in this system, cellular studies using gene knockdowns or small molecule inhibitors have an inherent limit in defining the sufficiency of precise molecular mechanisms as well as quantifying aspects of their mechanical performance. Thus, there exists a need for novel experimental approaches that reconstitute important aspects of the mitotic spindlein vitro. Here, we show that by microfabricating Cr electrodes on quartz substrates and micropatterning proteins on the electrode surfaces, AC electric fields can be used to assemble opposed bundles of aligned and uniformly oriented microtubules as found in the mitotic spindle. By immobilizing microtubule ends on each electrode, analogous to anchoring at centrosomes, solutions of motor or microtubule binding proteins can be introduced and their resulting dynamics analyzed. Using this “artificial mitotic spindle” we show that beads functionalized with plus-end kinesin motors move in an oscillatory manner analogous to the movements of chromosomes and severed chromosome arms during metaphase. Hence, features of directional instability, an established characteristic of metaphase chromosome dynamics, can be reconstituted in vitro using a pair of uniformly oriented microtubule bundles and a plus-end kinesin functionalized bead.
Insight, innovation, integrationMicrotubules that constitute the mitotic spindle have precise spatial organization that is essential for proper alignment of chromosomes at the cell equator preceding separation. Here, we develop a novel experimental platform, an “Artificial Mitotic Spindle”, that recapitulates this complex microtubule organization in vitro and thus enables novel experiments that test the sufficiency of specific molecular mechanisms underlying mitosis. We show that, with this proper microtubule geometry, plus-end directed kinesin motors (as found on the arms of chromosomes) are sufficient to reproduce features of directional instability seen for chromosomes and severed chromosome arms in cells. Hence, this system integrates microscale engineering and AC electrokinetics together with functional intracellular proteins to quantitatively characterize fundamental mechanisms underlying cell division. |
Introduction
During mitosis, duplicated chromosomes are properly segregated to the two daughter cells by the combination of microtubule dynamics and microtubule-based motors operating in the mitotic spindle.1,2Assembly and proper function of the mitotic spindle is essential for proper cell division, and chromosome missegregation can lead to a variety of problems including cell death, Down's Syndrome, and cancer.1,3,4 Furthermore, proteins involved in mitosis are important targets for novel anticancer therapeutics.5 However, despite years of intensive investigation, many of the molecular details underlying the proper segregation of duplicated chromosomes during cell division are not understood. One reason these important questions remain unanswered is that numerous redundant processes have evolved to ensure proper chromosome segregation. For instance, numerous kinesin and dynein motor proteins bound to both the kinetochore and the chromosome arms generate forces to push and pull the chromosomes along the mitotic spindle,1,2,6 and microtubule polymerization and depolymerization forces are also thought to play important roles.7 While drug inhibition and gene knockdown experiments have been vital to defining the specific molecules that carry out mitosis, the inherent redundancy in this system makes it difficult to define the precise molecular mechanisms underlying cell division.Due to the inherent complexity of the mitotic spindle as well as its importance in biology and medicine, novel experimental approaches are needed to uncover the fundamental principles underlying spindle function. Microscale engineering tools such as photolithography, microfabrication, and microfluidics provide excellent opportunities for gaining insight into mitosis because they enable manipulation of microtubules and motor proteins at subcellular length scales. We recently developed a system that uses MHz-range AC electric fields generated by microfabricated electrodes to achieve unprecedented control in manipulating populations of taxol-stabilized microtubulesin vitro.8 Due to differences in the dielectric properties of microtubules and the buffer solution in which they are suspended, MHz AC fields cause microtubules to be attracted to regions of maximum electric field gradient (positive dielectrophoresis), and the accompanying fluid flows serve to align the microtubules into parallel bundles. This novel manipulation tool works on populations of filaments and it provides micron-scale resolution, making it an attractive approach for generating mitotic spindle-like organizations of microtubulesin vitro (Fig. 1).
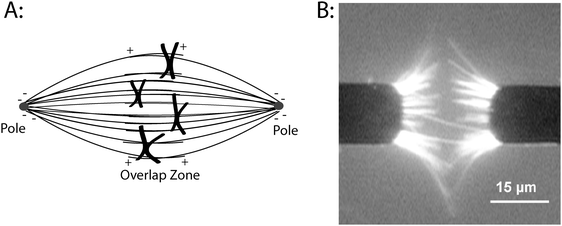 | ||
| Fig. 1 Organizing microtubules into spindle-like assembliesin vitro. (A) Diagram of microtubule organization in the metaphase mitotic spindle. Microtubule minus-ends are anchored at each pole and the plus-ends extend into the overlap zone at the equator of the cell. During mitosis, chromosomes move along spindle microtubules, eventually reaching the equator of the cell before separating and moving toward each pole. (B) Organizing microtubulesin vitro using AC electrokinetics. Cr microelectrodes were patterned on a quartz substrate and a 30 Vp–p, 5 MHz electric field was applied. Microtubules are attracted to the electrode edges where the field gradients are maximal, and the combination of dielectrophoretic forces and electrohydrodynamic fluid forces align the microtubules. Image was taken while the electric field was on. Image taken from Uppalapati et al.8 with permission from Wiley-VCH. | ||
The goal of the present work is to create an “artificial mitotic spindle” consisting of two opposed bundles of parallel microtubules, each with uniform polarity and having their ends anchored at each pole to create an overlap zone. By developing an in vitro system that replicates the complex geometry of microtubules found in dividing eukaryotic cells, we seek to develop a novel experimental platform for testing specific molecular mechanisms underlying the coordinated behavior of microtubules and chromosome during mitosis. This system is intended to complement ongoing studies in cells and provide an avenue to test more biophysical questions while eliminating many of the unknowns inherent in studying intact cells. As a first test of a specific molecular mechanism underlying chromosome dynamics, we use this system to test the centering behavior of a bead coated with plus-end directed kinesin motors on the artificial mitotic spindle. Interestingly, the bead demonstrates directional instability as seen for chromosomes and severed chromosome arms in mitosis, demonstrating that proper microtubule geometry and plus-ended kinesins alone are sufficient for achieving directional instability.
Results and discussion
The overarching goal of this work is to use dielectrophoresis to organize taxol-stabilized microtubules into two opposing bundles of uniformly oriented filaments—an “artificial mitotic spindle”—and use this system to understand the molecular mechanisms underlying cell division. In conducting buffers, AC electric fields impose dielectrophoretic forces on polarizable objects such as cells and microtubules; these field also generate fluid flows in the form of AC electroosmotic and electrothermal flows.8 By minimizing the buffer ionic strength to reduce its conductivity (<12 mM PIPES) and identifying the optimal frequency range where the flows are balanced (∼5 MHz), we previously found that microtubules accumulate at the tips of microfabricated electrode pairs (Fig. 1). The microtubules remain there as long as the field remains, but when the field is turned off they diffuse away. This ability to organize microtubulesin vitro, with geometries defined by the placement of the microfabricated electrodes, provides the starting point for this investigation.For this system to be of utility in mimicking a real mitotic spindle, two hurdles must be overcome. First, while the bundled microtubules are aligned, their orientations (plus- and minus-ends) are mixed. Because microtubule dynamics and chromosome transport depend on spindle microtubules having proper polarity, the microtubules on each electrode need to be uniformly oriented for this system to be of utility. Secondly, in each bundle the microtubule ends must be anchored to the electrode in a similar way to the anchoring of microtubule minus-ends at the centrosome, or the bundling of minus-ends at each pole by motor and microtubule binding proteins. Below, we develop two approaches for achieving these goals, motor patterning and the use of biotinylated microtubules and patterned neutravidin.
Sorting by patterned motor proteins
The first approach we took for orienting and immobilizing the microtubules was to pattern kinesin motors on the electrodes and use these patterned motors to actively sort the microtubules by polarity. When microtubules land on a kinesin-coated surface, their direction of movement is determined by their initial orientation—because conventional kinesin walks to the plus-ends of microtubules, the filaments move with their minus-ends leading. We previously showed that full-length kinesin motors retain their functional activity when adsorbed to gold electrode surfaces, but they are inactive when adsorbed to hydrophobic surfaces, presumably because the heads adsorb, preventing their interaction with the microtubules.9,10 For this approach, 10 μm wide Cr electrodes were fabricated by depositing a continuous 100 nm thick layer of Cr on a quartz substrate, spinning on a layer of Shipley 1811 photoresist, exposing the photoresist through a mask, and etching away the unprotected Cr to expose the underlying substrate. The entire surface was then treated with a hydrophobic silane (octadecyl trichlorosilane), and the remaining photoresist was then stripped from the electrodes, creating a pair of hydrophilic Cr electrodes surrounded by a hydrophobic quartz substrate. This patterned substrate was then incorporated into a flow cell by attaching a cover-slip and spacers, and when motors were adsorbed to the surface in the presence of casein, microtubule gliding activity was observed selectively on the electrode surfaces and not on the hydrophobic silane-treated quartz (data not shown).When a 5 MHz electric field (20 VP–P at the amplifier) was applied to the electrodes, microtubules were pulled out of solution to the electrode tips, and oriented predominantly parallel to the long axis of the electrodes. Kinesin motors on the electrodes engaged the microtubules and transported them either toward or away from the electrode gap, depending on their orientation (Fig. 2 and Supplementary Movie 1†). Microtubules transported toward the gap accumulated there, held by a balance of dielectrophoretic forces pulling them to the electrode tips and motor forces pushing them into the gap. Hence, this approach actively sorts microtubules by polarity, and the microtubules remaining at the tips are held in a dynamic balance by the motor forces pushing them toward the gap and dielectrophoretic forces pulling them toward the edges of the electrode where the field gradient is maximum.8
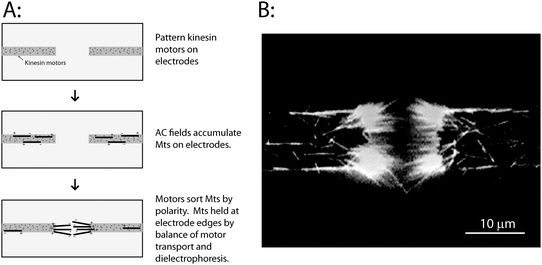 | ||
| Fig. 2 Sorting microtubules by polarity using patterned kinesin motors. (A) Conventional kinesin motors were selectively patterned on the electrodes as described in text. When the electric field was turned on, microtubules were attracted to the electrodes and were transported along the electrodes by the immobilized kinesin motors in a direction defined by their initial polarity. Over time, a bundle of microtubules accumulate at the electrode tips, and are held there by motor-driven forces that push them into the electrode gap, and dielectrophoretic forces that pull them back to the electrodes. (B) Image of the resulting microtubule bundles accumulated at electrode tips. Image was taken while the electric field was on. A movie of microtubule accumulation and sorting by the electrodes is available as Supplementary Movie 1.† | ||
This active sorting provides a novel approach for generating uniformly oriented microtubule bundles, and it could potentially be used as a platform to test the bundling of microtubule minus-ends by Kinesin-14 motors, which are thought to carry out this function in plant cells.11 However, for our artificial mitotic spindle, this method has three drawbacks. First, because it is a dynamic process with some microtubules being lost over time and replaced by new filaments from solution, it is impossible to actually achieve 100% uniform polarity for the microtubules in the electrode gap. Second, because the motors transport the filaments off of the end of the electrodes, they are not anchored there and when the electric field is turned off, they diffuse away. And third, because these are plus-end directed motors, the microtubules are oriented with their plus-ends at the electrodes and their minus-ends in the overlap zone, opposite to the geometry in cells. In theory, by patterning minus-end directed motors on the electrodes, the opposite microtubule polarity could be generated; however, we were unable to achieve similar patterning with the minus-end directed motor ncd, presumably because its tail domain and hence mechanism of surface adsorption differs from Drosophila conventional kinesin. Because of these drawbacks, we pursued a second strategy.
Minus-end immobilization by biotin–avidin
Our second approach to immobilizing microtubules on the electrodes was to employ the biotin–avidin system—electrodes were selectively functionalized with neutravidin and microtubules were bound to the electrodes through a biotinylated segment at their minus-ends. To achieve this, Cr electrodes were patterned on the quartz substrate using photolithography as described above, but in this case all of the photoresist was removed, exposing the bare electrodes. A drop of solution containing 1 mg ml−1 neutravidin was placed on the surface for 5 min, the sample washed in buffer, and then acetone, air dried, and then neutravidin was ablated from the quartz by exposing the back side of the sample to deep-UV illumination, as described previously.12 Using this novel neutravidin patterning approach, the electrode acts as a mask to protect the electrode-bound neutravidin, such that when the surface is assembled into a flow cell and hydrated, biotinylated proteins only bind to the electrodes and not to the surrounding quartz.The second tool we used were segmented microtubules, which were made by growing short microtubule seeds containing both biotinylated and Cy5 labeled tubulin, and selectively extending them from their plus-ends using rhodamine-labeled tubulin (see Methods).13,14 Hence, the microtubules have a short, Cy5 minus-end that can bind to the neutravidin-treated electrodes and a long, rhodamine plus-end that can extend out into the gap between the electrodes (Fig. 3A). When the AC electric field was turned on, the microtubules were drawn to the electrode tips and aligned parallel to the electrodes by the electric field (Fig. 3). When the electric field was turned off, they remained bound through their biotin–avidin attachments, and solutions could be exchanged into the flow cell to introduce alternate buffers or specific proteins into the system. Hence, this approach generates a pair of oppositely oriented microtubule bundles attached to the electrodes in a similar geometry as found in dividing cells.
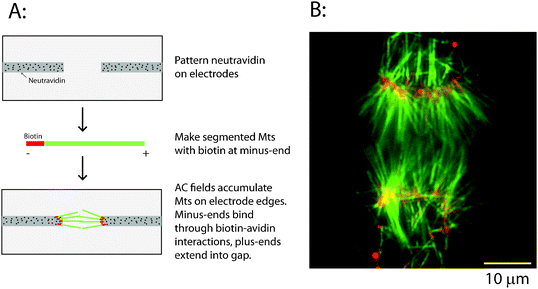 | ||
| Fig. 3 Immobilizing uniformly oriented microtubules using biotin–avidin. (A) Schematic for the immobilization process. Neutravidin is patterned on electrodes by UV ablation, and segmented microtubules containing biotinylated minus-ends are attracted to the electrode tips by dielectrophoretic forces and bound through biotin–avidin linkages. (B) Image of overlapping bipolar asters showing Cy5-labeled biotinylated microtubule minus-ends (pseudocolored red) and long rhodamine-labeled microtubule plus-end segments (pseudocolored green). Image was taken after the electric field was turned off. | ||
Extending microtubules by polymerization
Up to this point, all of the work was carried out with microtubules polymerized in vitro and stabilized with the drug taxol, which binds to microtubules and prevents depolymerization. While taxol-stabilized microtubules are much easier to work with because they don't depolymerize, they contrast with microtubules in dividing cells, which are dynamic, alternating between phases of growing and shrinking. Because these microtubule dynamics are integral both to the formation of the spindle and the movements of chromosomes during mitosis, any experimental platform developed to investigate mechanisms of mitosis needs to be able to incorporate and analyze microtubule dynamics. The second reason for incorporating microtubule dynamics into our artificial mitotic spindle system is as follows. Due to the spatial distribution of the attractive dielectrophoretic forces and the accompanying fluid flows generated by the AC electric fields,8 most of the microtubules that accumulate on the electrodes extend less than half way across the electrode gap, and hence little microtubule overlap at the midzone between the electrodes is achieved (for examples, see Fig. 1 and 2). This is particularly true when larger electrode gaps greater than 20 μm are used. Hence, to generate a spindle with a large electrode gap and overlapping microtubule plus-ends, the approach we chose was to first generate two oppositely oriented bundles of microtubules, and then polymerize off of the plus-ends to create an overlap in the midzone.While extending microtubules using free tubulin in the presence of taxol is problematic due to the nucleation of new filaments, we showed previously if the taxol containing solution is replaced with a solution of free tubulin, NEM-tubulin and GTP, the filaments will incorporate this new tubulin, and extend selectively from their plus-ends.14–16Fig. 4A shows an artificial spindle created using a 30 μm electrode gap, which results in minimal microtubule overlap. To extend the microtubule plus-ends, the AC electric field was turned off, the low ionic strength buffer containing taxol was washed out, and a microtubule extension solution containing free tubulin was introduced (see Methods). After a 20 min incubation, significant overlap was observed (Fig. 4B), and in principle by regularly replacing the solution with new free tubulin, the microtubules could be extended to even longer lengths. While studying microtubules dynamics is not the focus of the present study, this system could be used in future studies to investigate the interactions of microtubule binding proteins with bundles of dynamic plus-ends or dynamic overlapping microtubules, as is found in cells, or to investigate the ability of uniformly oriented bundles of filaments to exert mechanical forces on chromosomes and other objects.
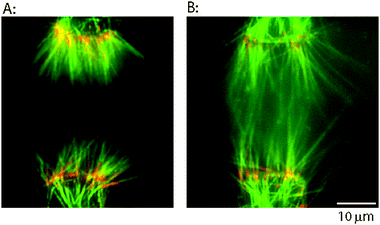 | ||
| Fig. 4 Extending immobilized microtubules by polymerization to create microtubule overlap zone. (A) Oriented bundles of microtubules accumulated at electrode edges by dielectrophoresis and bound through biotin–avidin linkages. When electrodes with >20 μm spacing are used, microtubules attached through biotinylated minus-ends have minimal overlap. (B) Free tubulin was flowed into the chamber to extend the microtubules, and then a taxol solution was introduced to stabilize them. Microtubules are longer and are overlapped at the midzone. Colors are same as Fig. 3. Images were taken after the electric field was turned off. | ||
Movement of kinesin-functionalized beads
This ability to create opposed bundles of immobilized, parallel, uniformly oriented microtubules sets the stage to investigate the sufficiency of specific mechanisms proposed to drive mitotic chromosome movements. Chromosomes contain numerous motor activities—proteins at the kinetochore include (but are not necessarily limited to) dynein, the Kinesin-7 CENP-E, and the Kinesin-13 MCAK, which provide minus-end directed movement, plus-end directed movement, and microtubule depolymerizing activity, respectively.17–21 In addition, the chromosome arms contain plus-end directed kinesins (Kid),22,23 and microtubule polymerization dynamics can produce pushing forces to move chromosomes away from the poles.7 During prometaphase and metaphase, following nuclear envelope breakdown but preceding separation of the duplicated chromosomes, chromosomes undergo complex oscillatory movements toward and away from each pole that eventually result in every chromosome aligning at the cell equator. Achieving this proper alignment before sister chromatid separation is crucial to avoid aneuploidy (unequal division of chromosomes). Because of the fundamental biological and clinical importance of this process, the molecular basis of these complex chromosome dynamics has been investigated in detail in cells and cell extracts by depleting specific components and studying the resulting phenotype.6,24,25 While this work has been very fruitful, there are three inherent weaknesses in this strategy: (a) redundant mechanisms complicate interpretation of negative results, (b) interpreting changes in phenotypes in terms of molecular activities often requires assumptions that lack strong experimental support, and (c) interpreting molecular activities by depletion makes quantitation and physical modeling of these activities difficult.The artificial mitotic spindle developed here allows us to study molecular mechanisms of mitosis using a complementary approach—by assembling the complex geometry of the spindle from the bottom up and asking whether specific molecular mechanisms are sufficient for carrying out particular cellular activities. Here, we focus on one particular aspect of chromosome dynamics, namely transport along spindle microtubules by plus-end directed motors. In a dividing cell there are forces on the chromosome arms directed away from each pole, often termed “polar ejection forces”, which participate in transporting chromosomes to the cell equator.26–31 The molecular source of these polar ejection forces is an outstanding question—polymerizing microtubules are able to generate pushing forces,32–37 but the chromosome arms also contain plus-end directed kinesins that can interact with spindle microtubules and generate pushing forces away from the pole.38–40
Here we ask the question: is proper spindle microtubule orientation alone sufficient to drive oscillatory movement of beads coated with plus-ended kinesins? Chromosomes that are congressing to the cell equator during metaphase show characteristic oscillations having periods of constant velocity movement with amplitudes of a few microns punctuated by rapid directional switching.41,42 Interestingly, Keet al. found that when chromosome arms were severed by a laser, the arms continued these bidirectional movements. The authors attributed this behavior to steric forces in the crowded cytoplasm;42 however it is possible that motors on the chromosome arms are also contributing to this bidirectional movement. We set out to test whether the oscillatory behavior seen in cells can be recapitulated in our refined system, with the goal of testing the following questions. (1) Can motor activity be achieved on the immobilized microtubules in our artificial spindle? (2) Do beads move bidirectionally in the system, or do they instead seize up in the overlap zone or buckle the overlapped microtubules? (3) What amplitude oscillations are seen?
We assembled an artificial mitotic spindle using biotin–avidin to immobilize the minus-ends, and then introduced a solution of 0.5 μm diameter glass beads functionalized with full-length DrosophilaKinesin-1 at a density of 50 motors/bead. Initially, the beads diffused in solution, but occasionally a bead would land on a microtubule, move away from the nearest electrode (pole) and cross the midzone. When beads reached the end of a microtubule, they would briefly diffuse in solution, and then attach to another microtubule or microtubule bundle and generally reverse directions (Fig. 5). In each phase of unidirectional movement, the speed parallel to the axis of the electrodes was relatively constant at ∼700 nm s−1, which matches the unloaded speed of these motors in microtubule gliding assays.43 Hence, in the overlap zone where microtubules of both orientations are present, the bead (which can interact with multiple microtubules) neither seizes up, nor appears to be slowed at all due to interactions with oppositely oriented microtubules. This constant speed (independent of position in the spindle) is consistent with what is seen in cells (though due to differences in motor types and other factors, the speed is 20-fold higher)42 and it suggests that when the bead is moving in our in vitro system there are negligible viscous or motor-dependent loads resisting movement.
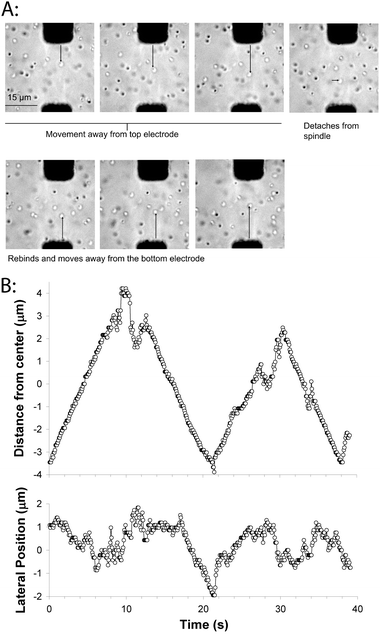 | ||
| Fig. 5 Movement of kinesin-coated beads on the artificial mitotic spindle. (A) Images of bead motility on the artificial spindle. Frames are 5 s apart, and one bead can be seen moving toward the top electrode, transiently detaching from the spindle, and reattaching and moving toward the bottom electrode. A movie corresponding to this sequence of images is available as Supplementary Movie 2.† (B) Plot of bead movement along the electrode axis, relative to the centerline separating the two electrodes (top), and perpendicular to the electrode axis (bottom). Movement along the electrode axis shows steady periods of movement punctuated by diffusive movements and directional switching. Electric fields were turned off during bead motility experiments. | ||
The second parameter that can be compared to cellular studies is the amplitude of the observed oscillations. In newt lung cells and HeLa cells, chromosome oscillations are in the range of 1–2 μm peak-to-peak,42,44 while in the bead example shown in Fig. 5, oscillations are in the range of 5–6 μm. The molecular mechanisms that determine the oscillations in cells are an active area of investigations; depletion of the chromokinesin Kid suppresses the oscillations, while inhibiting the Kinesin-8 KIF18A or cutting off a chromosome arm have been shown to enhance oscillation amplitudes.23,42,44 Directional switching of chromosome movement (which is the primary determinant of oscillation amplitudes) is generally assumed to be a force-dependent process, but the various force balances involved are complex—poleward kinetochore forces compete with away-from-pole directed forces on the chromosome arms, both poles need to be taken into consideration, and forces depend on the position of the chromosome within the spindle. In our reconstituted system, the oscillation amplitudes appear to be determined primarily by the width of the microtubule overlap zone—beads run to the end of the microtubules and transiently diffuse before binding to another microtubule and switching direction. One direction for future experiments will be to characterize the behavior for other motors that are less processive than the Kinesin-1 used here and hence detach from the microtubules before reaching the end—it is expected that the behavior will be a complex function of motor processivity and motor and microtubule densities.
Conclusion
Using microfabrication, AC electrodynamics, and microscale protein patterning, we show that microtubules can be assembled in vitro into architectures mimicking the mitotic spindle in dividing cells. By exchanging in free tubulin, the dynamics of these microtubules can be studied in this complex geometry. Further, plus-end kinesin-functionalized beads move along these immobilized microtubules in a similar manner to the oscillations seen for metaphase chromosomes and isolated chromosome fragments. These experiments using a single motor type set the stage for future investigations into the interplay between plus- and minus-end directed motors as well as the interplay of motor forces and microtubule polymerization forces. Because the geometry of the electrodes and the chamber geometry can both be optimized for specific experiments, this artificial mitotic spindle is a valuable tool for a range of future experiments testing the sufficiency of specific mechanism underlying mitotic chromosome dynamics.Material and methods
Microtubules and kinesin
Tubulin was purified from bovine brains and labeled with rhodamine, Cy5 and N-ethylmaleimide (NEM) using succinimidyl ester (NHS) derivatives as previously described.13,14,45Microtubules were polymerized by mixing 32 μM tubulin, 4 mM MgCl2, 1 mM GTP and 5% DMSO in BRB80 buffer (80 mM PIPES, 1 mM EGTA, 1 mM MgCl2, pH 6.9 with KOH), incubating at 37 °C for 20 min, and then diluting into a solution containing 10 μM taxol.To make segmented microtubules containing Cy5 and biotin on the minus-end, Cy5-labeled tubulin was polymerized as described above and then labeled in polymer form with biotin by adding a 20-fold excess of Biotin-XX-NHS ester (Invitrogen) for 12 min at 37 °C in BRB80 buffer. The reaction was quenched by adding 50 mM K-glutamate, and the labeled microtubules were separated from unreacted biotin by pelleting in a Beckman Airfuge for 10 min at 30 p.s.i. Pellets were resuspended in ice-cold BRB80 and incubated on ice for 1 h to depolymerize the microtubules, and the tubulin was aliquotted at 4 mg ml−1, flash frozen on liquid N2 and stored at −80 °C for later use. On the day of the experiment, biotin-Cy5-labeled tubulin was polymerized as described above and diluted 100-fold into BRB80 containing 10 μM taxol. 200 μl of this solution was pelleted in an Airfuge to remove excess taxol and resuspended in 100 μl of extension solution 1 (2.5 μM rhodamine-labeled tubulin, 6 μM NEM-tubulin, 4 mM MgCl2, 1 mM GTP and 5% DMSO in BRB80 buffer). The solution was then passed through a 29 gauge needle to shear the microtubules into short segments, and the solution was incubated at 37° C for 25 min to polymerize rhodamine tubulin off of the plus-ends of the short biotin-Cy5 seeds. Following polymerization, 10 μM taxol was added, resulting in stabilized microtubules with a short biotin-Cy5-labeled segment on their minus-ends and a long rhodamine-labeled segment on their plus-ends.
Full-length Drosophila conventional kinesin was bacterially expressed and purified by Ni column chromatography using previously published protocols.14,43
AC Electrokinetics-driven microtubule accumulation
Cr electrodes were patterned on quartz substrates using photolithography, as described in detail in Uppalapati et al.8 The electrode design was optimized for maximizing dielectrophoretic forces and optimizing electrohydrodynamic flows to achieve proper microtubule alignment on the electrodes. Electrodes used in the present study were 12 mm long, either 10 μm or 15 μm wide, and were separated by a gap ranging from 10–60 μm.Experimental chambers were assembled on the electrode-patterned substrates by attaching two parallel strips of double-sided tape and attaching a glass coverslip to create a flow cell in which solution could be introduced in one end and drawn out the other end by capillary action using filter paper or tissue. For microtubule accumulation experiments, electrode-patterned surfaces were blocked with 0.5 mg ml−1 casein in BRB12 buffer (identical to BRB80 but containing only 12 mM PIPES), and then flushed with microtubule solution (0.64 nM tubulin, 10 μM taxol, 20 mM D-glucose, 0.1 mg ml−1glucose oxidase, 0.04 mg ml−1catalase, 0.2 mg ml−1 casein, 1 mM MgATP and 70 mM β-mercaptoethanol in BRB12 buffer). The leads of the electrodes were connected to a function generator (BK precision, Yorba Linda, CA, USA) and AC electric fields with frequencies varying from 100 kHz to 5 MHz were applied with a maximum peak-peak voltage of 40 V. Microtubules were observed using epifluorescence microscopy (60×, 1.2 N.A. water immersion objective). For Fig. 1 and 2, fluorescent microtubules were visualized using a rhodamine filter, images captured using a Genwac GW-902H CCD camera, the resulting videos were recorded to videotape, and frames digitized using Scion Image (Scion Corporation). For Fig. 3 and 4, segmented microtubules were visualized by taking sequential images using a rhodamine filter and a Cy5 filter, capturing the images on a Cascade 512 CCD camera (Roper Scientific) connected to a PC, and combining the images using Meta-Vue software (Universal Imaging).
Kinesin and neutravidin patterning
For selectively patterning kinesin motors on electrodes, electrodes were patterned by photolithography on quartz substrates as described,8 but the final step of stripping the Shipley 1811 photoresist from the patterned electrodes was omitted. The entire substrate was treated with octadecyl trichlorosilane (OTS),9 followed by stripping the photoresist on the electrodes to create a surface consisting of hydrophilic electrodes surrounded by hydrophobic quartz. The surface was blocked with 0.5 mg ml−1 casein in BRB12 to optimize motor adsorption to the electrodes,46 and then flushed with a solution containing 0.2 mg ml−1 casein, 1% Triton X-100, 1 mM MgATP and 5 μg ml−1 of kinesin in BRB12. It has been shown previously that adsorbing kinesin to hydrophobic surfaces in the presence of 1% Triton results in negligible motor activity.47,48Neutravidin patterning on electrodes was carried out by back exposure to deep-UV, as described previously.12
Extending immobilized microtubules in the chamber
After accumulating microtubules on the electrodes and achieving attachment through biotin–avidin, flow chambers were flushed using at least two chamber volumes of extension solution 2 (10 μM rhodamine-tubulin, 6 μM NEM-tubulin, 0.2 mg ml−1 casein, 4 mM MgCl2, 1 mM GTP and 5% DMSO in BRB80 buffer) and incubated for 20 min at room temperature to extend the immobilized microtubules. The extension solution was then replaced by a taxol-containing antifade solution (10 μM taxol, 4 mM D-glucose, 0.02 mg ml−1glucose oxidase, 0.008 mg ml−1catalase, 0.2 mg ml−1 casein, 1 mM MgATP and 70 mM β-mercaptoethanol in BRB80), and the extended microtubules visualized by epifluorescence microscopy.Bead motility experiments
200 pM polystyrene beads (0.5 μm, Bang Laboratories) were suspended in BRB80 containing 5 mg ml−1 casein and incubated in an ultrasonic bath for 30 min to passivate the surfaces. Motors were adsorbed to beads by adding 20 pM sonicated beads, 0.2 mg ml−1 casein, 1 mM MgATP and 1 nM conventional kinesin motors in BRB80 and incubating on ice for 30 min. The motor-bead solution was then diluted 5-fold into bead motility solution (10 μM taxol, 4 mM D-glucose, 0.02 mg ml−1glucose oxidase, 0.008 mg ml−1catalase, 0.2 mg ml−1 casein, 1 mM MgATP and 70 mM β-mercaptoethanol in BRB80) and flushed into the experimental chamber. Bead motility on the artificial spindles was observed using transillumination on a Nikon TE2000 inverted microscope with a 60× 1.2 N.A. water immersion objective. Images were captured by a Cascade 512b CCD camera (Roper Scientific) controlled by Meta-Vue software (Universal Imaging).Acknowledgements
The authors thank members of the Hancock lab for help in tubulin and kinesin purification, and Richard Cyr for helpful discussions. This work was supported by the NIH (grant R21GM083297 to W.O.H.). M.U. and Y-.M.H. were also partially supported by the Penn State Center for Nanoscale Science (NSF MRSEC DMR0213623).References
- T. Wittmann, A. Hyman and A. Desai, Nat. Cell Biol., 2001, 3, E28–34 CrossRef CAS.
- R. Heald, Cell, 2000, 102, 399–402 CrossRef CAS.
- I. Hansmann, Environ. Health Perspect., 1979, 31, 23–25 CrossRef CAS.
- J. Marx, Science, 2002, 297, 544–546 CrossRef CAS.
- J. R. Jackson, D. R. Patrick, M. M. Dar and P. S. Huang, Nat. Rev. Cancer, 2007, 7, 107–117 CrossRef CAS.
- G. Goshima and R. D. Vale, J. Cell Biol., 2003, 162, 1003–1016 CrossRef CAS.
- S. Inoue and E. D. Salmon, Mol. Biol. Cell, 1995, 6, 1619–1640 CAS.
- M. Uppalapati, Y. M. Huang, T. N. Jackson and W. O. Hancock, Small, 2008, 4, 1371–1381 CrossRef CAS.
- Y. M. Huang, M. Uppalapati, W. O. Hancock and T. N. Jackson, IEEE Trans. Adv. Packag., 2005, 28, 564–570 CrossRef CAS.
- L. Jia, S. G. Moorjani, T. N. Jackson and W. O. Hancock, Biomed. Microdevices, 2004, 6, 67–74 CrossRef CAS.
- A. Merdes and D. W. Cleveland, J. Cell Biol., 1997, 138, 953–956 CrossRef CAS.
- Y. M. Huang, M. Uppalapati, W. O. Hancock and T. N. Jackson, Lab Chip, 2008, 8, 1745–1747 RSC.
- A. Hyman, D. Drechsel, D. Kellogg, S. Salser, K. Sawin, P. Steffen, L. Wordeman and T. Mitchison, Methods Enzymol., 1991, 196, 478–485 CAS.
- M. Uppalapati, Y.-M. Huang, S. Shastry, T. N. Jackson and W. O. Hancock, in Methods in Bioengineering: Microfabrication and Microfluidics, ed. J. D. Zahn, Artech House Publishers, Boston, MA, 2009, pp. 311–336 Search PubMed.
- T. Brown and W. Hancock, Nano Lett., 2002, 2, 1131–1135 CrossRef CAS.
- G. Muthukrishnan, C. A. Roberts, Y.-C. Chen, J. D. Zahn and W. O. Hancock, Nano Lett., 2004, 4, 2127–2132 CrossRef CAS.
- K. W. Wood, R. Sakowicz, L. S. Goldstein and D. W. Cleveland, Cell, 1997, 91, 357–366 CrossRef CAS.
- Y. Mao, D. Varma and R. Vallee, Cell Cycle, 2010, 9, 715–719 CAS.
- C. M. Pfarr, M. Coue, P. M. Grissom, T. S. Hays, M. E. Porter and J. R. McIntosh, Nature, 1990, 345, 263–265 CrossRef CAS.
- L. Wordeman and T. J. Mitchison, J. Cell Biol., 1995, 128, 95–104 CrossRef CAS.
- C. E. Walczak, T. J. Mitchison and A. Desai, Cell, 1996, 84, 37–47 CrossRef CAS.
- N. Tokai, A. Fujimoto-Nishiyama, Y. Toyoshima, S. Yonemura, S. Tsukita, J. Inoue and T. Yamamota, EMBO J., 1996, 15, 457–467 CAS.
- A. A. Levesque and D. A. Compton, J. Cell Biol., 2001, 154, 1135–1146 CrossRef CAS.
- M. Kwon and J. M. Scholey, Trends Cell Biol., 2004, 14, 194–205 CrossRef CAS.
- C. Zhu, J. Zhao, M. Bibikova, J. D. Leverson, E. Bossy-Wetzel, J. B. Fan, R. T. Abraham and W. Jiang, Mol. Biol. Cell, 2005, 16, 3187–3199 CrossRef CAS.
- M. Kirschner and T. Mitchison, Cell, 1986, 45, 329–342 CrossRef CAS.
- C. B. O'Connell and A. L. Khodjakov, J. Cell Sci., 2007, 120, 1717–1722 CrossRef CAS.
- C. L. Rieder, E. A. Davison, L. C. Jensen, L. Cassimeris and E. D. Salmon, J. Cell Biol., 1986, 103, 581–591 CrossRef CAS.
- C. L. Rieder and E. D. Salmon, J. Cell Biol., 1994, 124, 223–233 CrossRef CAS.
- T. M. Kapoor and D. A. Compton, J. Cell Biol., 2002, 157, 551–556 CrossRef CAS.
- J. R. McIntosh, E. L. Grishchuk and R. R. West, Annu. Rev. Cell Dev. Biol., 2002, 18, 193–219 CrossRef CAS.
- T. E. Holy, M. Dogterom, B. Yurke and S. Leibler, Proc. Natl. Acad. Sci. U. S. A., 1997, 94, 6228–6231 CrossRef CAS.
- A. J. Hunt and J. R. McIntosh, Mol. Biol. Cell, 1998, 9, 2857–2871 CAS.
- H. T. Schek, 3rd, M. K. Gardner, J. Cheng, D. J. Odde and A. J. Hunt, Curr. Biol., 2007, 17, 1445–1455 CrossRef CAS.
- J. W. Kerssemakers, E. L. Munteanu, L. Laan, T. L. Noetzel, M. E. Janson and M. Dogterom, Nature, 2006, 442, 709–712 CrossRef CAS.
- M. Dogterom, J. W. Kerssemakers, G. Romet-Lemonne and M. E. Janson, Curr. Opin. Cell Biol., 2005, 17, 67–74 CrossRef CAS.
- M. Dogterom and B. Yurke, Science, 1997, 278, 856–860 CrossRef CAS.
- G. J. Brouhard and A. J. Hunt, Proc. Natl. Acad. Sci. U. S. A., 2005, 102, 13903–13908 CrossRef CAS.
- I. Vernos, J. Raats, T. Hirano, J. Heasman, E. Karsenti and C. Wylie, Cell, 1995, 81, 117–127 CrossRef CAS.
- C. Antonio, I. Ferby, H. Wilhelm, M. Jones, E. Karsenti, A. R. Nebreda and I. Vernos, Cell, 2000, 102, 425–435 CrossRef CAS.
- J. C. Waters, R. V. Skibbens and E. D. Salmon, J. Cell Sci., 1996, 109(Pt 12), 2823–2831 CAS.
- K. Ke, J. Cheng and A. J. Hunt, Curr. Biol., 2009, 19, 807–815 CrossRef CAS.
- W. O. Hancock and J. Howard, J. Cell Biol., 1998, 140, 1395–1405 CrossRef CAS.
- J. Stumpff, G. von Dassow, M. Wagenbach, C. Asbury and L. Wordeman, Dev. Cell, 2008, 14, 252–262 CrossRef CAS.
- R. C. Williams, Jr. and J. C. Lee, Methods Enzymol., 1982, 85(Pt B), 376–385 Search PubMed.
- T. Ozeki, V. Verma, M. Uppalapati, Y. Suzuki, M. Nakamura, J. M. Catchmark and W. O. Hancock, Biophys. J., 2009, 96, 3305–3318 CrossRef CAS.
- Y. Hiratsuka, T. Tada, K. Oiwa, T. Kanayama and T. Q. Uyeda, Biophys. J., 2001, 81, 1555–1561 CrossRef CAS.
- S. Moorjani, L. Jia, T. N. Jackson and W. O. Hancock, Nano Lett., 2003, 3, 633–637 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Movie 1 and 2. See DOI: 10.1039/c0ib00065e |
| This journal is © The Royal Society of Chemistry 2011 |