Patterning osteogenesis by inducible gene expression in microfluidic culture systems†
Yue
Zhang
a,
Zulma
Gazit
bc,
Gadi
Pelled
bc,
Dan
Gazit
bc and
Gordana
Vunjak-Novakovic
a
aDepartment of Biomedical Engineering, Columbia University, 351 Engineering Terrace, 1210 Amsterdam Avenue, Mail Code: 8904, New York, NY 10027, USA. E-mail: gv2131@columbia.edu; Fax: 212-305-4692; Tel: 212-305-2304
bSkeletal Biotechnology Laboratory, Hebrew University—Hadassah Faculty of Dental Medicine, Jerusalem, Israel
cDepartment of Surgery and Cedars-Sinai Regenerative Medicine Institute (CS-RMI), Cedars-Sinai Medical Center, Los Angeles, California, USA
First published on 5th October 2010
Abstract
The development of transitional interfacial zones between adjacent tissues remains a significant challenge for developing tissue engineering and regenerative medicine strategies. Using osteogenic differentiation as a model, we describe a novel approach to spatially regulate expression and secretion of the bone morphogenetic protein (BMP-2) in a two-dimensional field of cultured cells, by flow patterning the modulators of inducible BMP-2 gene expression. We first demonstrate control of gene expression, and of osteogenic differentiation of the cell line with inducible expression of BMP-2. Then we design laminar flow systems, with patterned delivery of Doxycycline (Dox), the expression modulator of BMP-2. The patterned concentration profiles were verified by computational simulation and dye separation experiments. Patterned differentiation experiments conducted in the flow systems for a period of three weeks showed the Dox concentration dependent osteogenic differentiation, as evidenced by mineral deposition. In summary, by combining inducible gene expression with laminar flow technologies, this study provided an innovative way to engineer tissue interfaces.
Insight, innovation, integrationDuring native tissue development and remodeling, the differentiating cells form boundaries and interfaces, in response to spatial gradients of cytokines, whereas the formation of transitional zones in vitro remains a significant challenge. Using a bone–soft tissue interface as a model, we describe a novel approach to spatially regulate expression and secretion of the bone morphogenetic protein (BMP-2), by using flow patterning of inducible BMP-2 gene expression. To this end, we constructed microfludic systems for cultivation of a cell line transfected with the human BMP-2 gene under control of the inducible Tet-Off system, that enabled a patterned delivery of Doxycycline (Dox), the expression modulator of BMP-2. This way, cell differentiation was also patterned, into an osteogenic zone (Dox−), a fibroblastic zone (Dox+), and a transient zone (gradient of Dox concentration). In experimental and modeling studies, we show that inducible gene expression can be effectively used in conjunction with microfluidic devices to form tissue interfaces. |
Introduction
Highly organized structures are a defining feature of biological tissues. Among these structures, tissue interfaces play central roles in organ development and function. For example, in musculoskeletal systems, the interface between bone and soft tissue facilitates transmission of mechanical loads and minimizes stress concentration in junctions.1 Without adequate tissue interaction in such junctions, conventional soft tissue grafts fail at the bone insertion site.2 An important goal in cell and tissue engineering is therefore to recapitulate tissue interfaces, if the organization of the engineered tissue is to reflect that of native tissue.Numerous regeneration approaches have been investigated to reconstruct functional tissues interfaces. The layered structure combined with multiple-phase culture is commonly used to pattern different cells, and construct tissue interfaces.3 One strategy to engineer osteochondral tissue interface is to fabricate a polymer scaffold consisting of two layers of materials with distinct properties—such as porosity, mechanical strength and material microstructure, to mimic the natural environment for bone and cartilage development. These scaffolds are seeded with either human mesenchymal stem cells or pre-differentiated chondrocytes and osteoblasts and the tissue formation is investigated in vitro or in vivo studies.4–7 However this approach is restricted by inherent discontinuities across different materials.8Growth factors and chemokines can serve as powerful long-range mediators of cellular organization and function.9 Thus, constructing growth factor gradients within the structure could be a promising way to develop tissue interfaces. In some studies, sustained release of growth factors incorporated into polymer scaffolds facilitated bone and cartilage differentiation in vitro and in vivo.10–12 However the dose of growth factors in these scaffold systems was not well defined, due to the lack of optimal techniques to deliver and maintain growth factor signals in engineered constructs.13
Recent studies have focused on alternative strategies, by directly modulating spatial patterns of gene expression within a cell population, thereby modulating the functions of cells. Ziauddin and Sabatini demonstrated that gene expression could be patterned in culture via a reverse transfection procedure wherein plasmid DNA was immobilized on a substrate onto which a monolayer of cells was subsequently grown.14 But this approach has been limited by the low efficiency of plasmid transfection. To alleviate the limitations of plasmid-based transfection, more efficient viral vectors (which may present safety issues) have been used to construct tissue interface by spatial regulation of gene transfer.15
In the present study we proposed a new strategy to pattern cell differentiation by patterning gene expression modulator within laminar fluidic system. When parallel laminar streams of different liquids flow inside a channel at a low Reynolds number (Re < 1), there is no mixing between these adjacent streams, other than by diffusion. For this reason, laminar flow systems have been utilized for separations or patterning of the cells and factors.16–19 The generation of specific patterns by using such fluidic methods is rather straightforward, as the characteristics of the fluidic environment can be tightly controlled.
We chose an inducible BMP-2 expressing cell line (C9) under the control of Tet-off gene expression system. This model can provide efficient control of BMP-2 gene expression by modulating the concentration of BMP-2 expression modulator doxycycline (Dox) (Fig. 1). BMP-2 belongs to the transforming growth factor superfamily and plays an important role in osteogenesis and bone metabolism, serving as a powerful inducer of osteoblast differentiationin vitro and bone formationin vivo.20,21 Therefore, by patterning the delivery of Dox to the cultured cells in laminar flow system, we can effectively pattern the expression of BMP-2 and thereby modulate osteogenic differentiation (Fig. 2A).
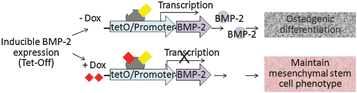 | ||
| Fig. 1 Biological model of inducible BMP-2 expression under Tet -off gene expression . Dox concentration dependent BMP-2 expression leads to concentration-dependent osteogenic differentiation. | ||
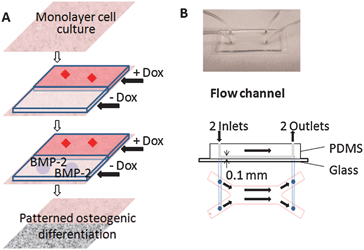 | ||
| Fig. 2 Experimental system. (A) Strategy for patterning osteogenic cell differentiation. Cultured cells with inducible BMP-2 expression are exposed to laminar flow with two streams (one with and one without Dox), generating a pattern of Dox concentration. The patterned Dox concentration profile leads to patterned BMP-2 expression, and thereby to patterned osteogenic differentiation. (B) Flow channel (main channel: 20 mm L × 6 mm W × 0.1 mm H). | ||
First, we characterized the inducible BMP-2 expression cell line (C9) in static culture, and proved that BMP-2 secretion was dependent on Dox concentration (0–1 ng ml−1). By modulating Dox concentration, we could modulate the BMP-2 mediated osteogenic differentiation, as verified by calcium deposition and alkaline phosphatase (ALP) analysis. Then we designed a laminar-fluidic system microfluidic channel (Fig. 2B). These fluidic systems can deliver different levels of Dox concentration to cultured monolayers of cells. In 3-week experiments of osteogenic differentiation, we demonstrated that patterning of osteogenesis in a mesenchymal stem cell line can be achieved by patterning BMP-2 gene expression modulator, Dox.
We also developed a larger fluidic chamber (Supplemental Fig. 1†) and successfully replicated the same method for patterned osteogensis over a larger field of cells. In addition to the scale up to larger cell numbers, this fluidic chamber can accommodate cultivation of cells in a scaffold sheet or a layer of hydrogel for generating a 3D tissue interface.
Materials and methods
Cell culture and osteogenic differentiation
The BMP-2 inducible expression cell line (C9 cell) was orginated from C3H10T1/2 murine MSCs cell line after transfection with the human BMP-2 gene under control of the inducible Tet-off system.22,23 The C9 cells were cultured in basic medium: Dulbecco's Modified Eagle's Medium (DMEM) (Fisher, Pittsburg, PA) supplemented with 10% fetal bovine serum (Fisher), 2 mM L-glutamine, 100 U ml−1penicillin, 100 μg ml−1streptomycin (Invitrogen, Carlsbad, CA), and 1 μg ml−1 Dox (Sigma-Aldrich, St Louis, MO) to repress BMP-2 secretion. Every 2–3 passages, C9 cells were subjected to selection medium: basic medium supplemented with 500 mg ml−1G418, 200 mg ml−1hygromycin (Sigma-Aldrich), and 1 μg ml−1 Dox to eliminate the cells which lost their genotype. For osteogenic differentiation, C9 cells were cultured in differentiation medium: basic medium supplemented with 10 mM sodium-b-glycerophosphate, 0.05 mM ascorbic acid-2-phophate (Sigma-Aldrich) and with or without Dox added. Medium was changed twice a week.BMP-2 and DNA
Secretion of BMP-2 into culture medium was measured by BMP-2 sandwich ELISA kit, according to the manufacturer's protocol (R&D Systems; Minneapolis, MN). For evaluating DNA content, the cells cultured in monolayer were washed in PBS, placed in digestion buffer (10 mM Tris, 1 mM EDTA, and 0.1% Triton X-100 with 0.1 mg mL−1proteinase K, Sigma-Aldrich) in centrifuge tubes, and incubated overnight at 50 °C. The supernatants were sampled, the Picogreen dye (Molecular Probes, Eugene, OR) was added to the samples in 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 ratio, and read in fluorescent plate reader at excitation of 485 nm, emission of 528 nm (BioTek, Winooski, VT). A standard curve was prepared from a solution of salmon testes DNA (Molecular Probes).
:1 ratio, and read in fluorescent plate reader at excitation of 485 nm, emission of 528 nm (BioTek, Winooski, VT). A standard curve was prepared from a solution of salmon testes DNA (Molecular Probes).
Alkaline prosphatase (ALP) measurement and staining
For alkaline phosphatase (ALP) measurement, cells were harvested in 100 μL of lysate buffer consisting of PBS (Fisher), 1% Triton X-100, 0.5% sodium deoxycholate, 0.1% sodium dodecyl sulfate, 0.1 mg mL−1phenylmethylsulfonylfluoride (PMSF) and 3% aprotinin (Sigma-Aldrich), that was maintained on ice and vortexed intermittently for 30 min to break down the cell membranes. The extracts were removed, centrifuged and the supernatants were stored at −20 °C. 50 μL of a sample was incubated with 50 μL of alkaline buffer and 50 μL nitrophenyl-phosphate substrate solution (Sigma-Aldrich) in micro-centrifuge tubes at 37 °C for 10–20 min. The reaction was stopped with 0.5 N NaOH (Sigma-Aldrich). The absorbance was read at 405 nm (Molecular Devices, Sunnyvale, CA) and compared to a standard curve obtained from p-nitrophenol solutions (Sigma-Aldrich) of known concentrations. For ALP staining, the cultured cell monolayer was fixed in citrate buffered acetone for 30 s, after rinsing in distilled water for 45 s, and the fixed monolayer was immersed into alkaline-dye mixture consisting of 4% AS-MX Phosphate Alkaline solution in diluted diazonium salt solution (Sigma-Aldrich), for 30 min in dark, and washed in de-ionized water. ALP staining was captured by microscope (Olympus IX81, Center Valley, PA).Calcium measurement and von Kossa staining
For calcium measurement, the cells were extracted by 5% trichloroacetic acid (Sigma-Aldrich), and diluted as necessary to be within tolerance ranges of assay. Ocresolphtalein complex was added (Calcium CPC LiquiColor Test, Stanbio Laboratory, Boerne, TX) and the calcium content was measured in spectrophotometer at 550 nm (Molecular Devices). For von Kossa staining, the cultured cells monolayers were washed with PBS and fixed in cold 10% buffered formalin (Fisher). The cells were then rinsed by distilled water and then immersed in 5% silver nitrate (Sigma-Aldrich) solution followed by exposure to bright light for 1 h. Mineralization was captured by microscope (Olympus X81).Bone sialoprotein (BSP) staining
For BSP staining, cell monolayers were washed with PBS and fixed in cold 10% buffered formalin (Fisher). The cells were then rinsed by distilled water quenched with 0.3% H2O2, and blocked with PBS containing 5% BSA and 0.02% (vol/vol) goat serum (Sigma-Aldrich) for 30 min at room temperature. After rinsing with PBS, slides were incubated with 1:1000 BSP antibody (Sigma-Aldrich) overnight. The fixed cells were washed with PBS and incubated with biotinylated goat anti-rabbit serum (Sigma-Aldrich) and Vectastain ABC Kit (Vector Laboratories, Burlingame, CA) for 30 min at room temperature. Brown color was developed with SIGMAFAST diaminobenzidine (Sigma-Aldrich) for 10 min at room temperature. Slides were then counter-stained with hematoxylin (Sigma-Aldrich).
Design and manufacture of fluidic systems
000 dpi resolution. Master molds patterned with 100 μm thick resist were made by patterning a negative photoresist on a silicon wafer (manufactured by Stanford Microfludics Foundry). PDMS molds were fabricated by curing prepolymer (Sylgard 184, Essex Chemical, Midland, MI) on silicon masters patterned with SU-8 photoresist. Briefly, PDMS prepolymer composed of elastomer and curing agent at 10:1 ratio, was poured on the silicon master that was patterned with photoresist and cured at 60 °C for 2 h. The PDMS molds were peeled off the silicon wafer. The inlet and outlet of the micro channel were created by a sharp punch (1 mm diameter, Fisher) for medium perfusion and cell seeding. The complete microfluidic device consisted of a top PDMS fluidic channel, a bottom glass slide, tygon tubing (0.79 mm inside diameter (ID), 2.38 mm outside diameter (OD), McMaster, Robbinsville, NJ), stainless steel tubing (0.64 mm ID, 0.81 mm OD, McMaster) and syringe pump (Harvard Apparatus, Boston, MA). The main fluidic channel was 100 μm high × 20 mm long × 6 mm wide, and the branches were 100 μm high × 10 mm long × 3 mm wide. The microfluidic layer was bonded to oxygen plasma treated glass slide (Harrick Scientific, Pleasantville, NY), with channels at the top, slide at the bottom).
For dye separation experiments, cells were subjected to 2 continuous streams of flow for 30–60 min, with one stream of media containing 2 μM Calcein-AM, and the other stream of unsupplemented medium. The stream with Calcein-AM was subsequently replaced with unsupplemented medium for a 30 min wash. For osteogenic differentiation experiments, cells were continuously perfused in differentiation medium, with 1 ng ml−1 or 0 ng ml−1 Dox for 3 weeks.
Mathematical model
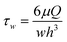 | (1) |
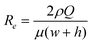 | (2) |
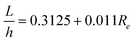 | (3) |
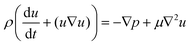 | (4) |
| ∇u = 0 | (5) |
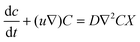 | (6) |
Image analysis
The sequential images were acquired using an inverted microscope (Olympus IX81), stored, assembled and analyzed digitally by Photoshop CS4 program (Adobe, San Jose, CA). For quantitative analysis of calcium deposition staining, the assembled color images were converted to black and white images, and the brightness and darkness of the images were inverted, to capture the dark stain of calcium deposition. Calcium deposition was quantified from ratio of the normalized sum of segmented image intensity and the minimum segmented image intensity, moving along the width of the channel/chamber, and using the Matlab R2007 program (Matlab, Natick, MA).Statistics/data analysis
Data were expressed as mean ± standard deviation (SD) of the measured values. Data were analyzed by ANOVA for multiple comparisons, and a post hoc test for group to group comparisons, with P < 0.05 considered statistically significant.Results
Regulation of BMP-2 expression and osteogenic differentiation in static culture
We first characterized the Dox-dependent BMP-2 secretion and Dox-dependent osteogenic differentiation in static culture, for a series of Dox concentrations (0–1000 ng ml−1, Fig. 3). For C9 cells grown in static culture, secretion of BMP-2 was induced when Dox concentration in osteogenic differentiation medium was <1 ng ml−1. The amount of secreted BMP-2 in medium was negatively correlated to the concentration of Dox, and reached ther maximum level when Dox was absent from medium. During 4 weeks of osteogenic differentiation, BMP-2 was mostly secreted within the first 2 weeks of culture, after which the secretion of BMP-2 significantly decreased, possibly due to the decrease in cell metabolism with time in culture. When Dox concentration was ≥1 ng ml−1, BMP-2 secretion could not be detected by ELISA throughout the culture period (Fig. 3A). The DNA measurements of the C9 cells showed that the variation of Dox concentration from 1000 ng ml−1 to 0 ng ml−1 had no significant effect on cell proliferation in the osteogenic-inductive culture (Fig. 3B).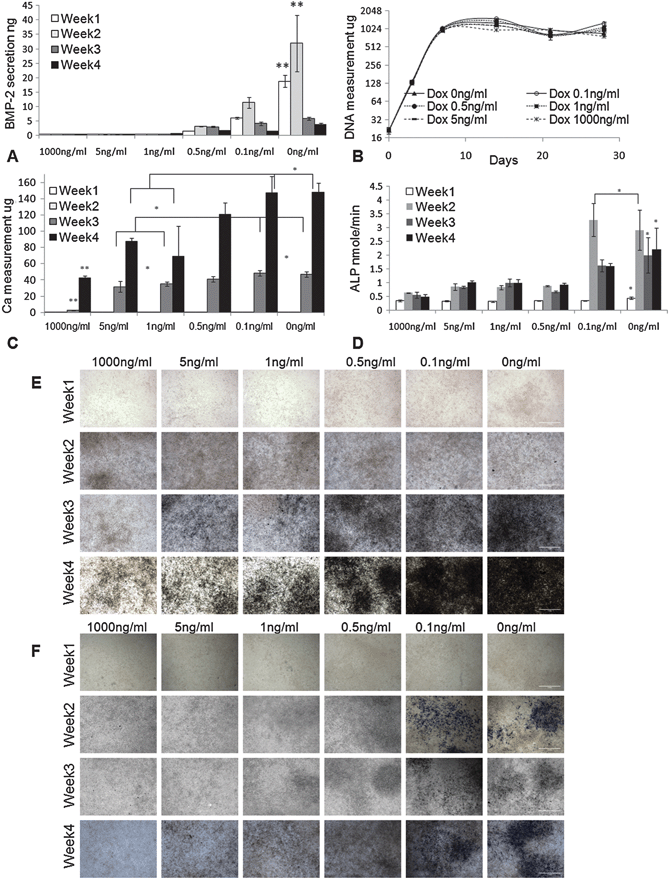 | ||
| Fig. 3 Characterization of inducible BMP-2 expressing cell line (C9). (A) ELISA measurement of the secreted BMP-2 in culture media (n = 3). (B) C9 cell proliferation profile by DNA measurement (n = 6). (C) Measurement of Calcium deposition (n = 4). (D) Measurement of alkaline phosphates (ALP) (n = 4). *: p < 0.05. **: p < 0.05. (E) von Kossa staining of the C9 cells. (F) ALP staining of the C9 cells. Original magnification: 200×. Scale bar: 1 mm. | ||
The Dox concentrations in the range 1 ng ml−1 to 0 ng ml−1 induced Dox-dependent expression of BMP-2, that in turn induced BMP-2-dependent osteogenic differentiation of C9 cells, as evidenced by calcium deposition and expression of ALP (Fig. 3C–F). Calcium measurement and von Kossa staining showed no detectable calcium deposition until the 3rd week of static culture. C9 cells cultured at lowest range of Dox concentrations (0 or 0.1 ng ml−1) had significantly higher deposition of calcium than C9 cells cultured at higher Dox concentrations (≥1 ng ml−1) in weeks 3–4 of culture (Fig. 3C and E). Consistently, the ALP expression in cells cultured at low Dox concentrations (0 or 0.1 ng ml−1) was high after one week of osteogenic induction culture (Fig. 3D and F), demonstrating the initiation of osteogenic differentiation.
In summary, the in vitro static culture suggest strong correlations between Dox concentration, BMP-2 secretion and osteogenic differentiation. These BMP-2 induced cells exhibited a clear “cutoff” of BMP-2 expression and osteogenic differentiation at Dox concentration of 1 ng ml−1, and the differentiation was detectable after 3 weeks of culture. Based on these findings, in subsequent flow experiments we used bi-laminar flows of CDox = 1 ng ml−1 and CDox = 0 ng ml−1 to generate osteoblast–fibroblast interface in the same construct.
Characterization of the flow channel
The velocity profiles and concentration profiles in the device were analyzed in detail using COMSOL Multiphysics®. Firstly, the flow and mixing in the fluidic channel were determined. In order to generate a concentration interface perpendicular to the flow inside the channel, it was necessary that the flow past the inlet tubes be fully developed and laminar.The flow was characterized by low Reynolds numbers, of 0.055–0.011 for the channel, for the range of flow rates studied (Fig. 4). The maximum flow rate of 5 μl min−1 × 2 corresponded to Re = 0.055 and the hydrodynamic development length of 32 μm in the flow channel. Thus, the flow in each system could be considered as fully developed, with negligible variation in the direction transverse to flow. The wall shear stress in the cell culture zone was 16.7–3.3 mPa.
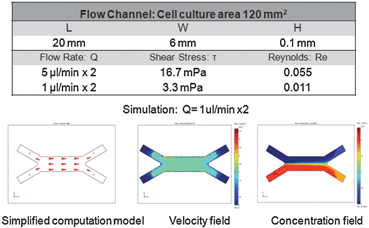 | ||
| Fig. 4 Flow analysis. Simplified flow model, the steady-state velocity and concentration profiles are shown for a flow rate of 1 μl min−1 × 2. Data were calculated for the diffusion coefficient DDox = 3.93 × 10−6 cm2s−1, fluid viscosity ν = 10−3 Pa s, fluid density ρ = 103 kg m−3, and inlet concentrations CDox = 1 ng ml−1 = 2.25 × 10−12 mol ml−1 and CDox = 0 ng ml−1. | ||
Simplified fluidic models were developed to study the flow and concentration profiles at steady state (Fig. 4). Different values of the flow rate were simulated, while maintaining at constant levels the other parameters, such as diffusion coefficient (DDox = 3.93 × 10−6 cm2s−1),25 flow viscosity (ν = 10−3 Pa s), fluid density (ρ = 103 kg m−3) and input concentrations (CDox = 1 ng ml−1 = 2.25 × 10−12 mol ml−1; CDox = 0). Simulation showed that two fluid flows meet in the device, creating a concentration interface. Complete mixing inside the channel was dependent on the flow rate.
To visualize the concentration profiles in each of the fluidic systems, Calcein-AM, (DCalcein = 2.6 × 10−6cm2s−1) was used as a model molecule, and the fluorescent images were acquired to show selective Calcein distribution only in cells within the channel (Fig. 5). The concentration profiles were additionally verified in experiments that used the trypan blue dye (Dtrypan_blue = 2.29 × 10−6 cm2s−1). In summary, the patterned Calcein/Trypan blue distribution verified the simulation result, indicating that the bi-laminar-fluidic system permitted the alignment of two flows with different concentration profiles.
 | ||
| Fig. 5 Dye separation studies. Separation of Calcein stain in the flow channel at the flow rates of 5 μl min−1 × 2 (A) and 1 μl min−1 × 2 (B). Data are for the diffusion coefficient of CalceinDCalcein = 2.6 × 10−6 cm2s−1, and inlet concentrations of Calcein C+ = 2 μM and C− = 0, diffusion coefficient of Trypan blue of DTrypan blue = 2.21 × 10−6 cm2s−1, and input concentrations of Trypan blue of C+ = 229 μM and C− = 0. Flow direction is left to right. Original magnification: 100×. Scale bar: 1 mm. | ||
Patterning of osteogenic differentiation
We examined the effect of patterning Dox concentration on osteogenic differentiation. Mineral deposition within cell culture constructs was analyzed after 3 weeks dynamic culture in osteogenic differentiation media as a functional marker of osteogenesis. The result showed C9 cells cultured with continuous Dox (1 ng ml−1) treatment displayed less mineral deposits (Fig. 6A1 and A2), whereas C9 cells cultured without Dox treatment displayed more and relatively uniform mineral deposits (Fig. 6C1 and C2). C9 cells cultured in a gradient of Dox concentration exhibited zonal organization of mineral deposition (Fig. 6B1 and B2). By pooling together the independent experiments, and quantifying the normalized intensity of mineralization in the flow chamber, we observed a gradual decrease in mineralization across the Dox 0 ng ml−1 and Dox 1 ng ml−1 interface (Fig. 6B3), that was not observed at continuous Dox concentrations of 1 ng ml−1 ((Fig. 6A3), or 0 ng ml−1 (Fig. 6C3). However, we also observed some scattering of mineralization across the interface, and close to edge areas, possibly due to molecular diffusion of Dox, intracellular signalling of BMP-2, edge effects, or disturbances of flow during medium exchange.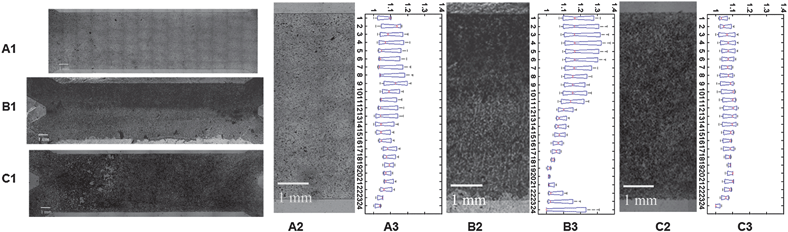 | ||
| Fig. 6 Dox dependent osteogenic differentiation in the flow channel . C9 cells were cultured in osteogenic differentiation medium in a perfused flow channel, at a flow rate of 1 μl min−1 × 2, for 3 weeks. Images show von Kossa staining of calcium deposition for selected conditions: (A1, A2) Perfusion medium with Dox 1 ng ml−1 (top)/1 ng ml−1 (bottom); (B1, B2) Perfusion medium: Dox 0 ng ml−1 (top)/1 ng ml−1 (bottom); (C1, C2) Perfusion medium: Dox 0 ng ml−1 (top)/0 ng ml−1 (bottom). (A3, B3, C3) Intensity of calcium staining, obtained by pooling repeated experiments (n = 3 per condition). The staining of the deposited calcium is expressed at the ratio of the normalized sum of segmented (∼0.25 mm) image intensity and the minimum intensity along the y axis (the width) of the chamber. Data were processed using Matlab R2007 program. Flow direction: left to right. Scale bar: 1 mm. | ||
After the flow channel was scaled up to the fluidic chamber (Supplemental Fig. 1†), we successfully replicated the computational simulation of the fluidic profiles, the dye separation test (Supplemental Fig. 2†), and the patterning of osteogenc differentiation (Supplemental Fig. 3†).
We observed significant effects of medium flow rate on osteogenic differentiation. Under high flow rate at 5 μl min−1 × 2 in fluidic channel (Supplemental Fig. 4A†), or 8 μl min−1 × 2 in fluidic chamber (Supplemental Fig. 4B†), the C9 cells exhibited a less differentiated phenotype, compared to the cells cultured at a flow rate of 1 μl min−1 × 2 in fluidic channel (Fig. 6), or 4 μl min−1 × 2 in fluidic chamber (Supplemental Fig. 3†). Osteogenic differentiation was further enhanced by lowering the flow rate to 0.5 μl min−1 × 2 in fluidic channel (Supplemental Fig. 4C†). These effects were likely due to the dilution of cell-secreted factors (such as BMP-2) at higher medium flow rates.
We were not able to generate an uniform gradient of ALP staining along the Dox concentration profile. In contrast, we observed a clear pattern of distribution of bone sialoprotein, a major component of the bone extracellular matrix, that followed the pattern of Dox concentration, both in flow channel and flow chamber (Supplemental Fig. 5B and C†). In addition, we observed relatively uniform distribution of cells after 3 weeks fluidic culture (Supplemental Fig. 5A†). Taken together, these results demonstrate that spatial control over Dox distribution can lead to spatial control over BMP-2 expression, and spatial control over osteogenic differentiation.
Discussion
We explored a novel strategy to patterning osteogenic differentiation to generate a stable interface between differentiated and undifferentiated cells. Osteogenic differentiation was patterned by BMP-2 gene expression, which in turn was patterned by the BMP-2 modulator Dox, using a bi-laminar microfluidic system. This strategy of patterning the expression of key master regulator genes within tissues mimics the mechanisms of the development that underlie patterned differentiation in vivo.26 Previous gene expression and immunohistochemical studies described distribution and localization of BMP-2 during bone formation,27,28 consistent with the known effect of BMP-2 gradients on osteogenesis. BMP-2 gradients were also implicated in chondrogenic differentiation in the growth plate29 and in pituitary organogenesis.30 The formation of BMP-2 gradients thus provides an interesting approach for application in tissue engineering.Characterization of the C9 cells with inducible BMP-2 gene expression in static culture showed that BMP-2 secretion was tightly controlled by Dox concentration. The C9 cells had clear “cutoff” of BMP-2 secretion at Dox of 1 ng ml−1. In the range of Dox concentrations of 1 ng ml−1 to 0 ng ml−1, the cells showed Dox-dependent BMP-2 secretion, leading to BMP-2-mediated osteogenic differentiation, that was clearly evidenced by calcium deposition and ALP activity. Therefore, by coupling the Dox-dependent BMP-2 secretion and BMP-2-dependent osteogenic differentiation, we were able to directly control osteogenic differentiation by the gene expression modulator, Dox. Notably, Dox is a simple semi-synthetic tetracycline (molecular weight of 444), widely used to treat a variety of infections,31 which could provides a cost-effective and efficient “turn on”–“turn off” functions for engineering tissue functionality. The “turn on”–“turn off” functions of the inducible gene expression could be further used in a temporary manner, and adapted to study the dynamics of gene expression in cell differentiation processes.
The microfluidic channel designed to conduct the present study is a simple device with two parallel laminar flow streams. The computational simulation and dye separation studies confirm that this microfluidic system can be used to create distinct regions of constant concentration and form time-invariant gradients across the cell culture area, due to the lack of convective mixing under low-Re number laminar flow. After 3 weeks of cell culture with perfusion that generated a patterned delivery of Dox, we observed patterned osteogenic differentiation that correlated to the pattern of Dox concentration. In the microfluidic chamber, after 3 weeks of osteogenic differentiation, a transient gradient of mineral deposition, with high calcium deposition at the top and low calcium deposition at the bottom, was established, in response to the Dox concentration profiles.
Compared to the fluidic channel, the fluidic chamber offers additional flexibility. First, it allows the cells to be attached to a glass slide before the chamber assembly, which can provide better control of cell seeding and distribution, a feature suitable to a variety of cell culture applications. Second, as the length of the flow chamber is increased, more mixing may occur via diffusion perpendicular to the direction of fluid flow, while keeping input flow rates and compositions constant. Therefore, we were able to create and maintain a range of gradients by carefully choosing the concentrations and flow rates of the fluid streams, for long periods of time. This feature may enable studies of more sophisticated cell functionality in response to gradients of signals. Third, the height of the fluidic chamber can be varied, by adjusting the thickness of the gasket, thereby adjusting shear stress in the system. Potentially, the microfluidic chamber could be eventually up-graded to hold slim 3-dimensional tissue constructs for study of higher order tissue interfaces.
In laminar-fluidic systems, the paracrine/autocrine signals released by the cells in response to the applied gradient could be quickly removed by medium perfusion and accumulated downstream. In this study we observed that flow diluted the extracellular concentration of BMP-2, leading to “less differentiated” phenotype compared to static culture. These effects could be modulated to some extent by adjusting the flow rates. By decreasing the flow rates, from 8 μl min−1 × 2 to 4 μl min−1 × 2 in fluidic chamber, or from 5 μl min−1 × 2 to 1 μl min−1 × 2 in fluidic channel osteogenic differentiation was markedly enhanced calcium deposition.
We also observed that the osteogenic differentiation increased at the channel walls, presumably due to the edge effects in laminar flow system. In a typical laminar flow profile, the highest velocity is found in the center of channel or chamber, while the lowest velocity (V = 0) is found along the wall. As discussed above, osteogenic differentiation is enhanced at lower fluid flow rates. Therefore, the zero velocity along the wall can lead to the accumulation of autocrine and paracrine signals, and result in a higher degree of osteogenic differentiation even with the presence of Dox. Therefore, we suggest that the analysis of cell differentiation in this flow system needs to take into account the edge effects.
In summary, we developed an interface between osteogenic and undifferentiated cells, as a prototype of our proposed patterned gene expression strategy. The 2D system investigated in this study presents a valuable tool for controlled in viro study of osteogenesis.
Patterned osteogenesis of mesenchymal stem cells, when scaled up to 3D, would enable generation bone-ligament or osteo–chondrogenic interface in engineered tissue constructs. Future work of interest includes patterning of osteogenesis–fibrogenesis interfaces, by modulation of inducible BMP-2 expression on one side of the interface, and modulation of other growth factors32 (connective tissue growth factor, CTGF, and TGF-beta1) on the other side of the interface, using the fluidic chamber. Likewise, osteogenic–chondrogenic interface could be patterned by constructing a super vector composed of two gene expression cassettes—inducible BMP-2 expression under Tet-on promoter and inducible Sox9 under RheoSwitch promoter. Such a vector would mimic a biological circuit controlling the expression of two separate genes (BMP-2 and Sox9) with combination of modulators.
Conclusions
In summary, this study demonstrates that a continuous, graded osteogenesis tissue interface can be established by patterning the BMP-2 gene expression, viapatterning of the BMP-2 modulator Dox, using a simple laminar-fluidic system. This approach provides an alternative way to develop transitional interface zones for enhanced tissue integration and biological function. The technology also provides an interesting model to study how a gradient of gene expression can modulate cell fate and expression of a specific phenotype or function. The use of alternative cell types and 3-dimensional scaffolds could enable regeneration of more sophisticated, higher-order tissue interfaces that mimic the structural and functional characteristics of native tissues.Acknowledgements
We gratefully acknowledge funding support of this work by the NIH (DE161525 and EB02520 to GVN).Notes and references
- S. Thomopoulos, G. R. Williams, J. A. Gimbel, M. Favata and L. J. Soslowsky, J. Orthop. Res., 2003, 21, 413–419 CrossRef.
- S. L. Woo, S. D. Abramowitch, R. Kilger and R. Liang, J. Biomech., 2006, 39(1), 1–20 CrossRef.
- Y. Mitsuhashi, Y. Mikami, H. Mikami, H. Ishikawa, K. Tamai and I. Hashimoto, J. Dermatol. Sci., 1993, 5(1), 3–13 CrossRef CAS.
- G. Chen, T. Sato, J. Tanaka and T. Tateishi, Mater. Eng., C, Biomim. Mater., Sens. Syst., 2006, 26, 118–123 Search PubMed.
- X. X. Shao, D. W. Hutmacher, S. T. Ho, J. C. Goh and E. H. Lee, Biomaterials, 2006, 27, 1071–1080 CrossRef CAS.
- C. H. Chang, F. H. Lin, C. C. Lin, C. H. Chou and H. C. Liu, J. Biomed. Mater. Res., 2004, 71b, 313–321 Search PubMed.
- R. Tuli, S. Nandi, W. J. Li, S. Tuli, X. Huang, P. A. Manner, P. Laquerriere, U. Noth, D. J. Hall and R. S. Tuan, Tissue Eng., 2004, 10, 1169–1179 CAS.
- J. P. Spalazzi, S. B. Doty, K. L. Moffat, W. N. Levine and H. H. Lu, Tissue Eng., 2006, 12, 3497–3508 CrossRef CAS.
- B. G. Chung, L. A. Flanagan, S. W. Rhee, P. H. Schwartz, A. P. Lee, E. S. Monuki and N. L. Jeon, Lab Chip, 2005, 5(4), 401–406 RSC.
- C. Li, C. Vepari, H. J. Jin, H. J. Kim and D. L. Kaplan, Biomaterials, 2006, 27, 3115–3124 CrossRef CAS.
- H. Hosseinkhani, M. Hosseinkhani, A. Khademhosseini and H. Kobayashi, J. Controlled Release, 2007, 117, 380–386 CrossRef CAS.
- B. D. Boyan, C. H. Lohmann, A. Somers, G. G. Niederauer, J. M. Wozney, D. D. Dean, D. L. Carnes Jr and Z. Schwartz, J. Biomed. Mater. Res., 1999, 46, 51–59 CrossRef CAS.
- R. Langer, Nature, 1998, 392, 5–10 CAS.
- J. Ziauddin and D. M. Sabatini, Nature, 2001, 411(6833), 107–110 CrossRef.
- J. E. Phillips, K. L. Burns, J. M. Le Doux, R. E. Guldberg and A. J. García, Proc. Natl. Acad. Sci. U. S. A., 2008, 105(34), 12170–12175 CrossRef CAS.
- S. Takayama, J. C. McDonald, E. Ostuni, M. N. Liang, J. A. Kenis, R. F. Ismagilov and G. M. Whitesides, Proc. Natl. Acad. Sci. U. S. A., 1999, 96, 5545–5548 CrossRef CAS.
- P. J. A. Kenis, R. F. Ismagilov and G. M. Whitesides, Science, 1999, 285, 83–85 CrossRef CAS.
- S. W. Rhee, A. M. Taylor, C. H. Tu, D. H. Cribbs, C. W. Cotman and N. L. Jeon, Lab Chip, 2005, 5(1), 102–107 RSC.
- S. Takayama, E. Ostuni, P. LeDuc, K. Naruse, D. E. Ingber and G. M. Whitesides, Chem. Biol., 2003, 10(2), 123–130 CrossRef CAS.
- E. Hay, E. M. Hott, A. M. Graulet, A. Lomri and P. J. Marie, J. Cell. Biochem., 1999, 72, 81–93 CrossRef CAS.
- P. Q. Ruhe, H. C. Kroese-Deutman, J. G. Wolke, P. H. Spauwen and J. A. Jansen, Biomaterials, 2004, 25, 2123–2132 CrossRef CAS.
- I. K. Moutsatsos, G. Turgeman, S. Zhou, B. G. Kurkalli, G. Pelled, L. Tzur, P. Kelley, N. Stumm, S. Mi, R. Müller, Y. Zilberman and D. Gazit, Mol. Ther., 2001, 3, 449–461 CrossRef CAS.
- D. Noël, D. Gazit, C. Bouquet, F. Apparailly, C. Bony, P. Plence, V. Millet, G. Turgeman, M. Perricaudet, J. Sany and C. Jorgensen, Stem Cells, 2004, 22(1), 74–85 Search PubMed.
- B. Atkinson, M. P. Brocklebank, C. C. H. Card and J. M. Smith, AIChE J., 1969, 15(4), 548–553 CrossRef CAS.
- C. R. Wilke and P. Chang, AIChE J., 1955, 1, 264–270 CrossRef CAS.
- A. E. Serls, S. Doherty, P. Parvatiyar, J. M. Wells and G. H. Deutsch, Development, 2005, 132(1), 35–47 CAS.
- K. A. McCullough, C. A. Waits, R. Garimella, S. E. Tague, J. B. Sipe and H. C. Anderson, J. Orthop. Res., 2007, 25(4), 465–472 CrossRef CAS.
- Y. Ohyama, A. Nifuji, Y. Maeda, T. Amagasa and M. Noda, Endocrinology, 2004, 145(10), 4685–4692 CrossRef CAS.
- O. Nilsson, E. A. Parker, A. Hegde, M. Chau, K. M. Barnes and J. Baron, J. Endocrinol., 2007, 193(1), 75–84 Search PubMed.
- M. Treier, A. S. Gleiberman, S. M. O'Connell, D. P. Szeto, J. A. McMahon, A. P. McMahon and M. G. Rosenfeld, Genes Dev., 1998, 12(11), 1691–1704 CrossRef CAS.
- R. L. Sweet, J. Schachter, D. V. Landers, M. Ohm-Smith and M. O. Robbie, Am. J. Obstet. Gynecol., 1988, 158(3 Pt 2), 736–741 CAS.
- C. H. Lee, B. Shah, E. K. Moioli and J. J. Mao, J. Clin. Invest., 2010, 120(9), 3340–3349 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Supplementary Fig. 1–5. See DOI: 10.1039/c0ib00053a |
| This journal is © The Royal Society of Chemistry 2011 |