The discovery of a novel prototype small molecule TLR7 agonist for the treatment of hepatitis C virus infection†
David C.
Pryde
*a,
Thien-Duc
Tran
a,
Peter
Jones
a,
Gemma C.
Parsons
a,
Gerwyn
Bish
a,
Fiona M.
Adam
a,
Mya C.
Smith
a,
Donald S.
Middleton
a,
Nick N.
Smith
a,
Frederick
Calo
a,
Duncan
Hay
a,
Michael
Paradowski
a,
Katie J. W.
Proctor
a,
Tanya
Parkinson
b,
Carl
Laxton
b,
David N. A.
Fox
a,
Nigel J.
Horscroft
b,
Giuseppe
Ciaramella
b,
Hannah M.
Jones
c,
Jonathan
Duckworth
c,
Neil
Benson
c,
Anthony
Harrison
c and
Rob
Webster
c
aWorldWide Medicinal Chemistry, Pfizer Global Research and Development, Ramsgate Road, Sandwich, Kent, England CT13 9NJ. E-mail: David.Pryde@pfizer.com; Tel: +44(1304) 643687
bDiscovery Biology, Pfizer Global Research and Development, Ramsgate Road, Sandwich, Kent, England CT13 9NJ
cPharmacokinetics, Dynamics and Metabolism, Pfizer Global Research and Development, Ramsgate Road, Sandwich, Kent, England CT13 9NJ
First published on 20th December 2010
Abstract
A series of heterocycle analogues of an adenine template were explored for TLR7 agonist potency and pharmacokinetics. One compound was identified with an excellent pharmacokinetic, in vitro potency and in vivo interferon induction profile in a mouse model, and was selected for further pre-clinical evaluation as a potential treatment for hepatitis C viral infection.
Introduction
Pattern recognition receptors (PRRs) are the sentinels of the innate immune system recognising components of invading pathogens which include various bacterial cell wall components such as lipopolysaccharide, peptidoglycan and lipopeptides, as well as flagellin and viral nucleic acids.1 The detection of foreign nucleic acid by Toll-like receptors (TLRs) is essential to mounting an innate response to viral infection.2TLRs are the best characterised PRRs and are evolutionarily conserved across a diverse range of species.3 They are homologues of the Drosophila Toll gene first identified as being essential for development and later, anti-fungal and anti-bacterial immunity.4 They are type I transmembrane proteins featuring an extracellular leucine-rich domain and a cytoplasmic tail that contains a conserved Toll/IL-1 receptor domain.5 TLRs are predominantly expressed in tissues involved in immune function as well as those exposed to the external environment such as lung and the skin.6 To date 11 human and 13 murine TLRs have been identified. They are mainly located on the plasma membrane with the exception of TLR3, TLR7, TLR8 and TLR9 which are expressed intracellularly in endosomes and recognise viral components by distinguishing dsRNA, ssRNA and CpG-containing DNA facilitating the recognition of all viral species and inducing the expression of type I interferons (IFNs), and thereby an antiviral state.7TLR7 is the best studied TLR using selective small molecule agonists for antiviral applications.8
Imiquimod (1) (Fig. 1) was initially launched in 1997 for the topical treatment of genital warts resulting from human papillomavirus (HPV) infection, through inducing type I interferons and other cytokines in various cell types, although the precise mechanism of action of the compound was only confirmed to be TLR7 agonism some years later.9Imiquimod is poorly tolerated when administered orally.10 Isatoribine (2) is the most clinically studied oral TLR7 agonist, having entered Phase II studies for the treatment of hepatitis C viral (HCV) infection.11 This study provided proof-of-principle that a systemically dosed TLR7 agonist can produce significant viral load reductions in hepatitis C patients, and offers the prospect of small molecule inducers of IFN replacing the injectable IFN which is the current standard of care for HCV treatment. Prodrugs of isatoribine have been investigated to mitigate the poor oral bioavailability of the compound.12 Dainippon Sumitomo Pharmaceuticals Company Ltd. have investigated a series of 8-hydroxy adenine derivatives which strongly activate the TLR7 receptor.13 SM-276001 (3) is one of a series of disclosed adenine derivatives which demonstrate high potency of interferon induction both in vitro and in vivo following oral dosing to mice.14
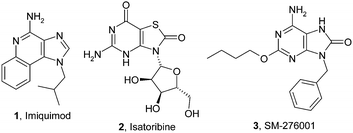 | ||
| Fig. 1 Literature TLR7 agonists | ||
In this paper, we introduce a series of novel, small molecule agonists of the TLR7 receptor intended as IFN-sparing treatment options for the treatment of HCV. Structure–activity relationship studies are presented, from which a lead candidate was identified incorporating many of the desirable features from across the known TLR7 chemical estate, into one molecule.
Design + synthesis strategy
Design targets
The premise of our work in this area was to aim to match the high levels of potency reported for the 8-oxo-purine series in a novel, non-purine structure, to minimise any potential risk of polypharmacology through purinergic or adenosine receptors. Our strategy was to target a range of novel purine analogue templates (Fig. 2), incorporating the equivalent of the 8-oxo functional motif which appeared to instil the potency advantages of 3.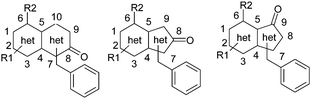 | ||
| Fig. 2 Generalised target structures | ||
There was no structural detail known of the TLR7 receptor to guide designs in this area, and instead we opted for a strategy of diverse exploration of core template structures, orientation of heteroatoms within the core and arrangement of substituents around the generalised target cores depicted below.
Synthetic chemistry
The synthesis of the target compounds was dependent on the exact structure of the template. For example, the synthetic route used to synthesise compound 18 is shown below and illustrates the general synthetic strategy of installing the C-2 and the N-9 substituents separately and then forming the bicyclic template. The commercially available amino-crotonate derivative A was reacted with ethyl malonyl chloride B to give the amidated product C. Cyclisation of C was effected using a strong base, followed by saponification and decarboxylation to give the diol D (Scheme 1).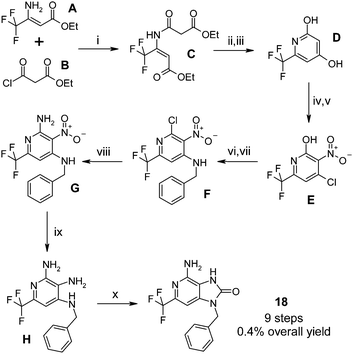 | ||
| Scheme 1 Initial synthesis of compound 18Reagents and conditions: i) Et3N, DCM, rt → 40 °C, 38%; ii) NaH, THF, reflux; iii) c.HCl, reflux, 75% over 2 steps; iv) c.HNO3, AcOH, EtOAc, 60 °C, 66%; v) POCl3, 100 °C, 58%; vi) BnNH2, THF, 50 °C, 58%; vii) POCl3, 100 °C, 70%; viii) 7M NH3 in MeOH, 70 °C, 95%; ix) Fe, AcOH, H2O, rt, 97%; x) CDI, MeCN, 80 °C, 10%, trace regioisomer observed but not isolated. | ||
Regioselective nitration and a further regioselective chlorination of the hydroxyl group at the 4-position gave E. The chloro group of E was displaced with benzylamine and the 6-hydroxyl chlorinated to give F which was then treated with ammonia solution in methanol to give the amino-pyridine G. Nitro group reduction took place very efficiently with iron filings to give the triamine H, which then cyclised smoothly with carbonyl-diimidazole in hot acetonitrile to give 18. This final step did produce a minor quantity of the regioisomeric imidazolone in which cyclisation took place between the 5- and 6-amino groups. A synthetic strategy to overcome this regioisomer issue will be disclosed elsewhere, but in the meantime, pure samples of the desired regioisomer were obtained by simple flash chromatography.
In the case of 18, the overall yield for the synthesis of this target was low over the 9 steps. Improvements in the overall synthetic route to this compound will be reported elsewhere. Syntheses of all other target compounds 4–19 contained in this manuscript are available in the ESI.†
Results
Selection of preferred core template
The initial targets shown in Fig. 3 featured small alkyl or amino groups in the C-2 position of a range of deazapurine targets, and were all compared to the C-2-methyl-8-oxo-adenine analogue 4. For example, 5 and 8 explored the 3-deazapurine and 1-deazapurine system respectively, while compound 6 replaced the N-7 moiety with a methylene group. Target 7 combined the 3-deazapurine design with an extra methyl substituent at the 3 position.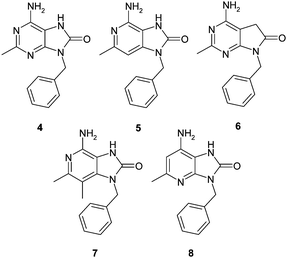 | ||
| Fig. 3 A range of non-purine targets based on the purine 4. | ||
A further set of targets are shown in Fig. 4 in which designs sought to explore template structures further removed from the purine ring system. In all cases the purine ring system was completely changed to a range of very different ring systems in terms of heteroatom electron density, overall dipole and positioning of polar functionality. Through the synthesis of pyrazolo-pyrimidine systems (11 and 13), a pyrazolo-triazine (9), a thiolated purine analogue (10) and a ring-expanded derivative (12), an investigation into the preferred template structure and orientation of polar groups was made.
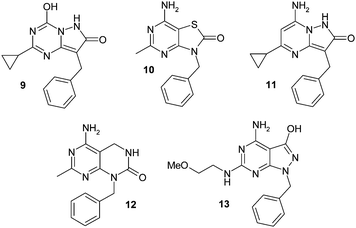 | ||
| Fig. 4 A further range of non-purine analogues of 4. | ||
All compounds were screened for microsomal stability and agonist potency (Table 1). The potency assay consisted of two steps; (a) the incubation of compounds with peripheral blood mononuclear cells (PBMCs) resulting in the secretion of interferon and other cytokines, and (b) the incubation of supernatants from compound-treated PBMCs with a HCV replicon cell line.7 None of the compounds had any effect on HCV replication at the concentrations tested when added directly to HCV replicon cells. All target compounds were low molecular weight (< 300), low-modest lipophilicity (Log D 1.6–2.6) and all had excellent microsomal stability.
| Compound | MWt. | Log Da (cLog P) | HLM b (μL min−1 mg−1) | EC50c (nM) |
|---|---|---|---|---|
| a Partition coefficient measured at pH 7.4 using mixtures of 1-octanol and neutral aqueous buffer. b Human liver microsomal stability intrinsic clearance values using 0.8 mg mL−1protein and 1 μM substrate concentration. c Potency values defined as the concentration of test compound required to be incubated with PBMCs to induce supernatant capable of inhibiting to 50% the replication of a HCV sub-genomic replicon cell-line. | ||||
| 4 | 255 | ND(2.3) | <7 | 829 |
| 5 | 254 | 2.3(2.7) | <7 | 1540 |
| 6 | 254 | 2.0 (1.0) | <7 | >4000 |
| 7 | 268 | 2.6 (3.2) | <7 | 266 |
| 8 | 254 | 2.3 (2.7) | <7 | >4000 |
| 9 | 282 | 1.7 (3.3) | <7 | >4000 |
| 10 | 272 | ND (1.8) | ND | >4000 |
| 11 | 280 | 1.6 (3.2) | <7 | >4000 |
| 12 | 269 | 1.6 (2.1) | 16 | >4000 |
| 13 | 300 | ND (1.8) | ND | >4000 |
Potency was a quite different matter, and the majority of compounds were very weak agonists of the TLR7 receptor, with only the 3-deazapurine compounds 5 and 7 showing EC50 values less than 2μM and similar to that of the benchmark adenine derivative 4.
From the initial tranche of targets made, a decision was made to focus the next round of targets on analogues of the 3-deazapurine core, and all work on further template variations terminated.
C-2/C-6 SAR
Further 3-alkyl substituents related to 7 were not explored due to synthetic complexity in installing diverse groups in this position. Instead, an investigation was made into the C-2 substituent SAR within a 3-deazapurine template related to compound 5 in compounds 14–18 (Fig. 5), with a single compound 19 exploring the deletion of the C-6 amino group.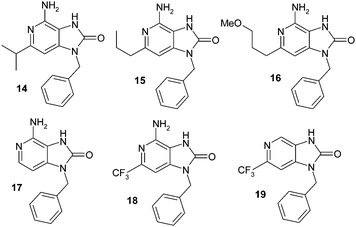 | ||
| Fig. 5 Literature TLR7 agonists. | ||
When this round of compounds was tested in the microsomal stability and potency assays (Table 2), a number of important SAR points emerged. Firstly, a larger branched alkyl substituent at C-2 (viz. 14) and n-alkyl substituent (viz. 15) increased agonist functional activity by some 3–5 fold. Completely deleting the N-6 amino group of 19 or the C-2 grouping as in 17, or extending out from the C-2 position with an alkyl ether group as in 16 was not tolerated. This latter result was interesting in that it offered a clear divergence of SAR compared to that of the adenine series cf.3.13,14
| Compound | MWt. | Log Da(cLog P) | HLMb(μL min−1 mg−1) | EC50c(nM) |
|---|---|---|---|---|
| a Partition coefficient measured at pH 7.4 using mixtures of 1-octanol and neutral aqueous buffer. b Human liver microsomal stability intrinsic clearance values using 0.8 mg mL−1protein and 1 μM substrate concentration. c Potency values defined as the concentration of test compound required to be incubated with PBMCs to induce supernatant capable of inhibiting to 50% the replication of a HCV sub-genomic replicon cell-line. | ||||
| 5 | 254 | 2.3 (2.7) | <7 | 1540 |
| 14 | 282 | 3.1 (3.7) | <7 | 304 |
| 15 | 282 | 3.1 (3.8) | 14 | 480 |
| 16 | 312 | ND (2.5) | ND | >4000 |
| 17 | 240 | 1.9 (2.2) | <7 | >4000 |
| 18 (PF-4171455) | 308 | 3.3 (3.3) | <7 | 102 |
| 19 | 293 | 3.1 (3.6) | <7 | >4000 |
Identification of 18, PF-4171455
Changing the methyl group of 5 for a trifluoromethyl group in 18 gave an unexpected and very pleasing 15 fold jump in potency to give an EC50 of approximately 100 nM. Despite 18 now being more lipophilic (Log D 3.3) than the other targets it maintained excellent stability in human hepatic microsomes.Pharmacokinetics
Based on its very encouraging early in vitro profile, 18 (PF-4171455) was progressed into a rat pharmacokinetic study, the data from which is shown in Table 3. 18 displayed pharmacokinetic properties consistent with its physicochemistry. It demonstrated moderate clearance with respect to liver blood flow in both rat and dog (33 and 6 mL min−1 kg−1 respectively) and modest effective half-life. Incomplete absorption was observed in both preclinical species and was therefore anticipated in man, perhaps as a result of the low aqueous solubility of 18 (approx. 1 μg mL−1), a result of its rigid structure and high melting point (Tm 365 °C) physicochemistry. Various oral formulations were assessed in an attempt to improve systemic exposures in the rat. Solution formulations were compared to suspension and nano-milled suspension (sub-micron particle size) formulations, with all formulations giving similar bioavailabilities in the range 50–70%.| Species | Routea | Clb(mL min−1 kg−1) | Vdssc(L kg−1) | T1/2d(h) | Fe(%) |
|---|---|---|---|---|---|
| a i.v.= intravenous, p.o.= oral. b Clearance. c Volume of distribution at steady state. d Terminal half-life of test compound in hours. e Oral bioavailability. f NC = not calculated. | |||||
| Rat 1mg kg−1 (n = 2) | i.v. | 33 | 2.7 | 1 | N/A |
| Rat 2mg kg−1 (n = 2) | p.o. (solution) | N/A | N/A | N/A | 48 |
| Rat 2mg kg−1 (n = 2) | p.o. (suspens.) | N/A | N/A | N/A | 69 |
| Rat 2mg kg−1 (n = 2) | p.o. (nano suspens.) | N/A | N/A | N/A | 71 |
| Dog 0.1mg kg−1 (n = 2) | i.v. | 6 | 2.3 | 5.5 | NCf |
Compound 18 was relatively highly bound to plasma proteins in rat, dog and human (94, 90, 93% plasma protein binding respectively) and distributed equally into blood and plasma. Based on predictions from allometric scaling of rat and dog parameters, 18 was expected to have an effective human half-life in the range 3 to 7 h, as shown in Table 4.
Selectivity
Compound 18 was shown to be highly selective against a panel of other TLRs (data shown in the ESI†), and highly selective when tested against a broad panel of enzymes, receptors and ion channels.15 The compound exhibited no inhibition or induction of P450 metabolism when tested at relevant concentrations.16Predicted human efficacy
A PK/PD model was constructed based on the observed clinical efficacy of IFNα2a in HCV patients and the measured IFNα2a induced by 18 in the mouse. The relationship between 18 pharmacokinetics and IFN levels was modelled in mouse using an indirect response model with 18 stimulating the production of IFN.17Fig. 6 shows the observed IFN levels versus those predicted using the model. The data points are dispersed around the line of identity indicating that the model described accurately the observed IFN data.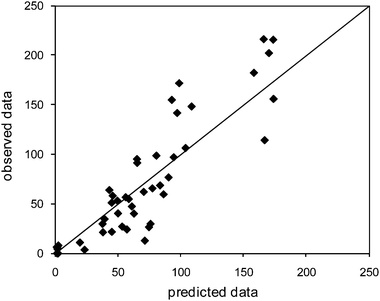 | ||
| Fig. 6 Observed versus predicted data from mouse IFN model fit. | ||
The EC50 (58nM, CV = 21%) from this model was coupled with a viral dynamic model18 and a physiologically based pharmacokinetic model19 to estimate potential efficacious doses in human. 18 was predicted to have comparable efficacy to exogenous IFN when administered to man at doses less than 50mg once daily.20
Conclusions
This paper describes our attempts to identify a potent, non-purine based agonist of the TLR7 receptor. Several designs were proposed and synthesised to explore a diverse range of purine analogues, from which a 3-deazapurine core template was selected for more detailed SAR investigation. From this work, a trifluoromethyl derivative 18 was identified and demonstrated a highly encouraging pharmacokinetic profile. Studies to assess the ability of 18 to induce IFN in the mouse allowed a prediction of human efficacy which was estimated to be competitive with that of exogenous IFNα2a at sub-50mg doses. Further details of our TLR7 agonist programme will be disclosed in due course.Notes and references
- C. A. Janeway and R. Medzhitov, Annu. Rev. Immunol., 2002, 20, 197 CrossRef CAS.
- S. S. Diebold, T. Kaisho, H. Hemmi, S. Akira, E. Reis and C. Sousa, Science, 2004, 303, 1529 CrossRef CAS.
- K. Takeda, T. Kaisho and S. Akira, Annu. Rev. Immunol., 2003, 21, 335 CrossRef CAS.
- B. Lemaitre, E. Nicolas, L. Michaut, J. M. Reichhart and J. A. Hoffmann, Cell, 1996, 86(6), 973 CrossRef CAS.
- J. Choe, M. S. Kelker and I. A. Wilson, Science, 2005, 309, 581 CrossRef CAS.
- T. Nishiya and A. L. DeFranco, J. Biol. Chem., 2004, 279(18), 19008 CrossRef CAS.
- A. Thomas, C. Laxton, J. Rodman, N. Myangar, N. Horscroft and T. Parkinson, Antimicrob. Agents Chemother., 2007, 51(8), 2969 CrossRef CAS.
- M. Czarniecki, J. Med. Chem., 2008, 51(21), 6621 CrossRef CAS.
- H. Hemmi, T. Kaisho and O. Takeuchi, Nat. Immunol., 2002, 3(2), 196 CrossRef CAS.
- N. L. Strominger, R. Brady, G. Gullikson and D. O. Carpenter, Brain Res. Bull., 2001, 55(3), 445 CrossRef CAS.
- D. Averett. SRI Hepatitis conference, March 12th, 2007 Search PubMed.
- S. Fletcher, K. Steffy and D. Averett, Curr. Op. Investig. Dr, 2006, 7(8), 702 Search PubMed.
- K. Hirota, K. Kazaoka, I. Niimoto, H. Kumihara, H. Sajiki, Y. Isobe, H. Takaku, M. Tobe, H. Ogita, T. Ogino, S. Ichii, A. Kurimoto and H. Kawakami, J. Med. Chem., 2002, 45(25), 5419 CrossRef CAS.
- Y. Isobe, K. Ayumu, T. Masanori, H. Kazuki, N. Tomoaki, N. Kei, O. Haruhisa and T. Haruo, J. Med. Chem., 2006, 49(6), 2088 CrossRef CAS.
- Compound 18 was tested against a broad panel of ion channels, receptors, kinases, transporters and other enzymes at CEREP. Weak hits were seen for human acetylcholinesterase (70% inhibition at 10μM) the adenosine A1 receptor (29% inhibition at 10μM) and the adenosine A2a receptor (32% inhibition at 10μM). All other targets assayed showed <15% inhibition.
- Compound 18 showed <15% inhibition when tested at 3μM concentration against CYP1A2, 3A4, 2D6 and 2C9.
- N. Benson, J. de Jongh, J. D. Duckworth, H. M. Jones, H. E. Pertinez, J. K. Rawal, T. J. van Steeg and P. H. Van der Graaf, Antimicrob. Agents Chemother., 2010, 54(3), 1179 CrossRef CAS.
- H. M. Jones, N. Parrott, K. Jorga and T. A. Lave, Clin. Pharmacokinet., 2006, 45(5), 511 CrossRef CAS.
- A. U. Neumann, N. P. Lam, H. Dahari, D. R. Gretch, T. E. Wiley, T. J. Layden and A. S. Perelson, Science, 1998, 282, 103 CrossRef CAS.
- It should be noted that the TLR7 agonism mechanism can in theory induce all IFN subtypes, not just IFNα2a. How this would impact on clinical efficacy is not accommodated in the model.
Footnote |
| † Electronic supplementary information (ESI) available: Synthetic routes for all target compounds 4–19 described in the manuscript. See DOI: 10.1039/c0md00197j |
| This journal is © The Royal Society of Chemistry 2011 |