Following the self assembly of supramolecular MOFs using X-ray crystallography and cryospray mass spectrometry†
Georg
Seeber
a,
Geoffrey J. T.
Cooper
a,
Graham N.
Newton
a,
Mali H.
Rosnes
a,
De-Liang
Long
a,
Benson M.
Kariuki
b,
Paul
Kögerler
c and
Leroy
Cronin
*a
aSchool of Chemistry, The University of Glasgow, University Avenue, Glasgow, UK G12 8QQ. E-mail: L.Cronin@chem.gla.ac.uk; Web: http://www.croninlab.com
bSchool of Chemistry, Cardiff University, Main Building, Park Place, Cardiff, UK CF10 3AT
cInstitut für Anorganische Chemie, RWTH Aachen University, D-52074, Aachen, Germany
First published on 21st May 2010
Abstract
We describe the ligand directed self assembly of two mesoscopic supramolecular MOF architectures based upon the rigid ligand cis,trans-1,3,5-triaminocyclohexane with copper(II) salts. The use of copper fluoride results in the assembly of an unprecedented cubic 3D network with 1 nm void spaces while the use of copper sulfate yields a hexagonal 3D layered network containing nanotubes which are 1.5 nm in diameter. Further, experiments probing the self assembly of the structures after complexation using cryospray mass spectrometry show it is possible to follow the nucleation events that lead to the supramolecular MOFs, and this opens up a new avenue for the direct observation and control of the assembly of complex inorganic architectures.
Introduction
The design of metal organic cages1 and frameworks (MOFs)2 using a structural building unit (SBU) approach has been an unparalleled success3 due to the ability to use well defined geometrical principles to both design and control the assembly of the overall architecture, which is especially effective in the assembly of structures linked by coordinative interactions, and also the post-synthetic modification of MOF systems.4 In contrast, the self assembly of MOFs defined by a combination of coordinative and weaker hydrogen-bonded interactions results in greater intrinsic variability, but at the same time gives unprecedented scope to explore the assembly processes at work.5 Of course, the ability to unravel these ‘solid-state’ design principles is thanks to the tremendous advances in X-ray crystallography and NMR spectroscopy, which have revolutionized our understanding of weak interactions and the ability to design new architectures,6 functional superstructures7 and materials.8These techniques have allowed many of the problems associated with self assembly by design to now be overcome as we have the ability to select or even program intermolecular interactions that take advantage of the ‘self-correcting’ properties of weakly bound and labile architectures.9 In this respect, supramolecular concepts have been used to investigate the assembly of protein sized polyoxometalate clusters,10 soft materials,11 interlinked molecular devices,12 coordination polymers and MOFs with functional cavities,13 and the crystal engineering of optically interesting materials,14 virus models15 and sensors.16 Despite the immense activity in these areas the underlying processes that allow a correlation between the solid state and solution-based self-assembly are still far from being understood and there is a need to link techniques to give a complete picture of the assembly process to allow design concepts to be realized.17
In this work, we aimed to examine the interplay between coordinative and hydrogen-bonded self assembly18 of nanoscale architectures via anion control, using both crystallographic and mass spectrometric techniques. In particular, we hypothesised that it may be possible to directly probe the self assembly of the structures after complexation, in solution using cryospray (temperature controlled) electrospray mass spectrometry (CSI-MS).5 As such we thought it may even be possible to follow the nucleation events that lead to the supramolecular MOFs, and this opens up a new avenue for the direct observation and control of the assembly of complex inorganic architectures.19 We therefore selected cis,trans-1,3,5-triaminocyclohexane (trans-tach) to mediate the assembly of the coordination compounds and self assembly of extended supramolecular architectures; see Fig. 1.
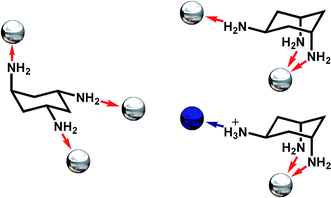 | ||
| Fig. 1 Possible coordination modes of trans-tach in the open (left) and ring-flipped (right) conformations. Coordinate bonds are shown as red arrows, hydrogen bonds as a blue arrow, while the possible positions for coordinated metal ions are represented as silver spheres, and hydrogen-bonded anions as blue spheres. | ||
This is because this triamine, trans-tach, has two distinct sites suitable for both metal ion coordination, and protonation, allowing the formation of polynuclear and extended hydrogen bonded architectures.20 Using this approach we demonstrate here that trans-tach complexed with copper(II) salts, and the acid of those salts, mediates the self assembly of two nanoscale networks of unique structure, through a combination of both coordinative and hydrogen bonded interactions. By adopting this approach we are able to explore the nanoscale structures produced using conventional crystallographic techniques,21 and are also able to follow the initial steps of the organization of the building blocks underpinning the assembly process using cryospray mass spectrometry (CSI-MS).
Results and discussion
These architectures are simply constructed from the complexation of trans-tach with CuX2 followed by control of the pH of the resulting solution with HnXn− (in the case of compounds 1 and 2 X is fluoride and sulfate respectively). For 1, the reaction of trans-tach with copper(II) fluoride at pH 7.7 via addition of hydrofluoric acid results in the formation of cubic crystals of the tetrameric cluster [Cu4O4(trans-tachH)4F4]·8(CH3OH)·20(H2O) (1), giving an unprecedented 3D supramolecular array containing interlinked 3D voids, crystallising in the cubic space group F![[4 with combining macron]](https://www.rsc.org/images/entities/char_0034_0304.gif) 3c with 96 equivalent positions of the asymmetric mononuclear copper(II) units assembling to 24 cubane clusters within the unit cell. Whereas for 2, the reaction of trans-tach with copper(II) sulfate at pH 8.8, via the addition of sulfuric acid, results in the formation of hexagonal crystals of the diligand complex {[Cu(trans-tachH)2(SO4)]SO4}·(MeOH)·4(H2O) (2), giving a unique 3D structure containing 1D nanotube or cylindrical cavities, crystallizing in the trigonal space group P
3c with 96 equivalent positions of the asymmetric mononuclear copper(II) units assembling to 24 cubane clusters within the unit cell. Whereas for 2, the reaction of trans-tach with copper(II) sulfate at pH 8.8, via the addition of sulfuric acid, results in the formation of hexagonal crystals of the diligand complex {[Cu(trans-tachH)2(SO4)]SO4}·(MeOH)·4(H2O) (2), giving a unique 3D structure containing 1D nanotube or cylindrical cavities, crystallizing in the trigonal space group P![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) c1. The topologies of the structures are shown in Fig. 2.
c1. The topologies of the structures are shown in Fig. 2.
![Schematic showing the assembly of 1 (left) and 2 (right) from trans-tach with CuX2 followed by acidification with HnXn−. The resultant cryospray mass spectrum for each of the complexes that form the superstructures are also shown (red line isotopic fit, black line is the actual spectrum) for 1: [(Cu4O4(OH)2(CH3O)(trans-tachH)4)]+ and 2: [Cu(trans-tach)2(SO4)H]+. The middle part shows the topology of the structures (left: grey polyhedra represent the hydrogen-bonded octahedron defined by the pendant amines of the tach which are hydrogen-bonded to the fluoride anions; the blue polyhedra represent the {Cu4O4} units. Right: grey polyhedra correspond to the trigonal prismatic arrangement of the mononuclear tach complexes and the yellow polyhedra correspond to the sulfate counterions). The bottom part shows space filling representations with solvent accessible surfaces of the structures of 1 (left) and 2 (right) showing void spaces of 1.1 and 1.5 nm respectively which are occupied by solvent.](/image/article/2010/SC/c0sc00264j/c0sc00264j-f2.gif) | ||
| Fig. 2 Schematic showing the assembly of 1 (left) and 2 (right) from trans-tach with CuX2 followed by acidification with HnXn−. The resultant cryospray mass spectrum for each of the complexes that form the superstructures are also shown (red line isotopic fit, black line is the actual spectrum) for 1: [(Cu4O4(OH)2(CH3O)(trans-tachH)4)]+ and 2: [Cu(trans-tach)2(SO4)H]+. The middle part shows the topology of the structures (left: grey polyhedra represent the hydrogen-bonded octahedron defined by the pendant amines of the tach which are hydrogen-bonded to the fluoride anions; the blue polyhedra represent the {Cu4O4} units. Right: grey polyhedra correspond to the trigonal prismatic arrangement of the mononuclear tach complexes and the yellow polyhedra correspond to the sulfate counterions). The bottom part shows space filling representations with solvent accessible surfaces of the structures of 1 (left) and 2 (right) showing void spaces of 1.1 and 1.5 nm respectively which are occupied by solvent. | ||
Structural analysis of compound 1 shows that the asymmetric unit of the complex comprises one protonated trans-tach ligand, one CuII centre and one oxo ligand. These assemble into the tetranuclear cubane-type structure 1, in which four trans-tach capped CuII ions and four μ3-bridging oxo ligands are alternately positioned on the vertices of the cube. The cubane structure is slightly distorted, with Cu–O–Cu angles of between 95.7(1) and 98.9(2)°, and O–Cu–O angles of between 80.8(2) and 83.6(1)°. All CuII centers have square-based pyramidal coordination geometries, with Cu–Obasal bond lengths of 1.96(1) and 1.98(1) Å and Cu–N distances of 1.99(1) and 2.01(1) Å. The Cu–Oapical bonding distance is elongated to 2.31(1) Å, resulting in overall distortion of the cube motif. In the crystal packing of 1, each protonated pendant amine ‘tail’ forms hydrogen bonded interactions with three fluoride counterions with N⋯F distances of between 2.65(1) and 2.73(1) Å. Overall, six cubane complexes with twelve pendant amine groups form hydrogen bonded interactions with twelve fluoride counterions. The 24 pendant nitrogen atoms and fluoride counterions are positioned on the vertices of a truncated octahedron, which incarcerates a crystallographically disordered methanol solvent molecule (Scheme 1, left). This templating methanol molecule is disordered over four positions and is stabilized in each by hydrogen bonded interactions with three protonated amine groups of the truncated octahedron with a N⋯O distance of 3.17(1) Å. The truncated octahedra each represent the core of an octahedral junction of cubanes, which leads to the formation of a complex supramolecular architecture with cubic symmetry and channels running along all axes (Scheme 1, right). The accessible voids in these channels are ca. 11 Å in diameter and are occupied by solvent molecules. 1 crystallizes in the highly symmetric cubic space group F3c (219) with 96 equivalent positions of the asymmetric mononuclear copper(II) units assembling to 24 cubane clusters within the unit cell.
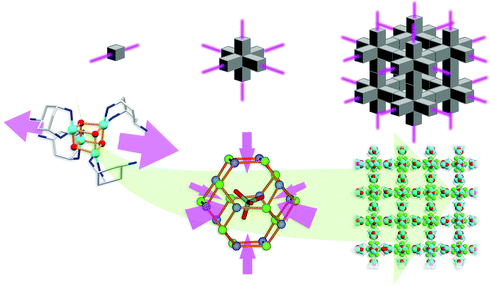 | ||
| Scheme 1 Formation of compound 1 showing the key structural components. Six single cubane units (left) incarcerate a disordered methanol molecule in a truncated octahedral cage of fluoride and nitrogen atoms (center, the orange lines do not represent chemical bonds). Assembly of these octahedral building units in three dimensions leads to the overall MOF architecture (right). Carbon atoms are shown in light grey, copper in sky blue, fluoride in green, nitrogen in dark blue and oxygen in red. Hydrogen atoms are omitted for clarity. | ||
Structural analysis of compound 2 shows that the asymmetric unit contains a diligand complex where the pendant amino residues are protonated. The tetragonal pyramidal coordination sphere of the Cu2+ ion is formed by basal coordination to two bidentate trans-tach ligands and apical coordination to one sulfato ligand (dCu–O = 2.32(1) Å) (Scheme 2, left). Despite the relative simplicity of the building blocks in 1 and 2, the extended structures display great complexity. In the crystal lattice the units of compound 2 arrange in hexagonal layers along the crystallographic c-axis. Hydrogen bonded interactions form a 3D network comprising 1D channels of ca. 15 Å in diameter. The channels are arranged hexagonally with a distance of approximately 28 Å separating the centers (Scheme 2, right). The self-assembly of 2 can best be understood as three complex units arranging in a trigonal fashion. These subunits are held together by hydrogen-bonded interactions between the protonated amino groups, sulfate counterions and methanol solvent molecules. Interestingly, the methanol molecule is rotationally disordered over three positions along the C3 axis located at the centre of the trigonal arrangement. Adjacent layers are rotated by 60° to each other, forming trigonal prismatic units throughout the crystal lattice (Scheme 2, center). These prismatic units are connected via their edges, forming the one-dimensional structure observed within the crystal lattice of 2 (Scheme 2, right). To understand the formation of the MOF architecture, it is informative to abbreviate the three-dimensional prismatic unit as a two-dimensional triangular unit. Edge sharing of each triangular unit through sulfate counterions consequently leads to the formation of the hexagonal structure (Scheme 2, right). It is crucial for the formation of the open structure that these triangular units ‘lack’ corners, resulting in a structure that exhibits holes. In the three-dimensional complex, the prismatic arrangement exhibits ‘rounded corners’ as well as spaces between the shared edges, which allow the formation of one-dimensional channels throughout the crystal lattice.
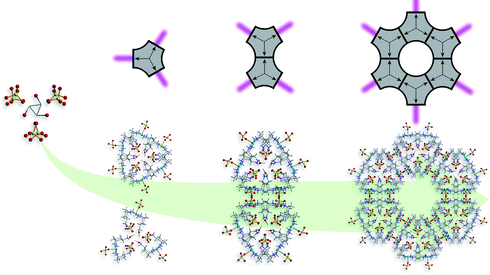 | ||
| Scheme 2 Formation of compound 2 showing the key structural components. Sets of three complex units arrange in a trigonal fashion around disordered methanol molecules and then further arrange into paired trigonal prismatic units (left). These units connect to one another via their edges (center), to give the final MOF architecture (right). Carbon atoms are shown in light grey, copper in sky blue, nitrogen in dark blue, oxygen in red and sulfur in yellow. Hydrogen atoms are omitted for clarity. | ||
Since the sulfate anion has four terminal oxygen atoms, it is able to simultaneously take part in more hydrogen bonded interactions compared to a single fluoride anion. As a result, in compound 2, sulfate is not only interacting with the uncoordinated protonated amino groups of trans-tach ligands, but also with the coordinated amino groups, leading to a considerably more dense packing around the copper centres. In compound 1, fluoride anions only form hydrogen bonded interactions with the uncoordinated protonated amino groups which are bound to one another at the octahedral junctions of the extended structure. In compound 1, the cubane building units are intrinsically linear, due to the syn arrangement of the trans-tach ligands, and in combination with the strong hydrogen-bonding with fluoride, this directs the overall structure towards a three-dimensional architecture.
Given the intricate and complex nature of the structures, especially given the extent of the weak, hydrogen-bonded interactions anchoring the building blocks together, we opted to examine the self assembly/disassembly process in solution using electrospray mass spectrometry. Initial studies using conventional electrospray mass spectrometry at a dissolvation temperature of ca. 453 K using both the mother liquor and the dissolved crystals showed a very complex set of spectra indicating only low nuclearity/monomeric with a large number of species. However, given the nature of the weak interactions we also did experiments at ca. 233 K using a cryospray source (this replaces the nebulizer and dry gas with cold gas). Similar to the high temperature experiments, the experiments on the dissolved crystals again gave complex spectra showing only low nuclearity. However, experiments using the solutions of 1 and 2 before crystallisation gave some clearly defined spectra which were amenable to quantitative isotopic fitting.
Analysis of 1 showed that the intact cubane fragments, capped by trans-tach, were present in the solution/gas phase (see ESI†). The CSI-MS spectrum can be fully assigned by m/z value and isotopic distribution to identify all major peaks. Importantly, the cubane found in the core of 1 with the addition of two hydroxo and one methoxo group, [Cu4O4(OH)2(OMe)(trans-tachH)4]+ at 903.2 m/z, can be unambiguously assigned (see Fig. 2 for the spectrum and fitting), which certainly indicates the possibility of the formation of the cubane in solution as a pivotal building block defining the assembly of compound 1. Further, we can observe cubane ‘nucleation’, without a full shell of trans-tach ligands, but only at the correct pH (as for the synthesis of 1), implying that the formation of the cubane, followed by the ligation of the tach ligand, results in the assembly of the cubane-based MOF, 1, see Fig. 3 (and Table S1 in the ESI†). By means of comparison the CSI-MS measurements of the mother liquor of 2 show a highly speciated, but well defined spectrum, with the {Cu1L2} core of 2 present at 418.1 m/z and fitted to the predicted spectrum with the formula [Cu(trans-tach)2SO4H]+ (Fig. 2). At higher m/z values larger species were identified with all corresponding to aggregations of simple monometallic units (see ESI†). It is interesting from the MS data that the only species which is seen to have a Cu![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :ligand ratio of 1:2 is the peak at 418.1, the peak that corresponds to the crystallographic structural subunit. The other peaks show a consistent aggregation of what appear to be (in general) [Cu(trans-tach)(SO4)] units up to a Cu7 species. Indeed, further aggregation can also be seen, but low intensity begins to prevent accurate fitting of species larger than Cu8. These data suggest that there may be (at least) two different clustering events occurring in the solution/gas phase: the simple aggregation of [Cu(trans-tach)SO4] units and the formation of the diligand complex which ultimately crystallises as 2, but despite the lability of the system higher nuclearity architectures are observed. The CSI-MS spectra for compounds 1 and 2, along with some structural assignments are shown in Fig. 3.
:ligand ratio of 1:2 is the peak at 418.1, the peak that corresponds to the crystallographic structural subunit. The other peaks show a consistent aggregation of what appear to be (in general) [Cu(trans-tach)(SO4)] units up to a Cu7 species. Indeed, further aggregation can also be seen, but low intensity begins to prevent accurate fitting of species larger than Cu8. These data suggest that there may be (at least) two different clustering events occurring in the solution/gas phase: the simple aggregation of [Cu(trans-tach)SO4] units and the formation of the diligand complex which ultimately crystallises as 2, but despite the lability of the system higher nuclearity architectures are observed. The CSI-MS spectra for compounds 1 and 2, along with some structural assignments are shown in Fig. 3.
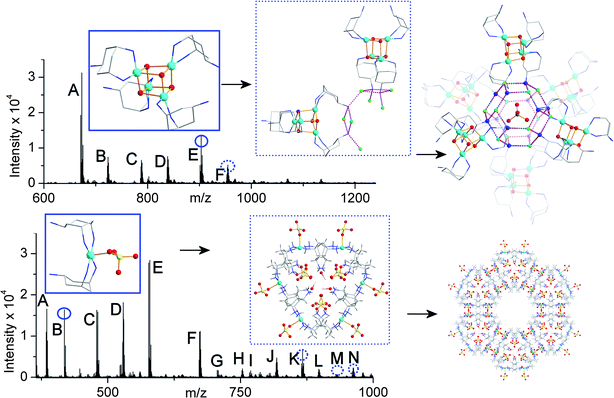 | ||
| Fig. 3 CSI-MS of the reaction solutions of compounds 1 (top) and 2 (bottom) before crystallization with segments of the crystal structures of compounds determined after crystallisation. The peaks labeled with letters have been fully assigned, see the ESI;† the species in the blue boxes (top – {Cu4}; bottom {Cu1}) are directly observed in the spectra (round circles) and the species in the dotted blue boxes (top – {Cu8 + Cu1}; bottom {Cu6}) are observed as more complex derivatives (dotted circles). Carbon atoms are shown in light grey, copper in sky blue, nitrogen in dark blue, oxygen in red, fluoride in green, sulfur in yellow. Hydrogen atoms are omitted for clarity. | ||
Overall we can tentatively suggest that CSI-MS has the potential to directly probe/give mechanistic information underpinning the assembly of complex solid state architectures such as MOFs. This is because in this work we are able to directly observe increasingly complex species in the CSI-MS experiments, which can be correlated with structural motifs seen as potential building blocks found in the structures of 1 and 2. Although it is very hard to use the CSI-MS experiments to directly prove aspects of the assembly/crystallisation processes at this point, it is worth bearing in mind that we were only able to observe ‘complex’ building blocks when conducting CSI-MS experiments directly on the reaction solution before crystallisation (experiments on the dissolved crystals did not result in any high observation of any high nuclearity species). In addition we conducted control studies as a function of pH, time, and metal:ligand ratios that showed that only those systems that closely mirrored the actual crystallisation conditions gave spectra where high nuclearity species could be clearly identified and correlated with the structural studies.
Conclusions
In conclusion, we have shown how the rigid ligand, trans-tach, can be used to mediate the formation of two gigantic supramolecular-MOF architectures and the topology of the resulting architectures can be radically altered as a function of the anion, going from a cubic architecture for compound 1 with fluoride to a trigonal architecture with compound 2 when sulfate is employed instead. Both structures have been extensively investigated using X-ray crystallography and we have successfully employed cryospray mass spectrometry to probe the self assembly of such MOF architectures for the first time. These studies showed that, only under conditions that closely mirror the crystallisation conditions, are complex and high nuclearity species that could be related to some of the structural building blocks found in the crystal structures directly observed. Furthermore, we showed that time, temperature, and pH control are critical parameters for the CSI-MS studies. These results are significant since this study paves the way for a more general approach, using CSI-MS, to be applied to investigations of the assembly and disassembly of MOF architectures. We are currently developing this technique to probe more traditional MOFs linked together exclusively by coordinative interactions.Experimental section
Synthesis
1: Caution: the toxic hydrofluoric acid (HF) must be treated with great care. All reactions with HF were conducted in plastic vessels. Copper(II) fluoride hydrate (29 mg, 0.29 mmol) was added to a solution of trans-tach (56 mg, 0.44 mmol) in methanol (20 ml) previously adjusted to pH 7.7 using 2 M hydrofluoric acid. The solution gradually became violet upon stirring as the copper(II) fluoride dissolved. Continued stirring overnight and reducing the volume to 4 ml resulted in an intense dark violet solution. Filtration through cotton wool and crystallisation by diffusion of ether yielded 1 as royal blue cubic crystals embedded in a white ‘crusty’ amorphous material. Isolation of the crystalline material was achieved by continuous cleansing of the combined crystallization samples by ultrasonication in ether after 2–3 days. Removal of the ether suspension resulted in royal blue crystalline material. Yield 28 mg (0.03 mmol, 39%). IR (Golden Gate) ν/cm−1 3071(s), 2932(s), 2758(s), 2530(m), 1639(m), 1593(s), 1574(s), 1462(w), 1389(w), 1207(m), 1150(w), 1111(w), 1045(w), 926(s), 876(s), 698(vs). Elemental analysis for C32H138Cu4F4N12O33, found (expected) %: C 28.65 (28.74), H 6.56 (6.43), N 16.62 (16.76). TGA analysis shows a mass reduction of 34.5% upon heating to 170 °C.2: A methanolic solution (3 ml) of copper(II) sulfate hexahydrate (90 mg, 0.36 mmol) was added to a solution of trans-tach (87 mg, 0.68 mmol) in methanol (30 ml) giving an instant colour change from colourless to dark violet. The pH of the solution was adjusted to pH 8.8 with three drops of 2 M sulfuric acid. After each drop, a precipitate was formed that redissolved for the first two drops but remained as a suspension after the third. The reaction mixture was stirred for 1.5 h, the violet precipitate filtered off and the volume reduced to ca. 2 ml. Filtration through cotton wool and microfibre filter paper was repeated until the filtrate ran clear. Crystallisation by diffusion of ether resulted in violet crystals of 2 suitable for single crystal X-ray analysis over the period of 3–4 days. Yield 37 mg (0.07 mmol, 21%). IR (Golden Gate) ν/cm−1 3212(m), 3112(m), 2916(m), 1593(s), 1456(w), 1367(m), 1178(m), 1049(bvs), 920(s), 764(w), 711(bw). Elemental analysis for C13H44CuN6O13S2, found (expected) %: C 27.64 (25.18), H 6.76 (7.15), N 14.76 (13.55).
X-Ray crystallography
Crystal data for 1: C32H138Cu4F4N12O33, M = 1549.70 g mol−1, cubic, space group F3c, a = 37.295(4) Å, V = 51872(10) Å3, Z = 24, μ(Cu-Kα) = 1.802 mm−1, 32868 reflections measured, 3022 unique that were used in all calculations. Final R1 = 0.0526 and wR2 = 0.1625 (all data).
Crystal data for 2: C13H44N6CuO13S2, M = 620.20 g mol−1, trigonal, space group Pc1, a = 28.3688(3) Å, c = 12.3891(2) Å, V = 8634.80(19) Å3, Z = 12, μ(Cu-Kα) = 3.001 mm−1, 29031 reflections measured, 4151 unique that were used in all calculations. Final R1 = 0.0909 and wR2 = 0.2694 (all data). Data were measured at 150(2) K on a Bruker AXS Smart 6000 diffractometer equipped with a copper rotating anode source (Cu-Kα= 1.54178 Å) and an Oxford Cryostream Cooler. Structure solution with SHELXS97 and refinement with SHELXL97 using WinGX.21
Cryospray mass spectrometric (CSI-MS) measurements on reaction solutions that crystallise compounds 1 and 2
Electrospray mass spectrometry measurements were conducted using the cryospray attachment at 233 K. Aliquots of the reaction solution were injected into the device at 180 μl h−1. The mass spectrometer used for the measurements was a Bruker microTOFQ and the data were collected in positive ion mode. The spectrometer was previously calibrated with the standard tune mix to give a precision of ca. 1.5 ppm in the region of 500–5000 m/z. The end plate voltage was set to −500 V and the capillary to −4500 V. The collision cell was set to a collision energy of −9.0 eV z−1 with a gas flow rate at 25% of the maximum and the cell RF was set at 1600 Vpp.Acknowledgements
We wish to thank the EPSRC, The Universities of Birmingham and Glasgow, WestCHEM, the Leverhulme Trust and the Royal Society for Funding. LC thanks the Royal Society/Wolfson Foundation for a merit award. We would like to acknowledge Narrinder Kaur for assistance in the characterisation of compound 2.Notes and references
- S. Sato, J. Iida, K. Sukuki, M. Kawano, T. Ozeki and M. Fujita, Science, 2006, 313, 1273 CrossRef CAS; P. Mal, B. Breiner, K. Rissanen and J. R. Nitschke, Science, 2009, 324, 1697 CrossRef CAS.
- O. M. Yaghi, M. O'Keeffe, N. W. Ockwig, H. K. Chae, M. Eddaoudi and J. Kim, Nature, 2003, 423, 705 CrossRef CAS; P. H. Dinolfo and J. T. Hupp, Chem. Mater., 2001, 13, 3113 CrossRef CAS.
- M. Eddaoudi, J. Kim, N. Rosi, D. Vodak, J. Wachter, M. O'Keeffe and O. M. Yaghi, Science, 2002, 295, 469 CrossRef; M. O'Keeffe, M. Eddaoudi, Hailian Li, T. Reineke and O. M. Yaghi, J. Solid State Chem., 2000, 152, 3 CrossRef CAS.
- Z. Wang and S. M. Cohen, J. Am. Chem. Soc., 2007, 129, 12368 CrossRef CAS; Y.-F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4635 CrossRef CAS.
- S. Kitagawa, R. Kitaura and S. Noro, Angew. Chem., Int. Ed., 2004, 43, 2334 CrossRef CAS.
- J. J. Pak, J. Greaves, D. J. McCord and K. J. Shea, Organometallics, 2002, 21, 3552 CrossRef CAS.
- C. P. Pradeep, D.-L. Long, G. N. Newton, Y.-F. Song and L. Cronin, Angew. Chem., Int. Ed., 2008, 47, 4388 CrossRef CAS.
- M. Kölbel and F. M. Menger, Langmuir, 2001, 17, 4490 CrossRef.
- A. M. Brouwer, C. Frochot, F. G. Gatti, D. A. Leigh, L. Mottier, F. Paolucci, S. Roffia and G. W. H. Wurpel, Science, 2001, 291, 2124 CrossRef CAS.
- G. W. Orr, L. J. Barbour and J. L. Atwood, Science, 1999, 285, 1049 CrossRef.
- C. Janiak, Dalton Trans., 2003, 2781 RSC.
- J. L. Atwood, L. J. Barbour, S. J. Dalgarno, M. J. Hardie, C. L. Raston and H. R. Webb, J. Am. Chem. Soc., 2004, 126, 13170 CrossRef CAS.
- P. D. Beer and P. A. Gale, Angew. Chem., Int. Ed., 2001, 40, 486 CrossRef.
- H. N. Miras, D.-L. Long, P. Kögerler and L. Cronin, Dalton Trans., 2008, 214 RSC.
- S. Leininger, B. Olenyuk and P. J. Stang, Chem. Rev., 2000, 100, 853 CrossRef CAS.
- E. F. Wilson, H. Abbas, B. J. Duncombe, C. Streb, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2008, 130, 13876 CrossRef CAS.
- H. N. Miras, E. F. Wilson and L. Cronin, Chem. Commun., 2009, 1297 RSC.
- Y. Aoyama, K. Endo, T. Anzai, Y. Yamaguchi, T. Sawaki, K. Kobayashi, N. Kanehisa, H. Hashimoto, Y. Kai and H. Masuda, J. Am. Chem. Soc., 1996, 118, 5562 CrossRef CAS.
- E. Kharlampieva, V. Kozlovskaya, J. Tyutina and S. A. Sukhishvili, Macromolecules, 2005, 38, 10523 CrossRef CAS.
- G. N. Newton, G. J. T. Cooper, P. Kögerler, D.-L. Long and L. Cronin, J. Am. Chem. Soc., 2008, 130, 790 CrossRef CAS; G. N. Newton, G. J. T. Cooper, D.-L. Long, P. Kögerler and L. Cronin, J. Mol. Struct., 2006, 796(1–3), 23 CrossRef CAS; G. J. T. Cooper, G. N. Newton, P. Kögerler, D.-L. Long, L. Engelhardt, M. Luban and L. Cronin, Angew. Chem., Int. Ed., 2007, 46, 1340 CrossRef CAS; G. Seeber, P. Kögerler, B. M. Kariuki and L. Cronin, Chem. Commun., 2004, 1580 RSC; G. Seeber, D.-L. Long, B. M. Kariuki and L. Cronin, Dalton Trans., 2003, 4498 RSC.
- L. J. Farrugia, J. Appl. Crystallogr., 1999, 32, 837 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available: Structural analysis and mass spectrometric measurements of 1 and 2. CCDC reference numbers 772923 and 772924. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/c0sc00264j |
| This journal is © The Royal Society of Chemistry 2010 |