Synthesis and biological evaluation of phosphatidylinositol phosphate affinity probes†
Stuart J.
Conway
a,
James
Gardiner
*b,
Simon J. A.
Grove
a,
Melloney K.
Johns
a,
Ze-Yi
Lim
a,
Gavin F.
Painter
a,
Diane E. J. E.
Robinson
*b,
Christine
Schieber
b,
Jan W.
Thuring
a,
Leon S.-M.
Wong
b,
Meng-Xin
Yin
b,
Antony W.
Burgess
c,
Bruno
Catimel
c,
Phillip T.
Hawkins
d,
Nicholas T.
Ktistakis
d,
Leonard R.
Stephens
d and
Andrew B.
Holmes
*b
aDepartment of Chemistry, University of Cambridge, Lensfield Road, Cambridge, CB2 1EW, UK
bSchool of Chemistry, Bio21 Institute, University of Melbourne, Building 102, 30 Flemington Road, Parkville, Victoria 3010, Australia
cLudwig Institute for Cancer Research, Melbourne Tumour Biology Branch, Royal Melbourne Hospital, Parkville, Victoria 3052, Australia
dBabraham Institute, Department of Signalling, Inositide Laboratory, Cambridge, CB2 4AT, UK
First published on 9th November 2009
Abstract
The synthesis of the complete family of phosphatidylinositol phosphate analogues (PIPs) from five key core intermediates A–E is described. These core compounds were obtained from myo-inositol orthoformate 1via regioselective DIBAL-H and trimethylaluminium-mediated cleavages and a resolution–protection process using camphor acetals 10. Coupling of cores A–E with phosphoramidites 34 and 38, derived from the requisite protected lipid side chains, afforded the fully-protected PIPs. Removal of the remaining protecting groups was achieved via hydrogenolysis using palladium black or palladium hydroxide on carbon in the presence of sodium bicarbonate to afford the complete family of dipalmitoyl- and amino-PIP analogues 42, 45, 50, 51, 58, 59, 67, 68, 76, 77, 82, 83, 92, 93, 99 and 100. Investigations using affinity probes incorporating these compounds have identified novel proteins involved in the PI3K intracellular signalling network and have allowed a comprehensive proteomic analysis of phosphoinositide interacting proteins.
Introduction
Phosphatidylinositol phosphates (PtdInsPs or PIPs) are an important class of membrane phospholipids.1,2 These lipids are involved in intracellular signalling mechanisms that are known to play a central role in fundamental cellular functions such as vesicle trafficking, apoptosis, cell proliferation and metabolism.3 Furthermore, lack of regulation of these pathways is known to result in a range of disease conditions, such as cancer, diabetes, Alzheimer's disease and autoimmune disorders.4PIPs are phosphorylated derivatives of myo-inositol. There are eight known naturally occurring classes of PIPs differing only in the degree of phosphorylation of the hydroxyl groups at the 3-, 4- and 5-positions of the inositol rings (Fig. 1). The natural materials carry a range of unsaturated fatty acid derivatives at the sn-1 and sn-2 positions. The nature and degree of saturation of the lipid chain is organism dependent. In this work we focus on saturated analogues and largely on palmitoyl derivatives, which, in our experience, have shown no marked influence on the binding properties of proteins to the synthetic PIPs. To understand the roles of each of these PIPs in their signalling pathways and their effect on downstream processes, it is necessary to identify proteins that exhibit binding specificity. However, the inability to isolate practically useful amounts of these PIP compounds from cells has led to a need for efficient syntheses.5-25 Furthermore, analogues of these compounds that can be used as affinity probes to investigate and isolate particular binding proteins, have become attractive targets1,5,7,12,13,24,26–28 Our approach has been the incorporation of an ω-amino group on the sn-1 position of a saturated lipid side chain (abbreviated as NH2-PIP) to facilitate immobilisation of the PIP by covalent linkage through an amide bond onto Affi-Gel® 10 beads.12,28
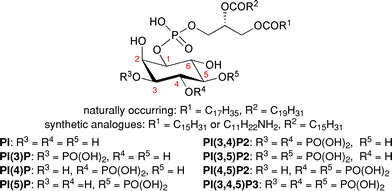 | ||
| Fig. 1 General structure of phosphatidylinositol phosphates. | ||
Herein, we describe our contribution over the last two decades to the synthesis of the complete family of PIP analogues, which we have used as probes for biological systems.28-35 We have previously reported the synthesis of PI(3,4,5)P3 and PI(4,5)P2 affinity beads.12,16
Synthetic approach
Our synthetic approach is simple and divergent and involves the late-stage coupling of phosphoramidites, derived from the requisite protected lipid side chains, to the suitably protected phosphorylated inositol cores followed by a global deprotection. All eight PIPs and their analogues can be synthesised from five inositol core compounds (A–E, Scheme 1).36 Each of these intermediates can in turn be synthesised from the readily available myo-inositol orthoformate 137 by judicial choice of protection, resolution and deprotection strategies. It should be noted that cores D and E can also be obtained from core C by suitable protecting group manipulation; furthermore, in principle all the PIPs can be obtained from core C, though this is not the most efficient route. This report focuses primarily on the most efficient routes to the target compounds.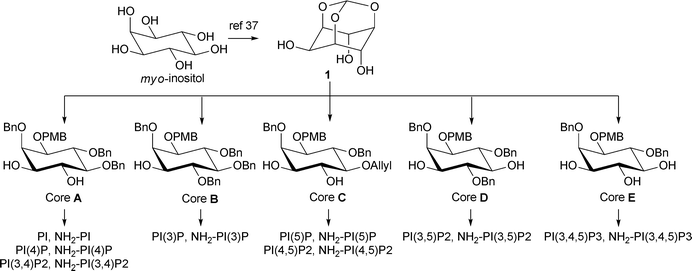 | ||
| Scheme 1 Core building blocks for the synthesis of all the PIPs. | ||
Synthesis of cores A–E
The syntheses of core molecules A–E are shown in Scheme 2 and have previously been reported.12,16,38,39 To gain access to cores A, C and E, selective mono PMB-protection of 1 followed by benzylation of the remaining hydroxyl groups provides orthoformate 2. Selective cleavage of the orthoformate by treatment with DIBAL-H affords 1,3-acetal 3. Benzylation or allylation and subsequent removal of the acetal and PMB protecting groups give the racemic triols 6 and 7. Resolution of the racemic triols 6 and 7 with simultaneous protection of the corresponding vicinal diols according to an elegant procedure previously reported by Bruzik40,41 afforded an efficient approach to the unnatural (1L-series) and natural (1D-series) of PIPs. Thus, treatment of triol 7 with the camphor acetal (1R)-10 afforded a mixture of four diastereomers of 3,4-acetal 8, one of which could be separated by column chromatography from the mixture of the other three; deprotection of the acetal allows preparation of the unnatural enantiomer of the triol 7 corresponding to the 1L-series (for the use of this enantiomer see Scheme 12). The remaining mixture is therefore enriched in the desired enantiomer, which when protected with dimethylacetal (1S)-10 provides diastereomer 8 with the correct absolute configuration corresponding to the natural 1D-series. It should be noted that direct resolution/protection of 6 or 7 with dimethylacetal (1S)-10 also provides the required diastereomer of 8 in comparable yields. This route also allows separation of the two enantiomers of the inositol ring and provides access to the unnatural enantiomers of the PIPs.21 Protection of the 1-position of the inositol ring of 8 as the p-methoxybenzyl ether, followed by deprotection of either the 3,4 and/or 5-positions afforded cores A, C and E.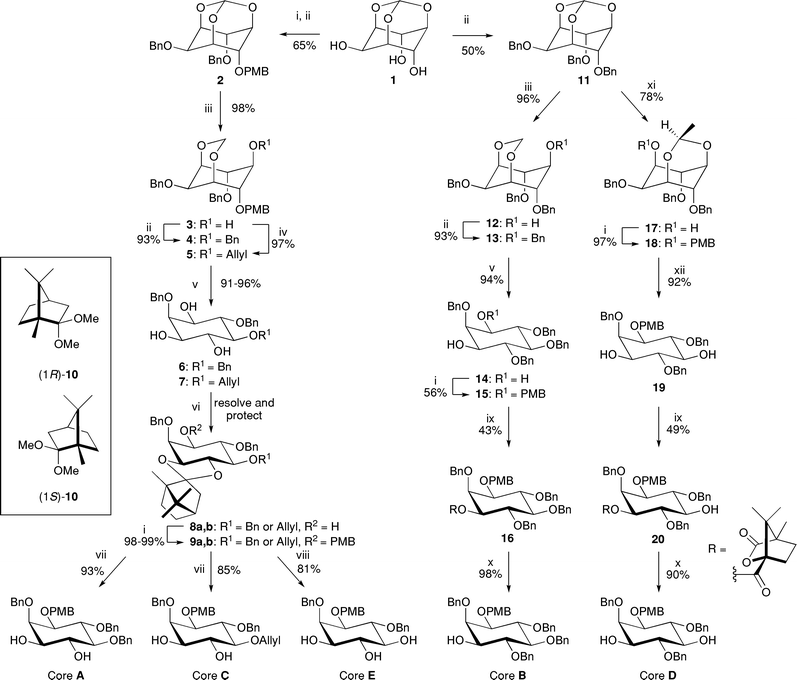 | ||
| Scheme 2 Synthesis of the core building blocks A–E. Reagents and conditions: i. NaH, PMBCl, DMF, 0 °C → rt; ii. NaH, BnBr, DMF, 0 °C → rt; iii. DIBAL-H (2.5 eq.), CH2Cl2/hexanes, 0 °C → rt; iv. NaH, allyl bromide, DMF, 0 °C → rt; v. HCl, MeOH, reflux; vi. (1R)-10, TsOH·H2O (cat.), CH2Cl2, reflux, separate, AcCl, CH2Cl2–MeOH (2/1 v/v), then (1S)-10, TsOH·H2O (cat.), CH2Cl2, reflux; vii. AcCl, CH2Cl2–MeOH (2/1 v/v), viii. From 9b: (Ph3P)3RhCl, DABCO, EtOH–toluene/H2O (7/3/1 v/v/v), reflux, then AcCl, CH2Cl2–MeOH (2/1 v/v); ix. (1S)-(−)-camphanic chloride, Et3N or pyridine, CH2Cl2, rt; x. LiOH, THF–H2O (10/1 v/v), rt; xi. Me3Al (2.5 eq.), CH2Cl2/hexanes, 0 °C → rt; xii. HCl, CH2Cl2–MeOH (2/1 v/v), rt. | ||
The synthesis of cores B and D begins with the complete benzylation of myo-inositol orthoformate 1 to form benzyl ether 11. The differentially protected inositol acetals 12 and 17 can be obtained via selective Lewis acid-mediated reductive or alkylative cleavage of trisbenzyl orthoformate 11.39,42 As seen in the syntheses of cores A, C and E, the use of DIBAL-H selectively afforded the 1,3-acetal, 12. In contrast, the use of trimethyl aluminium resulted in the 3,5-acetal, 17. Protection of the free hydroxyl groups in 12 and 17 followed by removal of the acetal groups gave diols 14 and 19 respectively. Subsequent mono-protection of the 1-position of 14 gave racemic p-methoxybenzyl ether 15. Racemic alcohols 15 and 19 were then resolved as their camphanate esters. Subsequent hydrolysis of esters 16 and 20 afforded enantiomerically pure cores B and D respectively.
The stereoselectivity of these Lewis acid-mediated reductive and alkylative cleavages of orthoformate 11 have been investigated and the mechanisms proposed (Scheme 3).42 In the DIBAL-H reduction of 2 to 3, and 11 to 12, it was determined that at least 2 molar equivalents of DIBAL-H are required for the reaction to proceed to completion. It is proposed that the first equivalent of DIBAL-H acts as a Lewis acid, coordinating to the C-5 oxygen, presumably the most sterically accessible oxygen. Subsequent cleavage of the orthoformate affords the oxocarbenium ion 22, the unfavourable 1,3-steric interactions in which can be accommodated by a ring flip to the boat conformation 23. Reduction of this oxocarbenium ion by a second equivalent of DIBAL-H from the less hindered face produces the 1,3-acetal 12 exclusively.
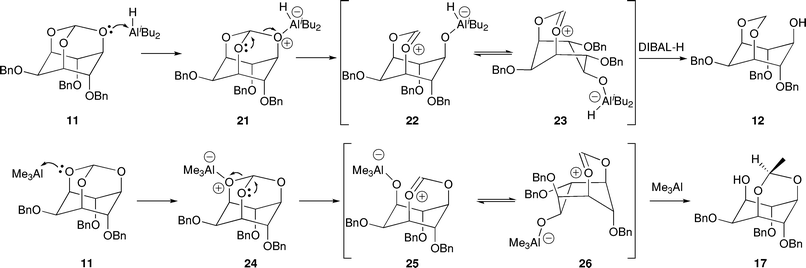 | ||
| Scheme 3 Proposed mechanisms of the Lewis acid-mediated regioselective cleavage of orthoformate 1142 | ||
The stereoselectivity of the hydride delivery to the orthoformate was established through two deuterium-labelling experiments (Scheme 4). Firstly, reduction of orthoformate 11 using 2.5 molar equivalents of diisobutylaluminium deuteride43 (DIBAL-D) resulted in acetals 28 and 29 in a 92![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :8 ratio. The reciprocal reaction in which deuterated orthoformate 2744 was treated with diisobutylaluminium hydride (DIBAL-H), afforded the two products with an inverted selectivity of 10:90 (28:29). The structures were confirmed by nOe experiments. This switch in selectivity between the two diastereomers 28 and 29, indicates that the reductive cleavage is stereoselective in the delivery of the hydride (or deuteride) occurring preferentially on the endo face, syn to the broken C–O bond. This retention of stereochemistry discounts a mechanism pathway in which the orthoformate in 21 (Scheme 3) is cleaved via inter- or intramolecular delivery of the hydride. Whilst these experiments do not allow the distinction between intramolecular delivery of the hydride (or deuteride) in 22 and intermolecular delivery in 23, the latter is a more feasible pathway when the unfavourable 1,3-steric interactions of 22 are considered.
:8 ratio. The reciprocal reaction in which deuterated orthoformate 2744 was treated with diisobutylaluminium hydride (DIBAL-H), afforded the two products with an inverted selectivity of 10:90 (28:29). The structures were confirmed by nOe experiments. This switch in selectivity between the two diastereomers 28 and 29, indicates that the reductive cleavage is stereoselective in the delivery of the hydride (or deuteride) occurring preferentially on the endo face, syn to the broken C–O bond. This retention of stereochemistry discounts a mechanism pathway in which the orthoformate in 21 (Scheme 3) is cleaved via inter- or intramolecular delivery of the hydride. Whilst these experiments do not allow the distinction between intramolecular delivery of the hydride (or deuteride) in 22 and intermolecular delivery in 23, the latter is a more feasible pathway when the unfavourable 1,3-steric interactions of 22 are considered.
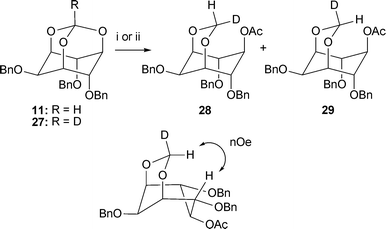 | ||
| Scheme 4 Deuterium labelling experiments for the cleavage of the orthoformates 11 and 27. Reagents and conditions: i. From 11: DIBAL-D (2.5 eq.), CH2Cl2, 0 °C → rt, then Ac2O, Et3N, DMAP, CH2Cl2, rt, 83%, 28:29: 92:8; ii. From 27: DIBAL-H (2.5 eq.), CH2Cl2, 0 °C → rt, then Ac2O, Et3N, DMAP, CH2Cl2, rt, 93%, 28:29: 10:90. | ||
In contrast, the trimethylaluminium-mediated cleavage of the orthoformate 11 exclusively affords the acetal 17, which results from the cleavage of one of two identical alternative C–O bonds in the orthoformate 24. This outcome indicates that trimethylaluminium is presumably sufficiently small to coordinate to one or both of the equivalent oxygens at the 1- or 3-positions (perhaps through intramolecular delivery by the equatorial benzyloxy group), allowing an alternative cleavage pathway resulting in the oxocarbenium ion 25. Delivery of a methyl group to the exo face of the oxocarbenium 26 affords the 3,5-acetal 17.
Synthesis of phosphoramidites
To install the lipid side chains, the phosphoramidites 3445 and 3812 were synthesised from (+)-1,2-O-isopropylidene-glycerol 30 (Scheme 5 and Scheme 6). The dipalmitoyl derivative 32 was obtained by esterification of the diol 31, which resulted from PMB protection of the free alcohol in 30 followed by methanolysis of the acetal (Scheme 5). Subsequent removal of the PMB group followed by a 1H-tetrazole mediated coupling of the alcohol 33 with (benzyloxy)bis(N,N-diisopropylamino)phosphine46 afforded the phosphoramidite 34, which is used for the synthesis of the dipalmitoyl PIP analogues.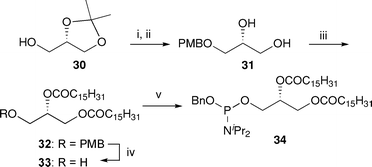 | ||
| Scheme 5 Synthesis of dipalmitoyl phosphoramidite 34. Reagents and conditions: i. NaH, PMBCl, DMF, 0 °C → rt, quant.; ii. p-TsOH, MeOH, reflux, 82%; iii. C15H31COCl, pyr., DMAP, CH2Cl2, 0 °C → rt, 88%; iv. DDQ, CH2Cl2–H2O (16/1, v/v), rt, 85%; v. (BnO)P(NiPr2)2, 1H-tetrazole, CH2Cl2, rt, 92%. | ||
In order to prepare the PIP derivatives that can be attached to beads for use in protein affinity experiments, side chains containing an ω-amino group at the sn-1-position were also synthesised (Scheme 6). A DCC-mediated coupling of diol 31 allowed selective esterification of the primary alcohol to afford the alcohol 35 in good yield. Esterification of the free alcohol with palmitoyl chloride and removal of the PMB group provided the primary alcohol 37 which when coupled with (benzyloxy)bis(N,N-diisopropylamino) phosphine gave phosphoramidite 38, providing access to the NH2-PIP analogues.
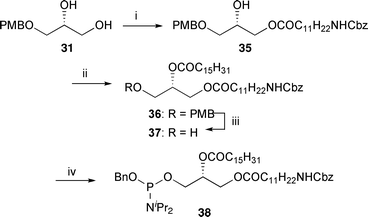 | ||
| Scheme 6 Synthesis of amino phosphoramidite 38. Reagents and conditions: i. CbzNHC11H22CO2H, DCC, DMAP, CH2Cl2, 0 °C → rt, 87%; ii. C15H31COCl, pyr., DMAP, CH2Cl2, 0 °C → rt, quant.; iii. DDQ, CH2Cl2–H2O (16/1, v/v), rt, 80%; iv. (BnO)P(NiPr2)2, 1H-tetrazole, CH2Cl2, rt, 92%. | ||
With all the necessary building blocks in hand, the whole family of the PIP analogues could be synthesised.
Synthesis of PI,7,8,10 PI(3)P,7,9,10,14–1619,23 PI(4)P10,19 and PI(5)P5,10,14,18 analogues
The simplest member of this family of compounds is phosphatidylinositol (PI), which contains the phospholipid side chain at the 1-position and none of the 3-, 4- or 5-positions are phosphorylated (Fig. 1).The inositol moiety of PI (40) was obtained from core A by benzylation of the 3- and 4-hydroxyl groups and removal of the PMB protecting group from the 1-position (Scheme 7). Coupling of the alcohol 40 with the amino-phosphoramidite 38 in the presence of an excess of 1H-tetrazole, followed by mCPBA oxidation of the intermediate phosphite (not isolated) provides the fully protected phosphate 41. A global deprotection via hydrogenolysis in the presence of palladium black and sodium hydrogen carbonate16,24 gave NH2-PI sodium salt 42,47 which was coupled to NHS-activated Affi-Gel® 10 beads in the presence of sodium hydrogen carbonate to give affinity probe 43.48
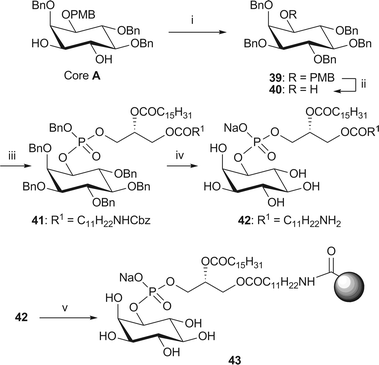 | ||
| Scheme 7 Synthesis of immobilised NH2-PI 43. Reagents and conditions: i. NaH (3 eq.), BnBr, (3 eq.), TBAI, DMF, 49%; ii. CAN, MeCN–H2O (4/1, v/v), 85%; iii. phosphoramidite 38, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 73%; iv. H2 (4.1 bar), Pd black, NaHCO3, tBuOH/H2O (6/1), 76%; v. Affi-Gel® 10, NaHCO3, CHCl3–MeOH/H2O (4/5/1, v/v/v), 0 °C → rt, 9% loading. | ||
To demonstrate the versatility of our synthetic strategy the enantiomer of core B was treated with dipalmitoyl phosphoramidite 34 and 1H-tetrazole followed by oxidation with mCPBA to give the fully protected dipalmitoyl-PI 44 (Scheme 8). Global deprotection via hydrogenolysis provides the dipalmitoyl-PI sodium salt 45 in a good yield.
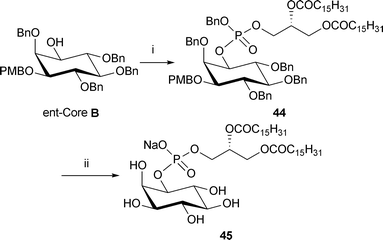 | ||
| Scheme 8 Synthesis of dipalmitoyl-PI analogue 45. Reagents and conditions: i. phosphoramidite 34, 1H-tetrazole, CH2Cl2, rt, then m-CPBA, −78 °C → rt, 55% (ii) Pd black, NaHCO3, tBuOH/H2O (6:1), H2 (3.1 bar), rt, 48 h, >99%. | ||
The next group of phosphatidylinositols to consider is that where, in addition to the phospholipid side chain at the 1-position, one of either the 3-, 4- or 5-positions is phosphorylated.
For example, PI(3)P is obtained by first phosphorylating core B with bis(benzyloxy)(N,N-diisopropylamino)phosphine49 followed by in situ oxidation with mCPBA, to install the phosphate group at the 3-position (Scheme 9). Removal of the PMB group in 46 and subsequent 1H-tetrazole-mediated coupling with the appropriate phosphoramidite afforded the fully protected dipalmitoyl-PI(3)P 4816 and NH2-PI(3)P 49. Standard global deprotection conditions afforded both analogues of PI(3)P (50 and 51). The affinity probe 52 was obtained by coupling of 51 with NHS-activated Affi-Gel® 10 beads.
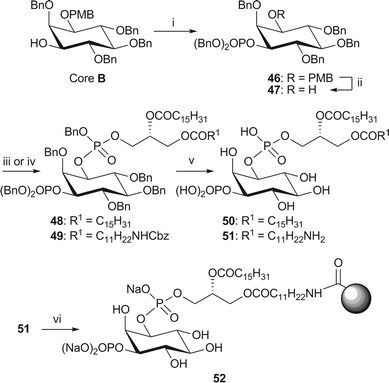 | ||
| Scheme 9 Synthesis of dipalmitoyl-PI(3)P 50 and immobilised NH2-PI(3)P 52. Reagents and conditions: i. (BnO)2P(NiPr2), 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 87%; ii. CAN, MeCN–H2O (4/1, v/v), 83%; iii. phosphoramidite 34, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 48: 90%; iv. phosphoramidite 38, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 49: 84%; v. H2 (4.1 or 3.8 bar), Pd black, tBuOH, 50: 89% from 48; 51: 62% from 49; vi. Affi-Gel® 10, NaHCO3, CHCl3–MeOH/H2O (4/5/1, v/v/v), 0 °C → rt, 2% loading. | ||
The analogues of PI(4)P were obtained from core A (Scheme 10). This synthesis required the selective protection of the hydroxyl group at the 3-position of the inositol ring, which was achieved using conditions developed by Gigg et al.50In situ generation of a stannane acetal in the presence of tetrabutylammonium bromide and benzyl bromide gave rise to the 3-benzylated derivative 53 and the corresponding 4-benzylated regioisomer in a 4:1 ratio (as judged by 1H NMR analysis).
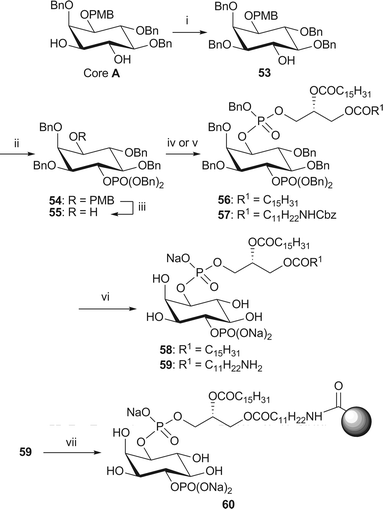 | ||
| Scheme 10 Synthesis of dipalmitoyl-PI(4)P 58 and immobilised NH2-PI(4)P 60. Reagents and conditions: i. Bu2SnO, BnBr, Bu4NBr, MeCN, 3 Å MS, reflux, 76%; ii. (BnO)2P(NiPr2), 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 93% iii. CAN, MeCN–H2O (4/1, v/v), 84%; iv. phosphoramidite 34, 1H-tetrazole, CH2Cl2, rt, then m-CPBA, −78 °C → rt, 56: 78%; v. phosphoramidite 38, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 57: 77%; vi. H2 (15 or 4.1 bar), Pd black, NaHCO3, tBuOH/H2O (6/1), 58: 59% from 56; 59: 75% from 57; vii. Affi-Gel® 10, NaHCO3, CHCl3–MeOH/H2O (4/5/1, v/v/v), 0 °C → rt, 16% loading. | ||
Subsequent phosphorylation of the remaining hydroxyl group at the 4-position of 53, followed by removal of the PMB group, coupling to the requisite phosphoramidite, oxidation, and global deprotection smoothly provided both the dipalmitoyl analogue 58 and the amino analogue 59 of PI(4)P. The immobilised PI(4)P affinity probe 60 was synthesised in the standard manner.
The synthesis of PI(5)P analogues 67 and 68 from core C first required protection of the 3- and 4-hydroxyl groups (Scheme 11). The allyl group at the 5-position was then removed to allow installation of the 5-phosphate. This deprotection was achieved through isomerisation of the allyl ether 61 with Wilkinson's catalyst followed by acid catalysed methanolysis of the resulting enol ether. The standard phosphorylation, PMB removal, phosphoramidite coupling with in situ oxidation and global deprotection methods were then employed to obtain both analogues of PI(5)P. Amino-PI(5)P 68 was then immobilised onto Affi-Gel® 10 beads in the same manner as above.
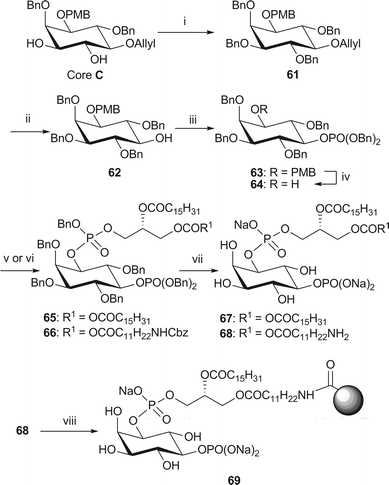 | ||
| Scheme 11 Synthesis of dipalmitoyl-PI(5)P 67 and immobilised NH2-PI(5)P 69. Reagents and conditions: i. NaH, BnBr, DMF, rt, 94%; ii. RhCl(PPh3)3, DIPEA, EtOH–toluene/H2O (7:3:1 v:v:v) then AcCl, MeOH–CH2Cl2, 83%; iii. (BnO)2P(NiPr2), 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 91%; iv. CAN, MeCN–H2O (4/1, v/v), 79%; v. phosphoramidite 34, 1H-tetrazole, CH2Cl2, rt, then m-CPBA, −78 °C → rt, 65: 62%; vi. phosphoramidite 38, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 66: 64%; vii. H2 (3.5 or 35 bar), Pd black, NaHCO3, tBuOH/H2O (6/1), 67: 86% from 65; 68: 83% from 66; viii. Affi-Gel® 10, NaHCO3, CHCl3–MeOH/H2O (4/5/1, v/v/v), 0 °C → rt, 8% loading. | ||
Intermediate 61 in the synthesis of the PI(5)P analogues, can also be accessed from the unnatural L-enantiomer of triol 7 through facile protecting group manipulation (Scheme 12). This enantiomer was obtained from diastereomer 70, resulting from the initial resolution step of racemic diol 7 with camphor (R)-10 (Scheme 2), by acid catalysed methanolysis of the acetal 70. Selective protection of the hydroxyl group at the 1-position as the PMB ether via the stannane acetal, followed by benzylation of the remaining hydroxyl groups afforded intermediate 61 in good yield with an optical rotation in agreement with an authentic sample (see Electronic Supporting Information for more details). Though this is not a direct route to PI(5)P, it makes good use of an otherwise unused by-product.
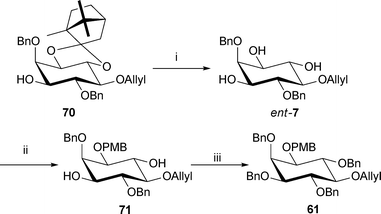 | ||
| Scheme 12 Alternative synthesis of intermediate 61. Reagents and conditions: i. AcCl, MeOH–CH2Cl2, (2/3 v/v), rt, 17 h, 97%; ii. Bu2SnO, PMBCl, NaBr, Bu4NBr, MeCN–toluene (2/1 v/v), 3 Å MS, reflux, 17 h, 72%; iii. NaH, BnBr, DMF, rt, 17 h, 66%. | ||
Synthesis of analogues of PI(3,4)P2,9,10,16,20,21,23,25 PI(3,5)P27,9,10,13–18 and PI(4,5)P210–12,22,25
PI(X,Y)P2, the third subclass of phosphatidylinositols contains those in which two of the 3-, 4- or 5-positions are phosphorylated. There are three possible permutations for the bisphosphates: PI(3,4)P2, PI(3,5)P2 and PI(4,5)P2. PI(3,4)P2 and PI(3,5)P2 are easily obtained from cores A and D respectively (Scheme 13). This route follows the standard methods for phosphorylation and deprotection to afford the dipalmitoyl (7616,21 and 8216) and amino (77 and 83) analogues of PI(3,4)P2 and PI(3,5)P2 respectively. It should be noted that alternative hydrogenolysis conditions using palladium hydroxide on carbon in butanol were employed for the global deprotection of both the dipalmitoyl analogues 75 and 81, affording 76 and 82 in comparable yields. Affinity probes 84 and 85 were then obtained in the manner as described above.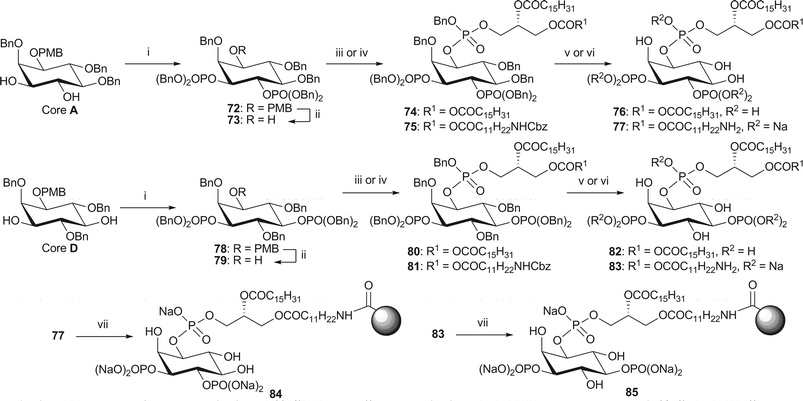 | ||
| Scheme 13 Synthesis of dipalmitoyl-PI(3,4)P2, 76, dipalmitoyl-PI(3,5)P2 82 and immobilised NH2-PI(3,4)P2 84 and NH2-PI(3,5)P2 85. Reagents and conditions: i. (BnO)2P(NiPr2), 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 72: 91%; 78: 71%; ii. CAN, MeCN–H2O (4/1, v/v), 73: 90%; 79: 72%; iii. phosphoramidite 34, 1H-tetrazole, CH2Cl2, rt, then m-CPBA, −78 °C → rt, 74: 91%; 80: 93%; iv. phosphoramidite 38, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 75: 83%; 81: 88%; v. H2 (3.5 or 4.1 bar), Pd(OH)2-C, tBuOH, 76: 97% from 74; 82: quant. from 80; vi. H2 (3.6 bar), Pd black, NaHCO3, tBuOH/H2O (6/1), 77: 80% from 75; 83: 88% from 81; vii. Affi-Gel® 10, H2O, 4 °C, 84: 3% loading; 85: 3% loading. | ||
The synthesis of the analogues of PI(4,5)P2, required some manipulation of the protecting groups to provide the correct substitution pattern (Scheme 14). Thus, the hydroxyl group in the 3-position of core C was selectively benzylated using the dibutyltin oxide strategy to afford alcohol 86. Removal of the allyl group was achieved via isomerisation with Wilkinson's catalyst and methanolysis of the resulting enol ether, affording diol 87. Subjection to the standard phosphorylation and deprotection conditions gave dipalmitoyl-PI(4,5)P2 92 and NH2-PI(4,5)P2 93.12 Affinity probe 9412 was synthesised in the manner described previously.
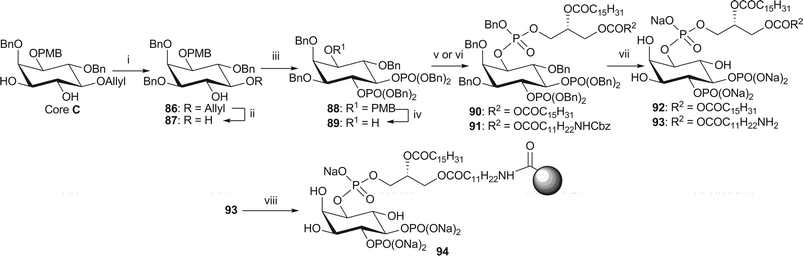 | ||
| Scheme 14 Synthesis of dipalmitoyl-PI(4,5)P2 92 and immobilised NH2-PI(4,5)P2 94. Reagents and conditions: i. Bu2SnO, BnBr, Bu4NBr, CH3CN, 3 Å MS, reflux, 75% ii, Rh(PPh3)3Cl, EtNiPr2, EtOH–toluene/H2O (7/3/1), reflux, then AcCl, CH2Cl2–MeOH (2/1), 58%; iii. (BnO)2P(NiPr2), 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 75%; iv. CAN, MeCN–H2O (4/1, v/v), 80%; v. phosphoramidite 34, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 90: 83%; vi. phosphoramidite 38, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 91: 82%; vii. H2 (25 or 15 bar), Pd black, NaHCO3, tBuOH/H2O (6/1), 92: 78% from 90; 93: 59% from 91; viii. Affi-Gel® 10, NaHCO3, H2O, 0 °C, 3% loading. | ||
Synthesis of PI(3,4,5)P3,6,9,10,16,21–25,28 and related analogues
Lastly, analogues of PI(3,4,5)P3 were obtained from core E by a simple global phosphorylation (Scheme 15). The standard methods for the coupling with the side chain phosphoramidites, oxidation and global deprotection were used to gain access to both the analogues 9916,21 and 10028 of PI(3,4,5)P3.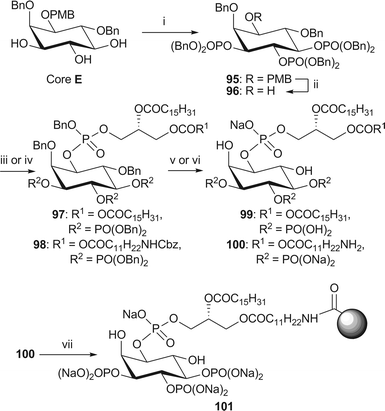 | ||
| Scheme 15 Synthesis of dipalmitoyl-PI(3,4,5)P3 99 and immobilised NH2-PI(3,4,5)P3 101. Reagents and conditions: i. (BnO)2P(NiPr2), 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 78%; ii. CAN, MeCN–H2O (4/1, v/v), 87%; iii. phosphoramidite 34, 1H-tetrazole, CH2Cl2, rt, then m-CPBA, −78 °C → rt, 97: 78%; iv. phosphoramidite 38, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 98: 63%; v. H2 (3.5 bar), Pd(OH)2-C, tBuOH, 99: 85% from 97; vi. H2 (4.1 or 3.5 bar), Pd black, NaHCO3, tBuOH/H2O (6/1), 100: 92% from 98; vii. Affi-Gel® 10, NaHCO3, H2O, 0 °C, 3% loading. | ||
To demonstrate the versatility of this approach to the synthesis of PIP analogues, shorter chain analogues of PI(3,4,5)P3 108 and 109, were synthesised using the corresponding hexanoyl51 and octanoyl24 phosphoramidites 102 and 103, respectively (Fig. 2,Scheme 16). An ω-amino analogue 110 with a hexanoyl chain at the secondary position of the glycerol side chain was also synthesised using the phosphoramidite 104.52 These shorter chain analogues are known to be more water-soluble than the longer chain PIPs.19,27
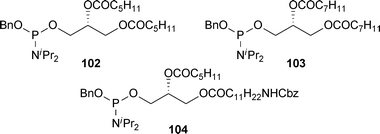 | ||
| Fig. 2 Short chain phosphoramidites 102–104. | ||
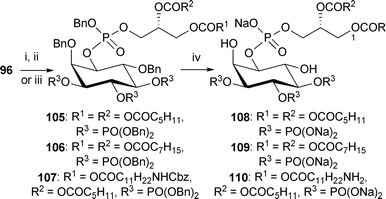 | ||
| Scheme 16 Synthesis of short chain analogues of PI(3,4,5)P3 108–110. Reagents and conditions: i. phosphoramidite 102, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 105: 67%; ii. phosphoramidite 103, 1H-tetrazole, CH2Cl2, rt, then m-CPBA, −78 °C → rt, 106: 75%; iii. phosphoramidite 104, 1H-tetrazole, CH2Cl2, rt, then mCPBA, −78 °C → rt, 107: 79%; iv. H2 (3.5, 3.1 or 25 bar), Pd black, NaHCO3, tBuOH/H2O (6/1), 108: 96% from 105; 109: 80% from 106; 110: 78% from 107. | ||
Biological evaluation
During the course of our investigations we have used both the dipalmitoyl and amino analogues of the PIPs in the investigation of the PI3K pathway.30-35 Our original experiments used cytosolic and membrane extracts from sheep brain or porcine neutrophils and leukocytes. Initial investigations using the dipalmitoyl analogue of PI(3,4,5)P3 revealed that Akt-1/PKB interacts directly with PI(3,4,5)P3 via the PH domain.35 PI(3,4,5)P3-binding to Akt/PKB was shown to be necessary for upstream phosphorylation by directly activating upstream kinases. These investigations confirmed the role of Akt/PKB in PI3K-signalling.Work using PI(3)P affinity probes in 2001, led to the discovery of the interactions of phosphatidylinositol phosphates with the PX domain and its role in the formation of reactive oxygen species (ROS) by neutrophils.32 It was determined that PI(3)P specifically stimulates ROS formation by binding to the PX domain of p40phox which in turn can interact with p67phox. Furthermore, p40phox was shown to target PI(3)P-containing membranes. In other work, two novel proteins FENS1 and DFCP1, containing one and two FYVE domains, respectively, were shown to bind PI(3)P specifically.33 These proteins are involved in intracellular trafficking.53
In 2002, investigations with immobilised PI(3)P, PI(3,4)P2 and PI(3,5)P2 discovered five novel proteins and seven proteins previously uncharacterised as phosphoinositide-binding.31 Interestingly, three of the proteins that were not known to bind PIPs, ATTP, MEG2 and Cdc42GAP, have different functions, but all proved to have an SEC14-like domain in common. This domain had not previously been known to bind to PIPs. One of the novel PI(3,4,5)P3 and PI(3,4)P2-binding proteins, ARAP3, is a PI3K-dependent, GTPase activating protein and was shown to be a genuine effector of PI3K signalling responsible for PI3K-dependent changes in the cell cytoskeleton and shape.
Investigations into the role of PI(3,5)P2 in vesicle recycling from vacuole/lysosomal compartments, using immobilised PI(3,5)P2 identified Svp1p as a specific PI(3,5)P2-binding protein.30 This protein participates in the recycling of membrane proteins from the vacuole to the late endosome and at the time of discovery, represented a new family of phosphoinositide-binding proteins. It was shown that the binding of PI(3,5)P2 by Svp1p is necessary for normal function in vacuole membrane trafficking.
More recently we have used analogues of PI(3,5)P2, PI(4,5)P2 and PI(3,4,5)P3 to perform a comprehensive proteomic analysis of the phosphoinositide interactome in cell extracts from LIM1215 colorectal carcinoma cells.29 The procedure for these experiments is outlined in Fig. 3. Phosphoinositide interacting protein complexes were pulled down using analogues of PI(3,5)P2, PI(4,5)P2 and PI(3,4,5)P3, either incorporated into liposomes or immobilised onto Affi-Gel® 10 beads in affinity-based assays. Proteins were then identified using nano-RP-HPLC ESI MS/MS analysis. The affinity probes were characterised using Biosensor technology. Thus, the phosphoinositide-containing liposomes were characterised using recombinant GST-tagged PH domains of General Receptor 1, Phospholipase C delta 1 and Dynamin, whilst the loading of the NH2-PIP on the beads was assessed by the analysis of the supernatants from the immobilisation reactions and comparison with calibration curves generated by injecting various concentrations of each PIP over immobilised neomycin.29 The NH2-PIP analogues can also be immobilised onto a sensor chip allowing the characterisation of their interaction with their protein-binding partners.
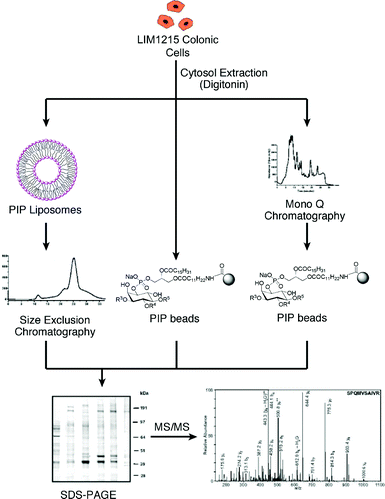 | ||
| Fig. 3 Schematic of procedure for pull down experiments with liposomes incorporating PIPs and immobilised PIPs.29 | ||
From the cytosolic extracts of LIM1215 colon cancer cells we identified 529 proteins/protein complexes that appeared to interact specifically with their phosphoinositide targets: 69 proteins interact specifically with PI(3,5)P2, 146 with PI(4,5)P2, 141 with PI(3,4,5)P3, 32 with both PI(3,5)P2 and PI(4,5)P2, 36 with both PI(3,5)P2 and PI(3,4,5)P3, 41 with both PI(4,5)P2 and PI(3,4,5)P3, and 64 with all 3 phosphoinositides (Fig. 4).
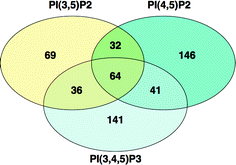 | ||
| Fig. 4 Venn diagram analysis of the purified phosphoinositide interacting proteins showing the number of common and unique proteins pulled down by PI(3,5)P2, PI(4,5)P2 and PI(3,4,5)P3. | ||
Our affinity method was validated by the purification of a large number of proteins that possess known phosphoinositide or phospholipid binding domains (for example: PH, PX, FERM, PTB/PID, Phosphatidylinositol 3- and 4-kinase, RANBP1, C1, C2, MARCKS, PDZ, PHD, Septin, Annexin, Clathrin adaptor, Calponin, Phosphatidylinositol transfer protein, HEAT, Inositol monophosphatase family, Cral-Trio, SPC2, START, SPFH). Furthermore, small GTPases of Ras, Rab, Arf and Rho families that are known to interact with phosphoinositide phosphates, were also purified. Thus at least 30% of the total number of proteins identified in our study were able to interact directly with their phosphoinositide targets, suggesting that the remaining proteins were presumably purified as part of interactome complexes.
Interestingly, the majority of purified proteins had molecular function and processes corresponding to previously well characterized cellular functions of phosphoinositide (e.g. molecular transport, protein trafficking, vesicle mediated transport, actin cytoskeletal regulation and GTPases regulated function, cell-to-cell signalling and interaction, cellular development, movement, assembly organization, growth and proliferation). A large number of purified proteins were also reported to have direct and indirect interactions with the PI3K/Akt signalling pathway.
Furthermore, proteins with different functions were found to bind to the liposomes and beads. Broadly speaking, those that require a membrane environment, such as GTPases and cell adhesion molecules, bound to the liposomes, whereas those involved in functions such as transport and trafficking were identified using beads. Phosphatases and kinases were identified in both pull-down experiments. This is depicted in Fig. 5 for PI(4,5)P2. Experiments are continuing in an effort to further our understanding of the PIP interactomes.
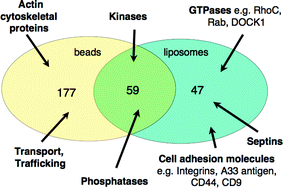 | ||
| Fig. 5 Venn diagram analysis of the purified PI(4,5)P2 interacting proteins showing the number of common and unique proteins pulled down by immobilised-NH2-PI(4,5)P2 and PI(4,5)P2-liposomes. | ||
Conclusions
The complete family of phosphatidylinositol phosphate analogues has been synthesised by a divergent strategy. Over the course of these investigations, affinity probes incorporating these analogues have been used to identify a range of important proteins in the PI3K signalling pathway, which were novel at the time of discovery.A comprehensive proteomic analysis of phosphoinositide interacting proteins in cell extracts from LIM1215 colorectal carcinoma cells has also been carried out. Our studies provide an initial detailed assessment of the phosphoinositide interactome which can be used in the study of the cellular responses mediated by phosphoinositides, to identify signalling pathways that are affected by a change of phosphoinositide cellular concentration (e.g. 3-phosphoinositide signalling cascade) and as a tool in drug discovery and biomarker development.
Acknowledgements
We thank the BBSRC and EPSRC for financial support and for provision of the Swansea Mass Spectrometry Service. This work was supported in part by the Australian Research Council, Discovery Project, Grant DP0770668 and an NHMRC Program Grant, 487922. General support was provided by CSIRO and VESKI (Victorian Endowment for Science, Knowledge and Innovation). We also thank Daiso Co., Ltd, Japan for the generous gift of (+)-1,2-O-isopropylidene-glycerol. SJC thanks Hughes Hall, Cambridge for a Research Fellowship. SJAG thanks GSK for a CASE studentship. Z-YL thanks Cambridge Commonwealth Trust, Universities UK (ORS award), New Hall, Cambridge and Tan Kar Kee Foundation, Singapore for generous financial support.Notes and references
- G. D. Prestwich, Chem. Biol., 2004, 11, 619 CrossRef CAS.
- T. F. J. Martin, Annu. Rev. Cell Dev. Biol., 1998, 14, 231 CrossRef CAS.
- A. Toker, Cell. Mol. Life Sci., 2002, 59, 761 CrossRef CAS; M. Overduin, M. L. Cheever and T. G. Kutateladze, Mol. Interv., 2001, 1, 150 CAS; L. V. Dekker and A. W. Segal, Science, 2000, 287, 982 CrossRef CAS; S. Corvera and M. P. Czech, Trends Cell Biol., 1998, 8, 442 CrossRef CAS; A. Toker and L. C. Cantley, Nature, 1997, 387, 673 CrossRef CAS; M. J. Berridge, Nature, 1993, 365, 388 CrossRef CAS.
- M. Vicinanza, G. D'Angelo, A. Di Campli and M. A. De Matteis, Cell. Mol. Life Sci., 2008, 65, 2833 CrossRef CAS; T. L. Yuan and L. C. Cantley, Oncogene, 2008, 27, 5497 CrossRef CAS; M. P. Wymann and R. Scheiter, Nat. Rev. Mol. Cell Biol., 2008, 9, 162 CrossRef CAS; A. G. Bader, S. Y. Kang and P. K. Vogt, Proc. Natl. Acad. Sci. U. S. A., 2006, 103, 1475 CrossRef CAS; D. W. Parsons, T. L. Wang, Y. Samuels, A. Bardelli, J. M. Cummins, L. DeLong, N. Silliman, J. Ptak, S. Szabo, J. K. V. Willson, S. Markowitz, K. Kinzler, B. Vogelstein, C. Lengauer and V. E. Velculescu, Nature, 2005, 436, 792 CrossRef CAS; B. T. Hennessy, D. L. Smith, P. T. Ram, Y. L. Lu and G. B. Mills, Nat. Rev. Drug Discovery, 2005, 4, 988 CrossRef CAS; C. Pendaries, H. Tronchere, M. Plantavid and B. Payrastre, FEBS Lett., 2003, 546, 25 CrossRef CAS; L. Shayesteh, Y. L. Lu, W. L. Kuo, R. Baldocchi, T. Godfrey, C. Collins, D. Pinkel, B. Powell, G. B. Mills and J. W. Gray, Nat. Genet., 1999, 21, 99 CrossRef CAS.
- K. J. Kayser-Bricker, P. A. Jordan and S. J. Miller, Tetrahedron, 2008, 64, 7015 CrossRef CAS.
- K. M. Sureshan, A. M. Riley and B. V. L. Potter, Tetrahedron Lett., 2007, 48, 1923 CrossRef CAS; D. S. Wang, A. L. Hsu and C. S. Chen, Bioorg. Med. Chem., 2001, 9, 133 CrossRef CAS; P. R. J. Gaffney and C. B. Reese, J. Chem. Soc., Perkin Trans. 1, 2001, 192 RSC; R. Shirai, K. Morita, A. Nishikawa, N. Nakatsu, Y. Fukui, N. Morisaki and Y. Hashimoto, Tetrahedron Lett., 1998, 39, 9485 CrossRef CAS; S. G. Aneja, A. Parra, C. Stoenescu, W. Y. Xia and R. Aneja, Tetrahedron Lett., 1997, 38, 803 CrossRef CAS.
- Y. J. Xu, B. R. Sculimbrene and S. J. Miller, J. Org. Chem., 2006, 71, 4919 CrossRef CAS.
- Y. Watanabe, Y. Kiyosawa, S. Hyodo and M. Hayashi, Tetrahedron Lett., 2005, 46, 281 CrossRef CAS; N. Morisaki, K. Morita, A. Nishikawa, N. Nakatsu, Y. Fukui, Y. Hashimoto and R. Shirai, Tetrahedron, 2000, 56, 2603 CrossRef CAS; R. Aneja and S. G. Aneja, Tetrahedron Lett., 2000, 41, 847 CrossRef CAS; J. G. Ward and R. C. Young, Tetrahedron Lett., 1988, 29, 6013 CrossRef CAS; P. A. Gent, R. Gigg and C. D. Warren, Tetrahedron Lett., 1970, 11, 2575 CrossRef.
- B. R. Sculimbrene, Y. Xu and S. J. Miller, J. Am. Chem. Soc., 2004, 126, 13182 CrossRef CAS; D. S. Wang and C. S. Chen, J. Org. Chem., 1996, 61, 5905 CrossRef CAS; K. S. Bruzik and R. J. Kubiak, Tetrahedron Lett., 1995, 36, 2415 CrossRef.
- R. J. Kubiak and K. S. Bruzik, J. Org. Chem., 2003, 68, 960 CrossRef CAS.
- F. H. Han, M. Hayashi and Y. Watanabe, Chem. Lett., 2003, 32, 46 CrossRef CAS; F. Han, M. Hayashi and Y. Watanabe, Tetrahedron, 2003, 59, 7703 CrossRef CAS; J. R. Falck, U. M. Krishna and J. H. Capdevila, Tetrahedron Lett., 1999, 40, 8771 CrossRef; C. E. Dreef, C. J. J. Elie, P. Hoogerhout, G. A. van der Marel and J. H. van Boom, Tetrahedron Lett., 1988, 29, 6513 CrossRef CAS.
- Z.-Y. Lim, J. W. Thuring, A. B. Holmes, M. Manifava and N. T. Ktistakis, J. Chem. Soc., Perkin Trans. 1, 2002, 1067 RSC.
- A. Nishikawa, S. Saito, Y. Hashimoto, K. Koga and R. Shirai, Tetrahedron Lett., 2001, 42, 9195 CrossRef CAS.
- J. R. Falck, U. M. Krishna, K. R. Katipally, J. H. Capdevila and E. T. Ulug, Tetrahedron Lett., 2000, 41, 4271 CrossRef CAS.
- J. R. Falck, U. M. Krishna and J. H. Capdevila, Bioorg. Med. Chem. Lett., 2000, 10, 1711 CrossRef.
- G. F. Painter, S. J. A. Grove, I. H. Gilbert, A. B. Holmes, P. R. Raithby, M. L. Hill, P. T. Hawkins and L. R. Stephens, J. Chem. Soc., Perkin Trans. 1, 1999, 923 RSC.
- A. M. Riley and B. V. L. Potter, Tetrahedron Lett., 1998, 39, 6769 CrossRef CAS.
- J. R. Peng and G. D. Prestwich, Tetrahedron Lett., 1998, 39, 3965 CrossRef CAS.
- J. Chen, L. Feng and G. D. Prestwich, J. Org. Chem., 1998, 63, 6511 CrossRef CAS.
- K. K. Reddy, J. H. Ye, J. R. Falck and J. H. Capdevila, Bioorg. Med. Chem. Lett., 1997, 7, 2115 CrossRef CAS; K. K. Reddy, J. Rizo and J. R. Falck, Tetrahedron Lett., 1997, 38, 4729 CrossRef CAS.
- S. J. A. Grove, A. B. Holmes, G. F. Painter, P. T. Hawkins and L. R. Stephens, Chem. Commun., 1997, 1635 RSC.
- Q. M. Gu and G. D. Prestwich, J. Org. Chem., 1996, 61, 8642 CrossRef CAS.
- T. Desai, J. Gigg, R. Gigg and E. Martin-Zamora, Carbohydr. Res., 1996, 296, 97 CrossRef CAS.
- K. K. Reddy, M. Saady, J. R. Falck and G. Whited, J. Org. Chem., 1995, 60, 3385 CrossRef CAS.
- L. X. Qiao, Y. H. Hu, F. J. Nan, G. Powis and A. P. Kozikowski, Org. Lett., 2000, 2, 115 CrossRef CAS.
- H. Zhang, Y. Xu, N. Markadieu, R. Beauwens, C. Erneux and G. D. Prestwich, Bioorg. Med. Chem. Lett., 2008, 18, 762 CrossRef CAS; S. J. Conway and G. J. Miller, Nat. Prod. Rep., 2007, 24, 687 RSC; Y. Xu, S. A. Lee, T. G. Kutateladze, D. Sbrissa, A. Shisheva and G. D. Prestwich, J. Am. Chem. Soc., 2006, 128, 885 CrossRef CAS; H. L. Zhang, Y. Xu, Z. Zhang, E. R. Liman and G. D. Prestwich, J. Am. Chem. Soc., 2006, 128, 5642 CrossRef CAS; P. W. Rzepecki and G. D. Prestwich, J. Org. Chem., 2002, 67, 5454 CrossRef CAS; G. D. Prestwich, Acc. Chem. Res., 1996, 29, 503 CrossRef CAS; T. L. Andresen, D. M. Skytte and R. Madsen, Org. Biomol. Chem., 2004, 2, 2951 RSC; J. Chen, A. A. Profit and G. D. Prestwich, J. Org. Chem., 1996, 61, 6305 CrossRef CAS; R. J. Kubiak and K. S. Bruzik, Bioorg. Med. Chem. Lett., 1997, 7, 1231 CrossRef CAS; C. Mihai, J. Mataka, S. Riddle, M. D. Tsai and K. S. Bruzik, Bioorg. Med. Chem. Lett., 1997, 7, 1235 CrossRef CAS; O. Thum, J. Chen and G. D. Prestwich, Tetrahedron Lett., 1996, 37, 9017 CrossRef CAS; H. L. Zhang, N. Markadieu, R. Beauwens, C. Erneux and G. D. Prestwich, J. Am. Chem. Soc., 2006, 128, 16464 CrossRef CAS; A. P. Kozikowski, L. X. Qiao, W. Tuckmantel and G. Powis, Tetrahedron, 1997, 53, 14903 CrossRef CAS.
- J. Chen and G. D. Prestwich, J. Org. Chem., 1998, 63, 430 CrossRef CAS.
- G. F. Painter, J. W. Thuring, Z.-Y. Lim, A. B. Holmes, P. T. Hawkins and L. R. Stephens, Chem. Commun., 2001, 645 RSC.
- B. Catimel, M.-X. Yin, C. Schieber, M. Condron, H. Patsiouras, J. Catimel, D. E. J. E. Robinson, L. S.-M. Wong, E. C. Nice, A. B. Holmes and A. W. Burgess, J. Proteome Res., 2009, 8, 3712 CrossRef CAS; B. Catimel, C. Schieber, M. Condron, H. Patsiouras, L. Connolly, J. Catimel, E. C. Nice, A. W. Burgess and A. B. Holmes, J. Proteome Res., 2008, 7, 5295 CrossRef CAS.
- S. K. Dove, R. C. Piper, R. K. McEwen, J. W. Yu, M. C. King, D. C. Hughes, J. Thuring, A. B. Holmes, F. T. Cooke, R. H. Michell, P. J. Parker and M. A. Lemmon, EMBO J., 2004, 23, 1922 CrossRef CAS.
- S. Krugmann, K. E. Anderson, S. H. Ridley, N. Risso, A. McGregor, J. Coadwell, K. Davidson, A. Eguinoa, C. D. Ellson, P. Lipp, M. Manifava, N. Ktistakis, G. Painter, J. W. Thuring, M. A. Cooper, Z. Y. Lim, A. B. Holmes, S. K. Dove, R. H. Michell, A. Grewal, A. Nazarian, H. Erdjument-Bromage, P. Tempst, L. R. Stephens and P. T. Hawkins, Mol. Cell, 2002, 9, 95 CrossRef CAS.
- C. D. Ellson, S. Gobert-Gosse, K. E. Anderson, K. Davidson, H. Erdjument-Bromage, P. Tempst, J. W. Thuring, M. A. Cooper, Z. Y. Lim, A. B. Holmes, P. R. J. Gaffney, J. Coadwell, E. R. Chilvers, P. T. Hawkins and L. R. Stephens, Nat. Cell Biol., 2001, 3, 679 CrossRef CAS.
- S. H. Ridley, N. Ktistakis, K. Davidson, K. E. Anderson, M. Manifava, C. D. Ellson, P. Lipp, M. Bootman, J. Coadwell, A. Nazarian, H. Erdjument-Bromage, P. Tempst, M. A. Cooper, J. Thuring, Z. Y. Lim, A. B. Holmes, L. R. Stephens and P. T. Hawkins, J. Cell Sci., 2001, 114, 3991 CAS.
- M. Manifava, J. W. J. F. Thuring, Z.-Y. Lim, L. Packman, A. B. Holmes and N. T. Ktistakis, J. Biol. Chem., 2001, 276, 8987 CrossRef CAS; K. E. Anderson, P. Lipp, M. Bootman, S. H. Ridley, J. Coadwell, L. Ronnstrand, J. Lennartsson, A. B. Holmes, G. F. Painter, J. Thuring, Z. Y. Lim, H. Erdjument-Bromage, A. Grewal, P. Tempst, L. R. Stephens and P. T. Hawkins, Curr. Biol., 2000, 10, 1403 CrossRef CAS; R. K. McEwen, S. K. Dove, F. T. Cooke, G. F. Painter, A. B. Holmes, A. Shisheva, Y. Ohya, P. J. Parker and R. H. Michell, J. Biol. Chem., 1999, 274, 33905 CrossRef CAS.
- L. Stephens, K. Anderson, D. Stokoe, H. Erdjument-Bromage, G. F. Painter, A. B. Holmes, P. R. J. Gaffney, C. B. Reese, F. McCormick, P. Tempst, J. Coadwell and P. T. Hawkins, Science, 1998, 279, 710 CrossRef CAS; D. Stokoe, L. R. Stephens, T. Copeland, P. R. J. Gaffney, C. B. Reese, G. F. Painter, A. B. Holmes, F. McCormick and P. T. Hawkins, Science, 1997, 277, 567 CrossRef CAS; S. R. James, C. P. Downes, R. Gigg, S. J. A. Grove, A. B. Holmes and D. R. Alessi, Biochem. J, 1996, 315, 709 CAS.
- Stereochemical representation: myo-inositol (Scheme 1), structures 1, 11–14 and 27–29 are meso-compounds. Structures 2–7, 15 and 17–19 refer to racemic compounds. Structures in Fig. 1, cores A–E, structures 8–10, 16, 20 and 30–110 refer to single enantiomers.
- D. C. Billington, R. Baker, J. J. Kulagowski, I. M. Mawer, J. P. Vacca, S. J. Desolms and J. R. Huff, J. Chem. Soc., Perkin Trans. 1, 1989, 1423 RSC; H. W. Lee and Y. Kishi, J. Org. Chem., 1985, 50, 4402 CrossRef CAS.
- S. J. A. Grove, I. H. Gilbert, A. B. Holmes, G. F. Painter and M. L. Hill, Chem. Commun., 1997, 1633 RSC.
- I. H. Gilbert and A. B. Holmes, Tetrahedron Lett., 1990, 31, 2633 CrossRef CAS.
- K. S. Bruzik and M.-D. Tsai, J. Am. Chem. Soc., 1992, 114, 6361 CrossRef CAS.
- Bruzik subsequently used camphor dimethyl acetal for the early resolution of myo-inositol as a strategy for the synthesis of analogues of all eight naturally occurring PIPs. See reference 10 for more details.
- I. H. Gilbert, A. B. Holmes, M. J. Pestchanker and R. C. Young, Carbohydr. Res., 1992, 234, 117 CrossRef CAS.
- Organometallic Syntheses, ed. J. J. Eisch, and R. B. King, Academic Press, New York, Vol. 2, p. 137 Search PubMed; J. J. Eisch, and S. G. Rhee, J. Am. Chem. Soc., 1974, 96, 7276 Search PubMed.
- Synthesised from myo-inositol and deuterated triethyl orthoformate, see Electronic Supporting Information for more details.
- M. K. Johns, M.-X. Yin, S. J. Conway, D. E. J. E. Robinson, L. S.-M. Wong, R. Bamert, R. E. H. Wettenhall and A. B. Holmes, Org. Biomol. Chem., 2009, 7, 3691 RSC.
- Synthesised from phosphorus trichloride by the method of W. Bannwarth and A. Trzeciak, Helv. Chim. Acta, 1987, 70, 175 Search PubMed.
- These compounds are notoriously difficult to characterise because of problems with solubility and/or micelle formation (also see reference 19). Characterisation data is available in the Electronic Supporting Information.
- The percentage loading was determined by either 1H NMR analysis of the supernatant (reference 12) or Biocore analysis (reference 29).
- J. W. Perich and R. B. Johns, Tetrahedron Lett., 1987, 28, 101 CrossRef CAS.
- T. Desai, J. Gigg, R. Gigg, E. Martin-Zamora and N. Schnetz, Carbohydr. Res., 1994, 258, 135 CrossRef CAS.
- Synthesised in a similar manner to dipalmitoyl phosphoramidite 34, see Electronic Supporting Information.
- Phosphoramidite 104 was synthesised from 35 following the procedure for the synthesis of 38, see Electronic Supporting Information for more details.
- E. L. Axe, S. A. Walker, M. Manifava, P. Chandra, H. L. Roderick, A. Habermann, G. Griffiths and N. T. Ktistakis, J. Cell Biol., 2008, 182, 685 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedures for 27–29, 39–45, 49, 51–69, 71, 75, 77, 81, 83–85, 90, 92, 102, 104–110. See DOI: 10.1039/b913399b |
| This journal is © The Royal Society of Chemistry 2010 |