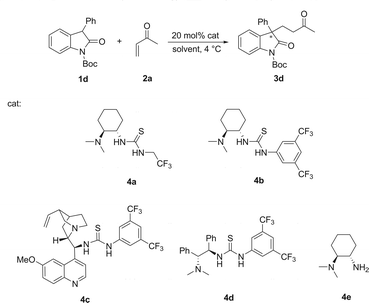
Asymmetric Michael addition reaction of 3-substituted-N-Boc oxindoles to activated terminal alkenes catalyzed by a bifunctional tertiary-amine thiourea catalyst†
Received 9th September 2009, Accepted 7th October 2009
First published on 29th October 2009
Abstract
The current article reports an organocatalytic strategy for the asymmetric catalysis of chiral oxindoles bearing 3-position all-carbon quaternary stereocenters. Accordingly, highly enantioselective Michael addition reactions of 3-substituted oxindoles to terminal alkenes have been developed by utilizing a bifunctional tertiary-amine thiourea catalyst. The reactions accommodate a number of Michael donor compounds (different substituted 3-aryl or methyl oxindoles), and Michael acceptor compounds (vinyl ketones and vinyl sulfones) to give the desired oxindole products with moderate to excellent yields (up to 99%) and moderate to excellent enantioselectivities (up to 91% ee).
Introduction
Asymmetric construction of all-carbon quaternary stereocenters has attracted a great deal of effort over recent decades and remains a challenging synthetic task.1 One particular context is in the synthesis of biologically and pharmaceutically active oxindole alkaloids wherein 3-position quaternary centers are a common structural motif.2 Accordingly, a number of synthetic transformations such as allylic alkylation, aldol reaction, Heck reaction, Michael addition reaction and cyanoamidation reaction3–8 have been developed in order to address this synthetic challenge. A straightforward approach for the construction of oxindoles bearing a 3-quaternary center would be the direct Michael addition of 3-substituted oxindoles to terminal activated alkenes (Fig. 1). Though easily conceived, an asymmetric catalytic method for such reactions has not been achieved until recently. Maruoka reported an enantioselective Michael addition reaction of 3-aryloxindoles to methyl vinyl ketone (MVK) catalyzed by a chiral quaternary tetraalkylphosphonium salt.9 Herein, we present a distinctive organocatalytic approach for the Michael addition of 3-substituted oxindoles to activated terminal alkenes by utilizing the well-proved bifunctional catalytic features of tertiary amine-thiourea catalysts.10 A number of 3-substituted oxindoles (R2 = Ar and CH3) and terminal alkenes (vinyl ketones and vinyl sulfones) are incorporated into this synthetic strategy and the detailed results are presented here.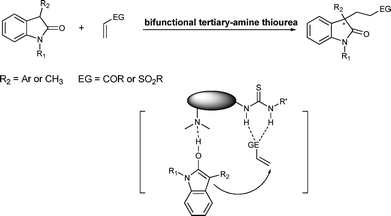 |
| | Fig. 1 Strategy of bifunctional tertiary-amine thiourea catalyzed Michael reactions of oxindoles to terminal alkenes. | |
Results and discussion
Bifunctional tertiary-amine thiourea catalyzed Michael addition reaction of 3-aryl oxindoles to vinyl ketones
The Michael addition reaction of oxindole 1a to MVK was first selected as our initial testing reaction. And four widely used bifunctional tertiary-amine thiourea catalysts 4a–4d11–14 (20 mol%) with different chiral scaffolds were screened in the current model reaction at room temperature. To our delight, all of the chiral thioureas 4a–4d exhibited high catalytic activity and the Michael addition products were cleanly isolated with quantitative yields (entries 2–5 in Table 1). No reaction was observed in the absence of catalyst (Table 1, entry 1). Among the four types of thiourea catalysts tested, catalyst 4d was found to give the optimal enantioselectivity (99% yield and 61% ee, entry 5 in Table 1). In addition, the racemic product obtained with a simple diamine catalyst 4e suggested an obvious bifunctional catalytic mode in the current reaction (Table 1, entry 6).
| Entry | Catalysts | Solvent | Time/h | Yield b(%) | eec(%) |
|---|
| The reaction was carried out on a 0.1 mmol scale in 200 μL solvent at 4 °C, and the molar ratio of oxindole 1d/2a is 1/2. Isolated yield. Determined by HPLC. Not determined. The reaction was carried out on a 0.1 mmol scale in 1 mL toluene with 4 Å molecular sieves at 4 °C, and the molar ratio of oxindole 1d/2a is 1/2. The reaction was carried out on a 0.1 mmol scale in 1 mL toluene at −60 °C, and the molar ratio of oxindole 1d/2a is 1/2. The reaction was carried out on a 0.1 mmol scale in 1 mL toluene with 4 Å molecular sieves at −60 °C, and the molar ratio of oxindole 1d/2a is 1/2. |
|---|
| 1 | no catalyst | Toluene | 24 | trace | ndd |
| 2 | 4a | Toluene | 2 | 98 | 40 |
| 3 | 4b | Toluene | 2 | 99 | 45 |
| 4 | 4c | Toluene | 2 | 97 | 37 |
| 5 | 4d | Toluene | 2 | 99 | 61 |
| 6 | 4e | Toluene | 2 | 96 | rac |
| 7 | 4d | CH2Cl2 | 2 | 99 | 56 |
| 8 | 4d | CHCl3 | 2 | 99 | 55 |
| 9 | 4d | ClCH2CH2Cl | 2 | 96 | 58 |
| 10 | 4d | Benzene | 2 | 97 | 58 |
| 11 | 4d | THF | 2 | 78 | 47 |
| 12 | 4de | Toluene | 2 | 99 | 67 |
| 13 | 4df | Toluene | 36 | 94 | 79 |
| 14 | 4dg | Toluene | 36 | 98 | 82 |
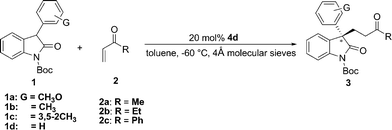
With 4d as the optimal catalyst, we next examined different solvents. As illustrated in Table 1, the initial selected toluene gave the best result among a range of screened solvents (Table 1, entries 5 and 7–11). The best result was achieved when the reaction was carried out at −60 °C in toluene in the presence of 4 Å molecular sieves (Table 1, entry 14). Under this condition, the yield was almost the same while the ee value was improved to 82%.
Under the optimal reaction conditions, the substrate scope was next explored (Table 2). A series of 3-aryl-N-Boc oxindoles 1a–1d (Michael donor compounds) and vinyl ketones 2a–2c (Michael acceptor compounds) were examined. As summarized in Table 2, vinyl ketones including methyl vinyl ketone (MVK), ethyl vinyl ketone (EVK) and phenyl vinyl ketone (PVK) demonstrated equally good activity in reacting with different substituted 3-aryl-N-Boc oxindoles and the reactions generally furnished the desired Michael products in quantitative yields. Inspection of the results in Table 2 suggested similar chiral inductions were normally observed with all three vinyl ketones with slightly better ees in cases of MVK and PVK. The best enantioselectivity (88% ee) was obtained in the reaction of MVK and 1c (Table 1, entry 3).
Table 2 Asymmetric Michael addition reaction of 3-aryl oxindoles to different vinyl ketonesa
| Entry | Terminal alkene | Oxindole | Time/h | Yield b(%) | eec(%) |
|---|
| The reaction was carried out on a 0.1 mmol scale in 1 mL toluene with 4 Å molecular sieves at −60 °C, and the molar ratio of oxindole 1/2 is 1/2. Isolated yield. Determined by HPLC. The absolute configuration of the Michael adduct 3d was determined to be S by comparison with literature.9 |
|---|
| 1 |  2a 2a | G = 4-CH3O (1a) | 48 | 3a: 92 | 65 |
| 2 | 4-CH3 (1b) | 36 | 3b: 99 | 81 |
| 3 | 3,5-(Me)2 (1c) | 36 | 3c: 98 | 88 |
| 4 | H (1d) | 36 | 3d: 98 | 82(S)d |
| 5 |  2b 2b | G = 4-CH3O (1a) | 72 | 3e: 93 | 46 |
| 6 | 4-CH3 (1b) | 36 | 3f: 99 | 63 |
| 7 | 3,5-(Me)2 (1c) | 36 | 3g: 99 | 76 |
| 8 | H (1d) | 36 | 3h: 99 | 61 |
| 9 |  2c 2c | G = 4-CH3O (1a) | 60 | 3i: 96 | 85 |
| 10 | 4-CH3 (1b) | 48 | 3j: 99 | 72 |
| 11 | 3,5-(Me)2 (1c) | 48 | 3k: 99 | 60 |
| 12 | H (1d) | 48 | 3l: 99 | 76 |
Bifunctional tertiary-amine thiourea catalyzed Michael addition reaction of 3-aryl oxindoles to vinyl sulfones
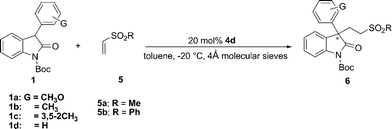
Vinyl sulfones, which bear two S![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O moieties that enable double H-bonding with the N–Hs of thiourea,15 were next attempted in the current reaction with the hope that better stereocontrol might be achieved with double H-bonding interactions. Indeed, the reactions of vinyl sulfone provided generally good enantioselectivity (Table 3, 82–91% ee), albeit with lower reactivity than those of vinyl ketones. Phenyl vinyl sulfone 5b demonstrated higher activity than the methyl vinyl sulfone 5a. Various 3-aromatic substituted oxindoles worked well with vinyl sulfone to give the desired product with yield ranging from 48% to 80%.
O moieties that enable double H-bonding with the N–Hs of thiourea,15 were next attempted in the current reaction with the hope that better stereocontrol might be achieved with double H-bonding interactions. Indeed, the reactions of vinyl sulfone provided generally good enantioselectivity (Table 3, 82–91% ee), albeit with lower reactivity than those of vinyl ketones. Phenyl vinyl sulfone 5b demonstrated higher activity than the methyl vinyl sulfone 5a. Various 3-aromatic substituted oxindoles worked well with vinyl sulfone to give the desired product with yield ranging from 48% to 80%.
Table 3 Asymmetric Michael addition reaction of 3-aryl oxindoles to different vinyl sulfonesa
| Entry | Terminal alkene | Oxindole | Time/d | Yield b(%) | eec(%) |
|---|
| The reaction was carried out on a 0.1 mmol scale in 200 μL toluene with 4 Å molecular sieves at −20 °C, and the molar ratio of oxindole 1/5 is 1/3. Isolated yield. Determined by HPLC. Not determined. |
|---|
| 1 |  5a 5a | G = 4-CH3O | 8 | trace | ndd |
| 2 | 4-CH3 | 8 | 6a: 48 | 86 |
| 3 | 3,5-2CH3 | 8 | 6b: 55 | 87 |
| 4 | H | 6 | 6c: 51 | 91 |
| 5 |  5b 5b | G = 4-CH3O | 6 | 6d: 60 | 88 |
| 6 | 4-CH3 | 6 | 6e: 75 | 82 |
| 7 | 3,5-2CH3 | 6 | 6f: 72 | 90 |
| 8 | H | 6 | 6g: 80 | 87 |
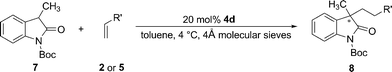
In addition to the 3-aromatic substituted oxindoles, 3-alkyl substituted oxindoles such as oxindole 7 have also been examined in the reactions and the results were summarized in Table 4. The reactions with vinyl ketones proceeded smoothly to give the desired Michael products in excellent yields, however, only low enantioselectivities were obtained in these cases (Table 4, entries 1–3). In accordance with previous observations, the vinyl sulfones reacted sluggishly with oxindole 7, but good ee (84%) could still be achieved in the reaction of phenyl vinyl sulfone 5b and oxindole (Table 4, entry 5).
| Entry | Terminal alkene | Time/h | Yield b(%) | eec(%) |
|---|
| The reaction was carried out on a 0.1 mmol scale in 200 μL toluene with 4 Å molecular sieves at 4 °C, and the molar ratio of oxindole/terminal alkene is 1/2. Isolated yield. Determined by HPLC. Not determined. |
|---|
| 1 | 2a | 72 | 8a: 90 | 22 |
| 2 | 2b | 72 | 8b: 99 | 17 |
| 3 | 2c | 72 | 8c: 99 | 48 |
| 4 | 5a | 168 | 8d: trace | ndd |
| 5 | 5b | 96 | 8e: 48 | 84 |
Conclusion
In summary, we have presented highly enantioselective Michael addition reactions of 3-substituted oxindoles to terminal alkenes using a bifunctional tertiary-amine thiourea organocatalyst. This study provided a rather mild procedure for the synthesis of multifunctional chiral oxindole compounds bearing all carbon-substituted quaternary stereocenters with moderate to excellent enantioselectivities. The reaction scope is substantial and a number of 3-aryl or methyl oxindoles could be successfully applied in current studied Michael addition system.Experimental section
General remarks
Commercial reagents were used as received, unless otherwise stated. Chemical shifts are reported in ppm from tetramethylsilane with the solvent resonance as the internal standard. The following abbreviations were used to designate chemical shift multiplicities: s = singlet, d = doublet, t = triplet, q = quartet, h = heptet, m = multiplet, br = broad. All first-order splitting patterns were assigned on the basis of the appearance of the multiplet. Splitting patterns that could not be easily interpreted are designated as multiplet (m) or broad (br). Elemental analysis was obtained from thermoQuest (Flash 1112EA, ITALY). Mass spectra were obtained using electron ionization (EI) mass spectrometer. Catalysts 4a,114b,124c,134d14 and 4e16 were synthesized from the literature methods. Michael products 3a, 3b, 3d and 3h were known compounds.9General experimental procedure for Michael reaction of 1 and 2
To a stirred solution of 3-aryl-N-Boc oxindole 1 (0.1 mmol) and vinyl ketones 2 (2.0 equiv.) in dry toluene (1 mL) was added thiourea-catalyst (0.2 equiv.) at −60 °C with 4 Å molecular sieves. After the reaction completed, the reaction solution was concentrated in vacuo and the crude was purified by flash chromatography to afford the product.3c. The Michael product was synthesized according to the general procedure as white solid in 98% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.94 (1H, d, J = 8.51 Hz), 7.37 (1H, td, J = 8.51, 1.65 Hz), 7.23–7.15 (2H, m), 6.89 (3H, s), 2.77–2.68 (1H, m), 2.51–2.25 (8H, m), 2.11–2.01 (4H, m), 1.63 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 207.12, 176.69, 149.30, 139.77, 139.38, 138.20, 130.82, 129.45, 128.57, 124.74, 124.65, 115.21, 84.56, 55.80, 38.74, 31.63, 29.96, 28.11, 21.43 ppm; HRMS (EI+): calcd. for [C25H29NO4] 407.2097; found 407.2101. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :hexane = 1:9), 1.0 mL min−1; tR = 4.3 min (major), 5.3 min (minor).
:hexane = 1:9), 1.0 mL min−1; tR = 4.3 min (major), 5.3 min (minor). 3e. The Michael product was synthesized according to the general procedure as white solid in 93% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.94 (1H, d, J = 8.23 Hz), 7.40–7.34 (1H, m), 7.24–7.16 (4H, m), 6.83 (2H, d, J = 9.06 Hz), 3.76 (3H, s), 2.76–2.66 (1H, m), 2.52–2.43 (1H, m), 2.37–2.14 (3H, m), 2.07–1.97 (1H, m), 1.63 (9H, s), 0.95 (3H, t, J = 7.14 Hz) ppm; 13C NMR (CDCl3, 75 MHz): δ 209.80, 176.77, 159.08, 149.30, 139.85, 131.53, 130.56, 128.62, 128.13, 124.76, 124.68, 115.27, 114.07, 84.55, 55.39, 55.25, 37.48, 35.94, 31.92, 28.09, 7.63 ppm; HRMS (EI+): calcd. for [C25H29NO5] 423.2046; found 423.2049. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 7.8 min (major), 11.7 min (minor). 3f. The Michael product was synthesized according to the general procedure as colorless oil in 99% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.94 (1H, d, J = 7.96 Hz), 7.39–7.33 (1H, m), 7.24–7.09 (6H, m), 2.78–2.68 (1H, m), 2.55–2.45 (1H, m), 2.38–2.14 (6H, m), 2.10–1.99 (1H, m), 1.63 (9H, s), 0.95 (3H, t, J = 7.14 Hz) ppm; 13C NMR (CDCl3, 75 MHz): δ 209.80, 176.69, 149.29, 139.84, 137.48, 136.57, 130.61, 129.41, 128.61, 126.82, 124.75, 124.70, 115.25, 84.53, 55.75, 37.46, 35.46, 31.77, 28.09, 20.91, 7.64 ppm; HRMS (EI+): calcd. for [C25H29NO4] 407.2097; found 407.2101. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 6.0 min (major), 8.7 min (minor). 3g. The Michael product was synthesized according to the general procedure as white solid in 99% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.93 (1H, d, J = 7.96 Hz), 7.39–7.33 (1H, m), 7.23–7.15 (2H, m), 6.89 (3H, m), 2.78–2.68 (1H, m), 2.54–2.44 (1H, m), 2.37–2.16 (9H, m), 2.08–1.98 (1H, m), 1.64 (9H, s), 0.95 (3H, t, J = 7.14 Hz) ppm; 13C NMR (CDCl3, 75 MHz): δ 209.89, 176.73, 149.31, 139.78, 139.43, 138.18, 130.81, 129.43, 128.54, 124.74, 124.71, 124.66, 115.19, 84.54, 55.92, 37.43, 35.95, 31.74, 28.11, 21.43, 7.65 ppm; Anal. Calcd. for C26H31NO4: C, 74.08; H, 7.41; N, 3.32. Found: C, 73.83; H, 7.47; N, 3.14. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 4.2 min (major), 5.2 min (minor). 3i. The Michael product was synthesized according to the general procedure as white solid in 96% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.95 (1H, d, J = 8.23 Hz), 7.81–7.78 (2H, m), 7.53–7.19 (10H, m), 6.85–6.82 (2H, m), 3.76 (3H, s), 2.97–2.83 (2H, m), 2.67–2.55 (2H, m), 1.64 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 198.80, 176.81, 159.12, 149.33, 139.84, 136.60, 133.07, 131.50, 130.61, 128.70, 128.51, 128.19, 128.01, 124.74, 115.35, 114.11, 84.56, 55.52, 55.26, 33.86, 32.60, 28.12 ppm; Anal. Calcd. for C29H29NO5: C, 73.87; H, 6.20; N, 2.97. Found: C, 72.76; H, 6.21; N, 2.87. The enantiomeric excess was determined by HPLC with an OD-H column at 210 nm (2-propanol:hexane = 1:49), 1.0 mL min−1; tR = 16.1 min (minor), 23.9 min (major). 3j. The Michael product was synthesized according to the general procedure as white solid in 99% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.94 (1H, d, J = 8.23 Hz), 7.81–7.78 (2H, m), 7.50 (1H, t, J = 7.14 Hz), 7.41–7.33 (3H, m), 7.25–7.10 (6H, m), 2.98–2.84 (2H, m), 2.70–2.57 (2H, m), 2.30 (3H, s), 1.63 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 198.81, 176.72, 149.33, 139.83, 137.52, 136.61, 136.54, 133.07, 130.66, 129.46, 128.68, 128.51, 128.01, 126.87, 124.76, 124.72, 115.32, 84.55, 55.88, 33.84, 32.44, 28.12, 20.94 ppm; Anal. Calcd. for C29H29NO4: C, 76.46; H, 6.42; N, 3.07. Found: C, 76.58; H, 6.49; N, 2.93. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 7.6 min (minor), 8.7 min (major). 3k. The Michael product was synthesized according to the general procedure as white solid in 99% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.94 (1H, d, J = 8.23 Hz), 7.81–7.78 (2H, m), 7.50 (1H, t, J = 7.14 Hz), 7.41–7.33 (3H, m), 7.25–7.18 (2H, m), 6.94–6.89 (3H, m), 2.97–2.84 (2H, m), 2.68–2.56 (2H, m), 2.26 (6H, s), 1.64 (9H, m) ppm; 13C NMR (CDCl3, 75 MHz): δ 198.91, 176.77, 149.35, 139.76, 139.39, 138.24, 136.61, 133.08, 130.86, 129.48, 128.61, 128.52, 128.03, 124.77, 124.72, 115.27, 84.56, 56.06, 33.81, 32.44, 28.14, 21.46 ppm; Anal. Calcd. for C30H31NO4: C, 76.73; H, 6.65; N, 2.98. Found: C, 74.61; H, 6.64; N, 2.93. The enantiomeric excess was determined by HPLC with an OD-H column at 210 nm (2-propanol:hexane = 1:49), 1.0 mL min−1; tR = 7.9 min (minor), 8.8 min (major). 3l. The Michael product was synthesized according to the general procedure as white solid in 99% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.95 (1H, d, J = 8.23 Hz), 7.81–7.78 (2H, m), 7.51 (1H, t, J = 7.14 Hz), 7.41–7.20 (10H, m), 2.99–2.85 (2H, m), 2.72–2.58 (2H, m), 1.64 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 198.75, 176.60, 149.28, 139.84, 139.49, 136.59, 133.09, 130.46, 128.77, 128.52, 128.01, 127.75, 127.01, 124.79, 115.35, 84.64, 56.17, 33.81, 32.51, 28.12 ppm; HRMS (EI+): calcd. for [C28H27NO4] 441.1940; found 441.1944. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 7.4 min (minor), 8.1 min (major). General experimental procedure for Michael reaction of 1 and 5
To a stirred solution of 3-aryl-N-Boc oxindole 1 (0.1 mmol) and vinyl sulfones 5 (3.0 equiv.) in dry toluene (200 μL) was added thiourea-catalyst (0.2 equiv.) at −20 °C with 4 Å molecular sieves. After the reaction completed, the reaction solution was concentrated in vacuo and the crude was purified by flash chromatography to afford the product.6a. The Michael product was synthesized according to the general procedure as white solid in 48% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.92 (1H, d, J = 8.23 Hz), 7.42–7.35 (1H, m), 7.26–7.12 (6H, m), 2.98–2.86 (5H, m), 2.79–2.59 (2H, m), 2.31 (3H, s), 1.63 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 175.89, 148.94, 139.57, 138.08, 135.10, 129.72, 129.35, 129.23, 126.62, 125.10, 124.50, 115.59, 84.99, 55.02, 50.57, 40.62, 30.49, 28.07, 20.92 ppm; HRMS (EI+): calcd. for [C23H27NO5S] 429.1610; found 429.1613. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 13.3 min (minor), 16.1 min (major). 6b. The Michael product was synthesized according to the general procedure as white solid in 55% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.92 (1H, d, J = 7.96 Hz), 7.42–7.36 (1H, m), 7.26–7.24 (2H, m), 6.92–6.90 (3H, m), 2.97–2.87 (5H, m), 2.79–2.58 (2H, m), 2.27 (6H, s), 1.64 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 175.95, 148.94, 139.52, 138.60, 138.03, 129.88, 129.49, 129.18, 125.12, 124.51, 124.42, 115.54, 85.00, 55.20, 50.56, 40.61, 30.46, 28.09, 21.44 ppm; Anal. Calcd. for C23H27NO5S: C, 64.31; H, 6.34; N, 3.26. Found: C, 64.67; H, 6.62; N, 3.10. The enantiomeric excess was determined by HPLC with an OD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 7.4 min (minor), 8.1 min (major). 6c. The Michael product was synthesized according to the general procedure as white solid in 51% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.93 (1H, d, J = 8.23 Hz), 7.43–7.26 (8H, m), 2.99–2.62 (7H, m), 1.64 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 175.77, 148.89, 139.60, 138.08, 129.32, 129.14, 129.05, 128.20, 126.76, 125.14, 124.56, 115.63, 82.08, 55.31, 50.55, 40.64, 30.53, 28.07 ppm; HRMS (EI+): calcd. for [C22H25NO5S] 415.1453; found 415.1457. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:19), 1.0 mL min−1; tR = 26.4 min (minor), 36.3 min (major). 6d. The Michael product was synthesized according to the general procedure as colorless oil in 60% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.91–7.82 (3H, m), 7.69–7.53 (3H, m), 7.37 (1H, td, J = 7.96, 1.37 Hz), 7.23–7.09 (4H, m), 6.80 (2H, d, J = 8.78 Hz), 3.76 (3H, s), 3.12–3.02 (1H, m), 2.88–2.67 (2H, m), 2.55–2.45 (1H, m), 1.60 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 175.72, 159.33, 148.95, 139.60, 138.60, 133.88, 129.90, 129.39, 129.30, 129.14, 128.11, 128.04, 124.93, 124.47, 115.58, 114.30, 84.88, 55.28, 54.59, 51.87, 30.93, 28.05 ppm; HRMS (EI+): calcd. for [C28H29NO6S] 507.1716; found 507.1720. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 19.4 min (minor), 21.3 min (major). 6e. The Michael product was synthesized according to the general procedure as white solid in 75% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.91–7.82 (3H, m), 7.69–7.53 (3H, m), 7.37 (1H, td, J = 7.96, 1.37 Hz), 7.22–7.08 (6H, m), 3.12–3.02 (1H, m), 2.90–2.69 (2H, m), 2.55–2.45 (1H, m), 2.29 (3H, s), 1.60 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 175.63, 148.94, 139.59, 138.60, 137.97, 135.03, 133.87, 129.64, 129.38, 129.11, 128.12, 126.67, 124.93, 124.45, 115.55, 84.87, 54.94, 51.85, 30.79, 28.05, 20.91 ppm; Anal. Calcd. for C28H29NO5S: C, 68.41; H, 5.95; N, 2.85. Found: C, 67.44; H, 5.97; N, 2.66. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 13.3 min (minor), 16.2 min (major). 6f. The Michael product was synthesized according to the general procedure as white solid in 72% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.91–7.82 (3H, m), 7.69–7.53 (3H, m), 7.36 (1H, td, J = 7.96, 1.37 Hz), 7.22–7.07 (2H, m), 6.89 (1H, s), 6.78 (2H, s), 3.13–3.03 (1H, m), 2.91–2.70 (2H, m), 2.54–2.45 (1H, m), 2.22 (6H, s), 1.61 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 175.66, 148.97, 139.53, 138.65, 138.51, 137.85, 133.86, 129.79, 129.56, 129.38, 129.04, 128.10, 124.92, 124.50, 124.47, 115.50, 84.87, 55.10, 51.82, 30.75, 28.07, 21.41 ppm; Anal. Calcd. for C29H31NO5S: C, 68.89; H, 6.18; N, 2.77. Found: C, 68.04; H, 6.18; N, 2.60. The enantiomeric excess was determined by HPLC with an OD-H column at 210 nm (2-propanol:hexane = 1:19), 1.0 mL min−1; tR = 12.0 min (minor), 16.5 min (major). 6g. The Michael product was synthesized according to the general procedure as white solid in 80% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.84–7.75 (3H, s), 7.60 (1H, t, J = 7.14 Hz), 7.49 (2H, t, J = 7.41 Hz), 7.31 (1H, td, J = 7.96, 1.37 Hz), 7.22–7.12 (6H, m), 7.05–7.03 (1H, d, J = 7.41 Hz), 3.06–2.93 (1H, m), 2.82–2.65 (2H, m), 2.54–2.42 (1H, m), 1.54 (9H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 175.52, 148.89, 139.60, 138.54, 137.99, 133.93, 129.41, 129.22, 129.13, 128.97, 128.12, 126.82, 125.00, 124.52, 115.61, 84.97, 55.22, 51.82, 30.84, 28.06 ppm; Anal. Calcd. for C27H27NO5S: C, 67.90; H, 5.70; N, 2.93. Found: C, 67.82; H, 5.71; N, 2.73. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 11.3 min (minor), 13.8 min (major). General experimental procedure for Michael reaction of 7 and 2 (or 5)
To a stirred solution of 3-methyl-N-Boc oxindole 5 (0.1 mmol) and terminal alkene (2.0 equiv.) in dry toluene (200 μL) was added thiourea-catalyst (0.2 equiv.) at 4 °C with 4 Å molecular sieves. After the reaction completed, the reaction solution was concentrated in vacuo and the crude was purified by flash chromatography to afford the product.8a. The Michael product was synthesized according to the general procedure as white solid in 90% overall yield.1H NMR (300 MHz, CDCl3): δ 7.85 (1H, d, J = 8.23 Hz), 7.33–7.27 (1H, m), 7.18–7.16 (2H, m), 2.35–1.95 (7H, m), 1.66 (9H, s), 1.43 (3H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 207.25, 178.75, 149.28, 139.01, 132.16, 128.26, 124.71, 122.57, 115.04, 84.48, 47.72, 38.48, 32.47, 29.89, 28.11, 24.70 ppm; HRMS (EI+): calcd. for [C18H23NO4] 317.1627; found 317.1630. The enantiomeric excess was determined by HPLC with an OD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 5.3 min (minor), 5.7 min (major). 8b. The Michael product was synthesized according to the general procedure as colorless oil in 99% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.84 (1H, d, J = 8.23 Hz), 7.33–7.27 (1H, m), 7.17–7.16 (2H, m), 2.33–2.10 (5H, m), 2.02–1.92 (1H, m), 1.66 (9H, s), 1.43 (3H, s), 0.94 (3H, t, J = 7.41 Hz) ppm; 13C NMR (CDCl3, 75 MHz): δ 209.96, 178.79, 149.28, 139.01, 132.18, 128.22, 124.67, 122.65, 115.01, 84.45, 47.83, 37.19, 35.89, 32.54, 28.11, 24.72, 7.62 ppm; HRMS (EI+): calcd. for [C19H25NO4] 331.1784; found 331.1787. The enantiomeric excess was determined by HPLC with an OD-H column at 210 nm (2-propanol:hexane = 1:49), 1.0 mL min−1; tR = 7.9 min (minor), 9.1 min (major). 8c. The Michael product was synthesized according to the general procedure as white solid in 99% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.79–7.70 (3H, m), 7.46–7.41 (1H, m), 7.34–7.29 (2H, m), 7.25–7.08 (3H, m), 2.85–2.74 (1H, m), 2.53–2.42 (1H, m), 2.35–2.12 (2H, m), 1.59 (9H, m), 1.40 (3H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 198.94, 178.82, 149.33, 139.01, 136.59, 133.05, 132.28, 128.50, 128.30, 127.98, 124.75, 122.60, 115.09, 84.47, 47.93, 33.51, 33.08, 28.14, 24.85 ppm; Anal. Calcd. for C23H25NO4: C, 72.80; H, 6.64; N, 3.69. Found: C, 72.47; H, 6.67; N, 3.54. The enantiomeric excess was determined by HPLC with an OD-H column at 210 nm (2-propanol:hexane = 1:49), 1.0 mL min−1; tR = 9.6 min (minor), 10.5 min (major). 8e. The Michael product was synthesized according to the general procedure as yellow oil in 48% overall yield. 1H NMR (300 MHz, CDCl3): δ 7.85–7.80 (3H, m), 7.69–7.53 (3H, m), 7.34–7.29 (1H, m), 7.21–7.10 (2H, m), 2.98 (1H, td, J = 13.45, 4.94 Hz), 2.76 (1H, td, J = 13.45, 4.94 Hz), 2.34–2.10 (2H, m), 1.64 (9H, s), 1.41 (3H, s) ppm; 13C NMR (CDCl3, 75 MHz): δ 177.61, 148.93, 138.85, 138.67, 133.86, 130.93, 129.36, 128.79, 128.05, 125.03, 122.40, 125.30, 84.82, 51.64, 47.17, 31.16, 28.08, 24.45 ppm; HRMS (EI+): calcd. for [C22H25NO5S] 415.1453; found 415.1457. The enantiomeric excess was determined by HPLC with an AD-H column at 210 nm (2-propanol:hexane = 1:9), 1.0 mL min−1; tR = 11.0 min (minor), 12.6 min (major).
Acknowledgements
We would likely to thank the Natural Science Foundation (NSFC 20702052 and 20902091), MOST (2008CB617501, 2009ZX09501-018) and the Chinese Academy of Sciences for their support.Notes and references
- Reviews on the catalytic enantioselective construction of quaternary chiral centers:
(a) J. Christoffers and A. Baro, Angew. Chem., Int. Ed., 2003, 42, 1688–1690 CrossRef CAS;
(b) D. J. Ramon and M. Yus, Curr. Org. Chem., 2004, 8, 149–183 CrossRef CAS;
(c) E. A. Peterson and L. E. Overman, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 11943–11948 CrossRef CAS;
(d) J. Christoffers and A. Baro, Adv. Synth. Catal., 2005, 347, 1473–1482 CrossRef CAS.
- For reviews see:
(a) A. B. Dounay and L. E. Overman, Chem. Rev., 2003, 103, 2945–2963 CrossRef CAS;
(b) H. Lin and S. J. Danishefsky, Angew. Chem., Int. Ed., 2003, 42, 36–51 CrossRef CAS;
(c) C. V. Galliford and K. A. Scheidt, Angew. Chem., Int. Ed., 2007, 46, 8748–8758 CrossRef CAS. Other examples:
(d) X. Zhang and C. D. Smith, Mol. Pharmacol., 1996, 49, 288–294 CAS;
(e) H. C. Malinakova and L. S. Liebeskind, Org. Lett., 2000, 2, 4083–4086 CrossRef CAS;
(f) X. Z. Wearing and J. M. Cook, Org. Lett., 2002, 4, 4237–4240 CrossRef CAS;
(g) B. K. Albrecht and R. M. Williams, Org. Lett., 2003, 5, 197–200 CrossRef CAS;
(h) A. H. Abadi, S. M. Abou-Seri, D. E. Abdel-Rahman, C. Klein, O. Lozach and L. Meijer, Eur. J. Med. Chem., 2006, 41, 296–305 CrossRef CAS;
(i) S. E. Reisman, J. M. Ready, M. M. Weiss, A. Hasuoka, M. Hirata, K. Tamaki, T. V. Ovaska, C. J. Smith and J. L. Wood, J. Am. Chem. Soc., 2008, 130, 2087–2100 CrossRef CAS.
-
(a) B. M. Trost and Y. Zhang, J. Am. Chem. Soc., 2006, 128, 4590–4591 CrossRef CAS;
(b) B. M. Trost, N. Cramer and S. M. Silverman, J. Am. Chem. Soc., 2007, 129, 12396–12397 CrossRef CAS;
(c) B. M. Trost and Y. Zhang, J. Am. Chem. Soc., 2007, 129, 14548–14549 CrossRef CAS and references therein.
- S. Ogawa, N. Shibata, J. Inagaki, S. Nakamura, T. Toru and M. Shiro, Angew. Chem., Int. Ed., 2007, 46, 8666–8669 CrossRef CAS and references therein.
- A. B. Dounay, K. Hatanaka, J. J. Kodanko, M. Oestreich, L. E. Overman, L. A. Pfeifer and M. M. Weiss, J. Am. Chem. Soc., 2003, 125, 6261–6271 CrossRef CAS and references therein.
-
(a) T. Bui, S. Syed and C. F. III Barbas, J. Am. Chem. Soc., 2009, 131, 8758–8759 CrossRef CAS;
(b) Y. Kato, M. Furutachi, Z. Chen, H. Mitsunuma, S. Matsunaga and M. Shibasaki, J. Am. Chem. Soc., 2009, 131, 9168–9169 CrossRef CAS.
- Y. Yasui, H. Kamisaki and Y. Takemoto, Org. Lett., 2008, 10, 3303–3306 CrossRef CAS.
- Other selected examples of catalytic asymmetric synthesis of oxindoles with quaternary carbon stereocenters:
(a) S. Lee and J. F. Hartwig, J. Org. Chem., 2001, 66, 3402–3415 CrossRef CAS;
(b) I. D. Hills and G. C. Fu, Angew. Chem., Int. Ed., 2003, 42, 3921–3924 CrossRef CAS;
(c) S. A. Shaw, P. Aleman and E. Vedejs, J. Am. Chem. Soc., 2003, 125, 13368–13369 CrossRef CAS;
(d) E. P. Kündig, T. M. Seidel, Y.-X. Jia and G. Bernardinelli, Angew. Chem., Int. Ed., 2007, 46, 8484–8487 CrossRef;
(e) T. B. Poulsen, L. Bernardi, J. Alemán, J. Overgaard and K. A. Jørgensen, J. Am. Chem. Soc., 2007, 129, 441–449 CrossRef CAS;
(f) B. K. Corkey and F. D. Toste, J. Am. Chem. Soc., 2007, 129, 2764–2765 CrossRef CAS;
(g) X. Tian, K. Jiang, J. Peng, W. Du and Y.-C. Chen, Org. Lett., 2008, 10, 3583–3586 CrossRef CAS;
(h) E. C. Linton and M. C. Kozlowski, J. Am. Chem. Soc., 2008, 130, 16162–16163 CrossRef CAS;
(i) T. A. Duffey, S. A. Shaw and E. Vedejs, J. Am. Chem. Soc., 2009, 131, 14–15 CrossRef CAS;
(j) P. Galzerano, G. Bencivenni, F. Pesciaioli, A. Mazzanti, B. Giannichi, L. Sambri, G. Bartoli and P. Melchiorre, Chem.–Eur. J., 2009, 15, 7846–7849 CrossRef CAS.
- R. He, C. Ding and K. Maruoka, Angew. Chem., Int. Ed., 2009, 48, 4559–4561 CrossRef CAS.
- Reviews on bifunctional thiourea catalysis:
(a) M. S. Taylor and E. N. Jacobsen, Angew. Chem., Int. Ed., 2006, 45, 1520–1543 CrossRef CAS;
(b) A. G. Doyle and E. N. Jacobsen, Chem. Rev., 2007, 107, 5713–5743 CrossRef CAS;
(c) S. J. Connon, Chem. Commun., 2008, 2499–2510 RSC.
- X. Li, H. Deng, B. Zhang, S. Z. Luo and J.-P. Cheng, Chem.–Eur. J. DOI:10.1002/chem.200902430.
- For leading examples of catalyst 4b:
(a) T. Okino, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2003, 125, 12672–12673 CrossRef CAS;
(b) T. Okino, Y. Hoashi, T. Furukawa, X.-N. Xu and Y. Takemoto, J. Am. Chem. Soc., 2005, 127, 119–125 CrossRef CAS;
(c) T. Inokuma, Y. Hoashi and Y. Takemoto, J. Am. Chem. Soc., 2006, 128, 9413–9419 CrossRef CAS;
(d) T. Okino, S. Nakamura, T. Furukawa and Y. Takemoto, Org. Lett., 2004, 6, 625–627 CrossRef CAS;
(e) Y. Hoashi, T. Yabuta and Y. Takemoto, Tetrahedron Lett., 2004, 45, 9185–9188 CrossRef CAS;
(f) Y. Hoashi, T. Yabuta, P. Yuan, H. Miyabe and Y. Takemoto, Tetrahedron, 2006, 62, 365–374 CrossRef CAS.
- For leading examples of catalyst 4c:
(a) B. Vakulya, S. Varga, A. Csámpai and T. Soós, Org. Lett., 2005, 7, 1967–1969 CrossRef CAS;
(b) B. J. Li, L. Jiang, M. Liu, Y. C. Chen, L. S. Ding and Y. Wu, Synlett, 2005, 603–606 CAS;
(c) S. H. McCooey and S. J. Connon, Angew. Chem., Int. Ed., 2005, 44, 6367–6370 CrossRef CAS;
(d) J. Ye, D. J. Dixon and P. S. Hynes, Chem. Commun., 2005, 4481–4483 RSC;
(e) Y.-Q. Wang, J. Song, R. Hong, H. M. Li and L. Deng, J. Am. Chem. Soc., 2006, 128, 8156–8159 CrossRef CAS;
(f) J. Song, H.-W. Shi and L. Deng, Org. Lett., 2007, 9, 603–606 CrossRef CAS;
(g) J. Song, Y. Wang and L. Deng, J. Am. Chem. Soc., 2006, 128, 6048–6049 CrossRef CAS;
(h) J. Wang, H. Li, X. H. H. Yu, L. S. Zu and W. Wang, Org. Lett., 2005, 7, 4293–4296 CrossRef CAS.
- For leading examples of catalyst 4d: B. Han, Q.-P. Liu, R. Li, X. Tian, X.-F. Xiong, J.-G. Deng and Y.-C. Chen, Chem.–Eur. J., 2008, 14, 8094–8097 Search PubMed ; Also see ref. 8g and 15c.
- For organocatalyzed asymmetric Michael addition to vinyl sulfones, see:
(a) H. Li, J. Song, X. Liu and L. Deng, J. Am. Chem. Soc., 2005, 127, 8948–8949 CrossRef CAS;
(b) S. Mosśe and A. Alexakis, Org. Lett., 2005, 7, 4361–4364 CrossRef CAS;
(c) T.-Y. Liu, J. Long, B.-J. Li, L. Jiang, R. Li, Y. Wu, L.-S. Ding and Y.-C. Chen, Org. Biomol. Chem., 2006, 4, 2097–2099 RSC.
- S. Z. Luo, H. Xu, J. Y. Li, L. Zhang and J.-P. Cheng, J. Am. Chem. Soc., 2007, 129, 3074–3075 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2010 |