Preparation of functionalized gold nanoparticles as a targeted X-ray contrast agent for damaged bone tissue
Zhenyuan
Zhang†
,
Ryan D.
Ross
and
Ryan K.
Roeder
*
Department of Aerospace and Mechanical Engineering, University of Notre Dame, Notre Dame, Indiana 46556, USA. E-mail: rroeder@nd.edu; Fax: +1 (574) 631-2144; Tel: +1 (574) 631-7003
First published on 27th January 2010
Abstract
Conventional methods used to image and quantify microdamage in bone tissue are limited to thin histological sections. Therefore recent studies have begun to investigate methods for non-destructive, three-dimensional (3-D) detection and imaging of microdamage in bone tissue. The objective of this study was to investigate gold nanoparticles (Au NPs) as a potential damage-specific X-ray contrast agent due to their relative biocompatibility, ease of surface functionalization, colloidal stability, and high X-ray attenuation. Au NPs were prepared using a citrate reduction reaction to ∼15 or 40 nm diameter, and functionalized with glutamic acid for targeting damaged bone tissue. As-synthesized and functionalized Au NPs were spherical, relatively monodispersed, and exhibited aqueous colloidal stability. Functionalized Au NPs were demonstrated to target damaged bovine cortical bone tissue as visually evidenced by surface scratches turning a characteristic red color after soaking in functionalized Au NP solutions. Individual Au NPs were observed on the surface of damaged tissue using backscattered electron imaging and atomic force microscopy. Therefore, functionalized Au NPs are a promising candidate for a targeted X-ray contrast agent for damaged bone tissue.
Introduction
Repetitive loading of bone tissue can lead to the accumulation of microdamage, observed histologically as microcracks or diffuse damage, and the degradation of mechanical properties.1–4 Therefore, microdamage accumulation in bone has been implicated with clinical scenarios for increased risk of fracture, including stress fractures in active individuals and fragility fractures in the elderly. However, the role of microdamage in clinical bone fragility is not well understood due, in part, to limited capabilities for detecting microdamage non-destructively.Contrast agents are used to aid detection of microdamage and differentiate artefactual damage created during specimen preparation. Current imaging techniques – including transmitted light microscopy using stains,5–7 epifluorescence microscopy using fluorochrome stains or chelating agents,8–10 laser scanning confocal microscopy using fluorochromes,11,12 and backscattered electron microscopy using heavy metal stains13 – are limited to thin histological sections, which are inherently invasive, destructive, tedious and two-dimensional.14 These limitations inhibit evaluating the effects of microdamage on whole bone strength and prohibit detecting microdamage in vivo.
Recent studies have begun to investigate methods for non-destructive, three-dimensional (3-D) detection and imaging of microdamage in bone tissue. X-Ray tomography using high energy, monochromatic synchrotron radiation has sufficient resolution to directly image microcracks in bone,15 but is neither readily available nor amenable to imaging large numbers of tissue specimens that are relatively large in size. Therefore, contrast agents have been investigated to enable the use of lower resolution, but commercially available, micro-computed tomography (micro-CT) instruments. Micro-CT has been used to detect microdamage in vitro using iodinated,14,16,17 lead sulfide,18,19 and barium sulfate20–22 contrast agents. In particular, non-destructive, 3-D imaging of the presence, spatial location and accumulation of microdamage in bone tissue was recently demonstrated for the first time using contrast-enhanced micro-CT with a precipitated BaSO4 stain.20–22 While this approach is expected to find increased use in the study of mineralized tissues, precipitation staining is non-specific to damage and the staining solutions are not biocompatible, limiting use to in vitro studies. Positron emission tomography (PET) was able to detect regions of damaged tissue in vivo using a sodium fluoride tracer.23,24 However, the resolution of this technique is relatively low and tracer uptake is not specific to microcracks but rather cellular activity in the vicinity of microcracks.
A deliverable, biocompatible and damage-specific X-ray contrast agent could have potential for use in vivo. Typical microcracks in bone are less than 10 μm in width,12 and nutrients are transported to bone cells through Haversian canals (∼50 μm diameter) and canaliculi (∼0.1–1 μm diameter).25 Therefore, the contrast agent must be nanoscale in order to be delivered through vasculature to microcracks. Gold nanoparticles (Au NPs) were recently investigated as a vascular contrast agent, exhibiting high X-ray attenuation, colloidal stability and biocompatibility.26,27 Moreover, Au NPs are readily synthesized28 and functionalized through surface adsorption of molecules with thiols29 or amines.30,31 Fluorochromes with carboxylate functional groups (e.g., calcein) are well-known to target microcracks by chelating calcium ions on the surfaces of exposed bone mineral crystals.9,10 Therefore, glutamic acid, a natural amino acid with a primary amine opposite carboxylate groups, was considered a logical choice for surface functionalization of Au NPs to target microdamage in bone tissue (Scheme 1). Glutamic acid residues in proteins such as osteonectin were shown to promote binding affinity for hydroxyapatite,32,33 a synthetic analog for bone mineral.
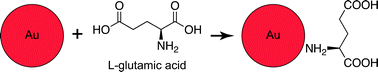 | ||
| Scheme 1 Schematic diagram showing a Au NP surface functionalized with glutamic acid (not to scale), which exhibits a primary amine for binding to the gold surface opposite carboxylate groups for targeting microcracks by chelating calcium ions on the surfaces of exposed bone mineral crystals. | ||
Therefore, the objective of this study was to prepare and characterize functionalized Au NPs as a targeted X-ray contrast agent for damaged bone tissue. Au NPs were prepared using a citrate reduction reaction, and functionalized with glutamic acid for targeting damaged bone tissue (Scheme 1). Alternatively, damaged bone surfaces were functionalized with glutamic acid for targeting Au NPs in order to enable imaging by atomic force microscopy (AFM).
Experimental methods
Synthesis of Au NPs
Monodispersed Au NPs, ∼15 nm in diameter, were synthesized by a citrate reduction reaction using the Turkevich method.28 Briefly, 0.1 g HAuCl4·3H2O (≥99.9%, Aldrich) was added to 400 mL of de-ionized (DI) water, heated to boiling, and 50 mL of a solution containing 1% trisodium citrate dihydrate (ACS reagent >99.0%, Aldrich) was added to the boiling solution under vigorous stirring. The solution was boiled for an additional 20 min, and the volume was adjusted to 500 mL with DI water after cooling. The resultant colloidal solution was wine red with a gold concentration of ∼0.5 mM. Larger Au NPs, ∼40 nm in diameter, were prepared using a modification of the Turkevich method with a lower [citrate]/[Au] ratio.34 0.1021 g trisodium citrate dihydrate in 20 mL water was added to the 400 mL boiling solution containing 0.1018 g HAuCl4·3H2O under vigorous stirring. For reproducible results, the [citrate]/[Au] molar ratio was controlled at ∼1.34.Functionalization of Au NPs
14 mL 2% polyvinyl alcohol (PVA 10–98, MW = 61![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000, Fluka) was added to 236 mL of the as-synthesized 0.5 mM solution containing 15 or 40 nm Au NPs. 5.8 g or 1.8 g ion exchange resin (Amberlite MB-150, Sigma) was added to remove citrate ions from the 15 nm or 40 nm Au sol, respectively. The solution was stirred overnight and subsequently filtered (grade 3 filter paper, Whatman) to remove the ion exchange resin. 4 mL or 2 mL of an aqueous solution containing 10 mM L-glutamic acid (≥99.5%, Fluka) were added to the 15 or 40 nm Au sol, respectively, and the solution was stirred for 2 days. Note that this was a large excess of glutamic acid relative to the amount calculated to be required for one full monolayer on the surface of Au NPs for their size and concentration. Excess glutamic acid was removed using dialysis membrane tubing (Spectra/Por, MWCO = 3500 kDa, Spectrum Laboratories) in a 3000 mL beaker containing DI water, which was changed every 2–3 h for a total of 12 cycles. The functionalized Au NPs were filtered (grade 3 filter paper, Whatman) to remove particle coagulation that may have occurred during dialysis.
000, Fluka) was added to 236 mL of the as-synthesized 0.5 mM solution containing 15 or 40 nm Au NPs. 5.8 g or 1.8 g ion exchange resin (Amberlite MB-150, Sigma) was added to remove citrate ions from the 15 nm or 40 nm Au sol, respectively. The solution was stirred overnight and subsequently filtered (grade 3 filter paper, Whatman) to remove the ion exchange resin. 4 mL or 2 mL of an aqueous solution containing 10 mM L-glutamic acid (≥99.5%, Fluka) were added to the 15 or 40 nm Au sol, respectively, and the solution was stirred for 2 days. Note that this was a large excess of glutamic acid relative to the amount calculated to be required for one full monolayer on the surface of Au NPs for their size and concentration. Excess glutamic acid was removed using dialysis membrane tubing (Spectra/Por, MWCO = 3500 kDa, Spectrum Laboratories) in a 3000 mL beaker containing DI water, which was changed every 2–3 h for a total of 12 cycles. The functionalized Au NPs were filtered (grade 3 filter paper, Whatman) to remove particle coagulation that may have occurred during dialysis.
Labeling damaged bone tissue
Bovine cortical bone specimens were sectioned on a diamond wafer saw to ∼5 × 5 × 2 mm, soaked in 0.5 mM calcein (ICN Biomedicals) for 20 min under vacuum (∼50 mmHg) to mask machining damage, and rinsed in DI water. Specimens were scratched with a scalpel to induce controlled surface damage,10 exposing calcium ions in the mineral phase, and labeled using one of two methods. Damaged bone specimens were soaked for 2 days under vacuum (∼50 mmHg) in a solution containing functionalized Au NPs (Scheme 1), 15 or 40 nm in size. After labeling damage with Au NPs, specimens were rinsed with DI water and dried under ambient conditions.Alternatively, in order to enable imaging by AFM, damaged bone surfaces, were functionalized with glutamic acid for targeting Au NPs. 15 nm Au NPs were mixed with PVA, ion exchange resin was added to remove citrate ions, and the mixture was stirred overnight, as described above. Upon filtration, the Au NP solution was dialyzed and filtered again to further remove citrate ions. Damaged bone specimens were soaked in a solution containing 10 mM glutamic acid for 5 h. Specimens were rinsed with DI water, soaked 2 days under vacuum (∼50 mm Hg) in a solution containing Au NPs, rinsed with DI water, and dried under ambient conditions.
Characterization
The mean particle diameter, particle size distribution, and morphology before and after functionalization were measured by transmission electron microscopy (TEM, Hitachi H-600) at 75 kV accelerating voltage (Fig. 1). Specimens were prepared by immersing carbon-coated grids in Au NP solutions. The maximum and minimum particle diameters were measured for a sample size of 116 particles per group. The mean particle diameter and aspect ratio were calculated as the mean and ratio, respectively, of the average of maximum and minimum diameter. The particle size distribution was measured using both TEM and dynamic light scattering (DLS, Zetasizer Nano-ZS, Malvern Instruments) (Fig. 2). Ultraviolet-visible (UV-Vis) absorption spectra (Varian Cary 50 Spectrophotometer) were collected before and after functionalization to verify colloidal stability and the concentration of Au NPs in solution using Beer's Law (Fig. 3). | ||
| Fig. 1 TEM micrographs of as-synthesized (a) 15 nm and (c) 40 nm Au NPs, and functionalized (b) 15 nm and (d) 40 nm Au NPs, showing that the particle size did not change after functionalization. | ||
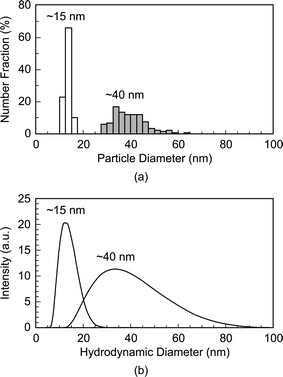 | ||
| Fig. 2 Particle size distributions of as-synthesized 15 and 40 nm Au NPs measured by (a) TEM and (b) dynamic light scattering (DLS). Note that DLS measures the hydrodynamic particle diameter, which was in close agreement with TEM. | ||
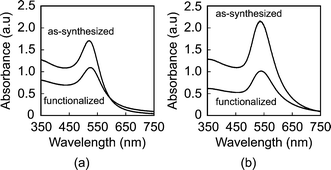 | ||
| Fig. 3 UV-vis spectra of (a) 15 nm and (b) 40 nm Au NPs, showing no change in the position of plasmon bands between as-synthesized and functionalized Au NPs. Absorbance was normalized to a 1 cm optical path. | ||
Damaged bone tissue labeled by Au NPs was characterized using reflected light microscopy (SMZ 800, Nikon Instruments Inc.), and scanning electron microscopy (SEM, Evo 50, LEO Electron Microscopy Ltd.) with backscattered electron imaging (BEI) at an accelerating voltage of 20 kV and working distance of 6.5 mm (Fig. 4). Note that image contrast in BEI is primarily due to compositional differences in atomic number, with increasing atomic number resulting in increased intensity. Damaged bone tissue labeled by Au NPs was also imaged using tapping mode AFM (AFM, Nanoscope® IIIa, Digital Instruments) (Fig. 5).
 | ||
| Fig. 4 (a) Optical micrograph of scratches on the surface of a bovine cortical bone specimen labeled by 15 nm functionalized Au NPs, as shown by the characteristic red color. The width of the image is approximately 7 mm. (b) Backscattered SEM micrograph showing the surface of a scratch labeled by 15 nm functionalized Au NPs (bright spots). | ||
 | ||
| Fig. 5 Tapping mode AFM images of the (a,c) damaged and (b,d) undamaged tissue surfaces on bovine cortical bone specimens labeled by (a,b) 15 nm and (c,d) 40 nm functionalized Au NPs. Note that images of undamaged tissue surface showed little or no presence of Au NPs. | ||
Results and discussion
As-synthesized and functionalized Au NPs were spherical, relatively monodispersed, and exhibited aqueous colloidal stability. The mean (± standard deviation) particle diameter of as-synthesized Au NPs measured from TEM micrographs was 13.4 (± 1.2) and 39.5 (± 7.1) nm (Fig. 1 and Fig. 2a). The particle size distributions measured by TEM and DLS were in close agreement, showing a narrow size distribution for 15 nm Au NPs and a broader size distribution for 40 nm Au NPs (Fig. 2). As-synthesized Au NPs were mostly spherical with some evidence of faceting, which was more pronounced for 40 nm Au NPs (Fig. 1). The mean (± standard deviation) aspect ratio of as-synthesized Au NPs measured from TEM micrographs was 1.1 (± 0.1) and 1.3 (± 0.2) for 15 and 40 nm particles, respectively. There was no apparent change in the particle size or morphology observed by TEM after functionalization (Fig. 1). Moreover, characteristic plasmon bands were observed at 520 and 536 nm for as-synthesized 15 and 40 nm Au NPs, respectively, and exhibited no apparent changes after functionalization, indicating colloidal stability (Fig. 3).As-synthesized Au NPs were surface functionalized with L-glutamic acid in order to target damaged bone tissue (Scheme 1). However, citrate ions, which functioned as both a reducing agent and stabilizer in the preparation of Au NPs,28 have three carboxylate groups which exhibit binding affinity for damaged bone tissue. Therefore, citrate ions were first removed in order to prevent interference with functionalized Au NPs for targeting damaged bone tissue. PVA was added to sterically stabilize the as-synthesized Au NPs while removing citrate ions by ion exchange resin. PVA is uncharged and was therefore not expected to interfere with the functionalization step or the binding of functionalized Au NPs to damage tissue. (Note that subsequent work has demonstrated the ability to add glutamic acid in the presence of citrate and remove both citrate and excess glutamic acid by dialysis, avoiding the addition of PVA.) After adding a large excess of glutamic acid relative to the amount required for one full monolayer coverage on Au NP surfaces, solutions were dialyzed to remove excess glutamic acid. Note that glutamic acid has amine and carboxylate functional groups which both exhibit affinity for gold surfaces. However, amines exhibit a stronger affinity to gold surfaces, displacing carboxylates.35,36 Therefore, the primary amine group was expected to adsorb to the Au NP surface, leaving carboxylate groups available for chelating calcium ions on the surfaces of exposed bone mineral crystals (Scheme 1). The concentration of Au NPs remaining in solution after functionalization was 64 and 47% of the as-synthesized concentration (0.5 mM) for 15 and 40 nm Au NPs, respectively (Fig. 3). After the final dialysis and filtration steps, 47 and 39% of the as-synthesized concentration remained for 15 and 40 nm Au NPs, respectively (not shown).
Specificity for damaged bone tissue was demonstrated with bovine cortical bone specimens by masking machining damage with calcein, scratching the surface with a scalpel, and soaking the specimen in functionalized Au NP solutions. Functionalized Au NPs were able to target surface scratches as visually evidenced by a characteristic red color (Fig. 4a). Similar results were obtained for either 15 or 40 nm functionalized Au NPs. Backscattered electron imaging also detected high contrast between the scratch and adjacent tissue, where increased brightness in the scratch was indicative of the higher atomic number of Au NPs compared to calcium phosphate. At higher magnification, individual Au NPs bound to the damaged tissue were imaged as bright spots on the surface of scratched tissue (Fig. 4b). AFM of damaged tissue labeled by functionalized Au NPs was unsuccessful for reasons that remained unclear. Possible reasons included a relatively greater variability in surface roughness or lower density in surface coverage of functionalized Au NPs.
Damaged bone surfaces were also functionalized with glutamic acid prior to labeling with Au NPs in order to enable high resolution imaging by AFM. The carboxylate groups of glutamic acid were expected to chelate calcium ions on the surfaces of exposed bone mineral crystals, similar to fluorochromes such as calcein. Bone specimens were subsequently soaked in Au NP solutions such that Au NPs were able to bind with amine groups on glutamic acid and thus target damaged bone surfaces. AFM images showed individual Au NPs bound to damaged tissue surfaces and little or no Au NPs on undamaged tissue surfaces (Fig. 5). The surface coverage on damaged tissue appeared to be more dense than labeling with functionalized Au NPs. However, the use of functionalized Au NPs is more desirable for a deliverable contrast agent. Furthermore, note that the glutamic acid solution exhibited pH∼3.5 which could demineralize the tissue over greater lengths of time than those utilized in this study.
This work is the first to our knowledge to demonstrate targeted labeling of damaged bone tissue using a nanoparticle X-ray contrast agent. A biocompatible, deliverable, and damage-specific contrast agent with greater X-ray attenuation than bone could enable non-destructive, three-dimensional and non-invasive (in vivo) imaging of microdamage in bone. Such a contrast agent would have the potential to enable clinical assessment of bone quality and damage accumulation, and scientific study of damage processes in situ. However, this ambitious goal will require much further work. The relative binding affinities of various potential functional groups, such as carboxylates and bisphosphonates, must be investigated. Preliminary efforts have highlighted the difficulty of obtaining sufficient signal for detection by commercially available, polychromatic micro-computed tomography (micro-CT) instruments with ∼10 μm resolution despite the relatively high X-ray attenuation of gold, due to the necessarily small size and concentration of Au NPs labeling damaged bone tissue. Therefore, the use of monochromatic synchrotron radiation may be advantageous in order to maximize contrast by edge subtraction imaging just above and below the energy of gold absorption peaks.
Conclusions
Au NPs were prepared using a citrate reduction reaction to ∼15 or 40 nm diameter, and functionalized with glutamic acid for targeting damaged bone tissue. As-synthesized and functionalized Au NPs were spherical, relatively monodispersed, and exhibited aqueous colloidal stability. Functionalized Au NPs were demonstrated to target damaged bone tissue, which was verified by visual staining, backscattered electron imaging and atomic force microscopy of surface scratches compared to undamaged tissue. Therefore, functionalized Au NPs are a promising candidate for a targeted X-ray contrast agent for damaged bone tissue.Acknowledgements
This research was supported by the U.S. Army Medical Research and Materiel Command (W81XWH-06-1-0196) through the Peer Reviewed Medical Research Program (PR054672). TEM was made available through the Notre Dame Integrated Image Facility (NDIIF). The Notre Dame Radiation Laboratory (NDRL) is gratefully acknowledged for use of the AFM and UV-vis spectroscopy.References
- D. B. Burr, M. R. Forwood, D. P. Fyhrie, R. B. Martin, M. B. Schaffler and C. H. Turner, J. Bone Miner. Res., 1997, 12, 6–15 CrossRef CAS.
- D. B. Burr, C. H. Turner, P. Naick, M. R. Forwood, W. Ambrosius, M. S. Hasan and R. Pidaparti, J. Biomech., 1998, 31, 337–345 CrossRef CAS.
- T. C. Lee, F. J. O'Brien and D. Taylor, Int. J. Fatigue, 2000, 22, 847–853 CrossRef CAS.
- R. B. Martin, Calcif. Tissue Int., 2003, 73, 101–107 CrossRef CAS.
- H. M. Frost, Henry Ford Hosp. Med. Bull., 1960, 8, 25–35 Search PubMed.
- D. B. Burr and T. Stafford, Clin. Orthop. Relat. Res., 1990, 305–308 CAS.
- D. B. Burr and M. Hooser, Bone, 1995, 17, 431–433 CrossRef CAS.
- T. C. Lee, E. R. Myers and W. C. Hayes, J. Anat., 1998, 193, 179–184 Search PubMed.
- T. C. Lee, T. L. Arthur, L. J. Gibson and W. C. Hayes, J. Orthop. Res., 2000, 18, 322–325 CrossRef CAS.
- F. J. O'Brien, D. Taylor and T. C. Lee, J. Biomech., 2002, 35, 523–526 CrossRef.
- P. Zioupos and J. D. Currey, J. Mater. Sci., 1994, 29, 978–986 CrossRef.
- F. J. O'Brien, D. Taylor, G. R. Dickson and T. C. Lee, J. Anat., 2000, 197, 413–420 Search PubMed.
- M. B. Schaffler, W. C. Pitchford, K. Choi and J. M. Riddle, Bone, 1994, 15, 483–488 CrossRef CAS.
- T. C. Lee, S. Mohsin, D. Taylor, R. Parkesh, T. Gunnlaugsson, F. J. O'Brien, M. Giehl and W. Gowin, J. Anat., 2003, 203, 161–172 Search PubMed.
- R. K. Nalla, J. S. Stölken, J. H. Kinney and R. O. Ritchie, J. Biomech., 2005, 38, 1517–1525 CrossRef CAS.
- R. Parkesh, T. C. Lee, T. Gunnlaugsson and W. Gowin, J. Biomech., 2006, 39, 1552–1556 CrossRef.
- R. Parkesh, W. Gowin, T. C. Lee and T. Gunnlaugsson, Org. Biomol. Chem., 2006, 4, 3611–3617 RSC.
- H. Leng, J. J. VanDersarl, G. L. Niebur and R. K. Roeder, Trans. Orthop. Res. Soc., 2005, 30, 665 Search PubMed.
- S. Y. Tang and D. Vashishth, Bone, 2007, 40, 1259–1264 CrossRef CAS.
- X. Wang, D. B. Masse, H. Leng, K. P. Hess, R. D. Ross, R. K. Roeder and G. L. Niebur, J. Biomech., 2007, 40, 3397–3403 CrossRef.
- H. Leng, X. Wang, R. D. Ross, G. L. Niebur and R. K. Roeder, J. Mech. Behav. Biomed. Mater., 2008, 1, 68–75 Search PubMed.
- M. D. Landrigan, G. L. Niebur and R. K. Roeder, Trans. Orthop. Res. Soc., 2009, 34, 332 Search PubMed.
- J. Li, M. A. Miller, G. D. Hutchins and D. B. Burr, Bone, 2005, 37, 819–824 CrossRef.
- M. J. Silva, B. A. Uthgenannt, J. R. Rutlin, G. R. Wohl, J. S. Lewis and M. J. Welch, Bone, 2006, 39, 229–236 CrossRef CAS.
- M. L. K. Tate, P. Niederer and U. Knothe, Bone, 1998, 22, 107–117 CrossRef.
- J. F. Hainfield, D. N. Slatkin and H. M. Smilowitz, Phys. Med. Biol., 2004, 49, N309–N315 CrossRef CAS.
- J. F. Hainfield, D. N. Slatkin, T. M. Focella and H. M. Smilowitz, Br. J. Radiol., 2006, 79, 248–253 Search PubMed.
- J. Turkevich, P. C. Stevenson and J. Hillier, Discuss. Faraday Soc., 1951, 11, 55–75 RSC.
- M. Brust, J. Fink, D. Bethell, D. J. Schiffrin and C. Kiely, J. Chem. Soc., Chem. Commun., 1995, 1655–1656 RSC.
- D. V. Leff, L. Brandt and J. R. Heath, Langmuir, 1996, 12, 4723–2730 CrossRef CAS.
- M. Aslam, L. Fu, M. Su, K. Vijayamohanan and V. P. Dravid, J. Mater. Chem., 2004, 14, 1795–1997 RSC.
- M. E. Bolander, M. F. Young, L. W. Fisher, Y. Yamada and J. D. Termine, Proc. Natl. Acad. Sci. U. S. A., 1988, 85, 2919–2923 CrossRef CAS.
- R. Fujisawa, Y. Wada, Y. Nodasaka and Y. Kuboki, Biochim. Biophys. Acta, Protein Struct. Mol. Enzymol., 1996, 1292, 53–60 Search PubMed.
- G. Frens, Nature Phys. Sci., 1973, 241, 20–22.
- L. M. Liz-Marzán, M. Giersig and P. Mulvaney, Langmuir, 1996, 12, 4329–4335 CrossRef CAS.
- Z. Y. Zhang, A. Berg, H. Levanon, R. W. Fessenden and D. Meisel, J. Am. Chem. Soc., 2003, 125, 7959–7963 CrossRef CAS.
Footnote |
| † Current address: Department of Chemistry, Duke University, Durham, NC 27708, USA |
| This journal is © The Royal Society of Chemistry 2010 |