Development of a new microwave-assisted cleavable backbone amide linker (BAL): a comparative study†
Stijn Claerhouta, Thibault Duchèneb, Dirk Tourwéb and Erik V. Van der Eycken*a
aLaboratory for Organic and Microwave-Assisted Chemistry (LOMAC), Department of Chemistry, Katholieke Universiteit Leuven, Celestijnenlaan 200F, B-3001 Leuven, Belgium. E-mail: erik.vandereycken@chem.kuleuven.be
bLaboratory for Organic Chemistry (ORGC), Vrije Universiteit Brussel (VUB), Pleinlaan 2, B-1050 Brussel
First published on 19th November 2009
Abstract
A thorough comparative study to demonstrate the properties of a new microwave labile backbone amide linker is presented. A cyclic pentapeptide, cyclo(Trp-Gln-Gly-β-Ala-Phe), was used as a model and synthesized following 16 different conditions. The new backbone amide linker is stable towards acid and base at r.t., and can be cleaved at elevated temperature in trifluoroacetic acid under microwave irradiation, avoiding the use of aggressive reagents like HF. The new linker is compatible with Boc- as well as Fmoc-strategy and allows the cleavage of acid labile side chain protective groups at r.t., prior to cleavage of the cyclic pentapeptide from the resin.
Introduction
Since the introduction of the concept of Solid Phase Peptide Synthesis (SPPS) by Merrifield in 1963,1 this technique has evolved to a very efficient procedure for the synthesis of peptides and even small proteins. The breakthrough of combinatorial chemistry has increased the need for solid phase synthesis, even outside the domain of peptides, e.g. for the synthesis of small organic molecules.2In the original design of Merrifield for SPPS, the growing peptide chain is attached to the resin through its C-terminal carboxyl group and the peptide chain is elongated towards its N-terminus. After cleavage from the resin, C-terminal carboxylic acids or amides are obtained. Nevertheless, within the context of pharmaceutical development of peptides, the demand for biologically relevant analogues of peptides where the C-terminus is modified to other functionalities such as alcohols, ethers, esters, N-alkylamides, hydrazides, trifluoromethyl ketones, aldehydes, mercaptoalkylamides, thioesters, thioamides and cyclic peptides is rising.3 Modifications of the C-terminus, as well as its use to form cyclic peptides, give access to new classes of peptides that represent potentially efficient therapeutic agents.3 These modifications increase the bioavailability of peptides by protecting them against enzymatic degradation and by increasing their ability to cross biological barriers. By fixing some conformations of peptides through cyclization, their receptor affinity and selectivity in comparison with their linear analogues, can be increased. Small cyclic peptides are also very good scaffolds for the introduction of pharmacophoric groups which can interact with the corresponding receptors.4 In this way, they form useful leads for the development of new drugs as was successfully demonstrated by the examples of Octreotide5 and Cyclosporin A.6
Linkers with an aldehyde functionality, to which the first amino acid is anchored via reductive amination, have become a widely used tool in SPPS, since their first description by Barany et al.7 in the mid 1990's. This so-called Backbone Amide Linker (BAL) approach, in which the growing peptide chain is anchored to a solid support via a backbone amide nitrogen instead of via its Cα-carboxy group, allows the limitations linked to C-terminal anchoring or side chain anchoring to be overcome and gives access to these C-terminal modified and cyclic peptides.8
Since the description of this first BAL-approach, numerous variations have been described, giving access to a variety of C-terminal modified peptides and allowing the use of several cleavage conditions.3
The acid sensitivity of the BAL is mainly determined by the stability of the formed carbenium ion after treatment with acid. The substitution pattern (e.g. the influence of methoxy groups on the benzene ring), the planarity of the aromatic core and the peri-effect define the ease in which the carbocation is formed.9 These factors have led to the development of many different BAL's with various acid sensitivities.3 Less electron-rich analogues than the original trisalkoxy BAL such as dialkoxy,10,11 alkoxy-hydroxy12 and monoalkoxy13–15 benzaldehyde, indole,16 naphthalene,9,17,18 thiophene19 and triazene20 based handles have also been used as core structures for SPPS by the BAL-strategy.3,21 Also photolabile BAL's have been developed which are not acid sensitive.22
However, these described linkers still show some limitations. Their synthesis is not always straightforward and the alkoxy substituents on the aromatic core often lead to steric hindrance which tends to make the acylation of the attached secondary amine more difficult.3,7,23
A comparison is given between the monoalkoxy-BAL 5-(4-formylphenoxy)pentanoic acid 114 and a new meta-dialkoxy-BAL 5-(4-formyl-2-methoxyphenoxy)pentanoic acid 2 which shows interesting properties in combination with microwave irradiation (Fig. 1). These properties were studied during the synthesis of a model cyclic peptide cyclo(Trp-Gln-Gly-β-Ala-Phe), which was previously prepared in a study using BAL linker 1.24
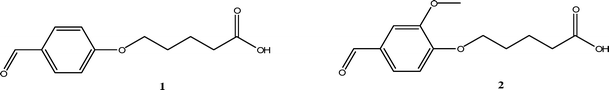 | ||
| Fig. 1 The monoalkoxy-BAL 1 and the meta-dialkoxy-BAL 2. | ||
The monoalkoxy-BAL 1 has the advantage of having a high acid stability, so that Boc- as well as Fmoc- and allyl-protective groups can be used. Due to this orthogonality, peptide cyclization as well as the removal of acid sensitive side-chain protective groups can be performed on-resin (Scheme 1). The disadvantage of this monoalkoxy-BAL 1 is the need of very strong acidic cleavage conditions as anhydrous HF or a mixture of HBr in trifluoroacetic acid (TFA),24 which is inappropriate for combinatorial issues. The long reaction times are also a major drawback. The acid sensitivity of BAL-linkers based on a benzaldehyde structure can be increased by the use of electron donating substituents, e.g. one or two methoxy groups.3 However, a general problem inherent to the BAL-strategy is the difficult coupling of the second amino acid to the secondary amine,7,23 leading to long reaction times. This problem is more severe when the electron donating substituent is in the ortho-position towards the aldehyde function, a position which assures the best resonance stabilization of the carbenium ion that is formed during cleavage. These problems can be addressed, for instance by using an ortho-phenolic hydroxyl which allows an intramolecular O–N acyl transfer reaction, but also the application of microwave irradiation19,25 has been reported.
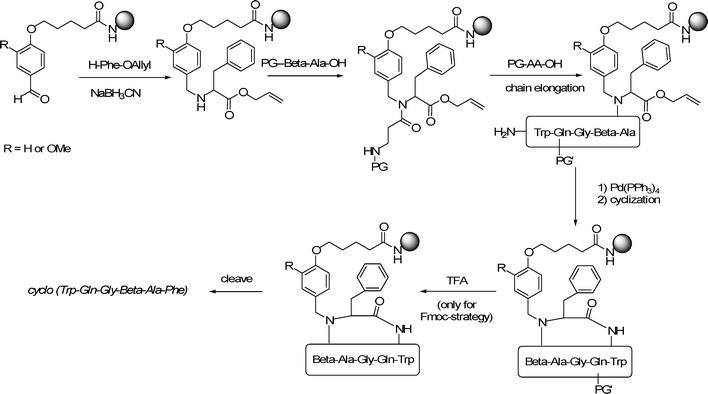 | ||
| Scheme 1 Synthesis of cyclo(Trp-Gln-Gly-β-Ala-Phe) applying the BAL-strategy. PG, PG′ = protective group; PG = Boc or Fmoc; PG′= trityl when Fmoc-strategy was used; no PG′ when Boc-strategy was used. | ||
We now propose a new linker 2 which offers the same advantages as linker 1, but whose acid sensitivity has been fine tuned by the introduction of a methoxy substituent in the meta position towards the aldehyde function, so that it is stable during TFA treatment at r.t., but it becomes sufficiently labile upon TFA treatment under microwave irradiation, and does not have an increased steric hindrance for the coupling of the second amino acid to the secondary amine.
Results and discussion
To demonstrate the properties of the new microwave labile BAL 2, a thorough comparative study was performed. A cyclic pentapeptide, cyclo(Trp-Gln-Gly-β-Ala-Phe), was used as a model and synthesized under 16 different conditions. This allowed a profound comparison of the applied conditions. This reference peptide has been prepared using BAL linker 1 and a Boc-strategy in 14% crude yield and 97% purity after HF cleavage from the solid support.24In order to investigate the acid sensitivity of the new BAL 2 and to compare it with that of the existing BAL 1, the cyclic peptide was assembled on both BAL-linkers as shown in Scheme 1. Microwave-assisted cleavage of the peptide from the resin applying linker 1 required a rather high temperature (Table 1, entry 6). Although the cleavage was complete at 120 °C, LCMS-analysis revealed that the peptide contains many impurities. Attempts to lower the cleavage temperature by increasing the TFA concentration resulted in incomplete or no cleavage of the peptide (Table 1 entries 1–5). In contrast, cleavage of the peptide linked with BAL 2, required only 80 °C to have complete cleavage (Table 1 entry 8), while linker 2 was stable towards TFA at r.t. (Table 1, entry 7)
| Entry | Linker | Conditionsa | Cleavageb? |
|---|---|---|---|
| a The peptide was synthesized following the Boc-strategy applying microwave irradiation for the coupling and cyclization reactions (see materials and methods).b The cleavage efficiency was determined by repeating the applied cleavage conditions twice and checking (HPLC) the filtrate for the presence of cleaved amino acid.c When the temperature and TFA concentration was lowered, incomplete cleavage was observed. | |||
| 1 | 1 | rt, TFA–DCM (95![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :5), 1 h :5), 1 h | No |
| 2 | 1 | MW, 80 °C, TFA–TIS (95:5), 50 W, 30 min | No |
| 3 | 1 | MW, 90 °C, TFA–TIS (95:5), 50 W, 30 min | No |
| 4 | 1 | MW, 100 °C, TFA–DCM (6:4), 80 W, 30 min | Traces |
| 5 | 1 | MW, 100 °C, TFA–TIS (95:5), 50 W, 30 min | Yes, incomplete |
| 6 | 1 | MW, 120 °C, TFA–DCM (1:4), 20 min | Yes, complete |
| 7 | 2 | rt, TFA–DCM (95:5), 1 h | No |
| 8 | 2 | MW, 80 °C, TFA–TIS (95:5), 50 W, 40 minc | Yes, completec |
The yields of the peptides, linked via linker 1 and cleaved upon microwave irradiation, were very low as were the purities (Table 2, entries 9, 11, 13, 15). Every synthesized peptide was also cleaved using HF at 0 °C, in order to make a profound comparison (Table 2, entries 2, 4, 6, 8, 10, 12, 14 and 16). These experiments showed that when the cyclic peptides were cleaved from the resin with HF, they were isolated with acceptable yields and purities (Table 2, entries 10, 12, 14, 16). Therefore we can conclude that the elevated temperature needed to cleave the cyclic peptide from the resin under microwave irradiation in TFA is detrimental to the peptide.
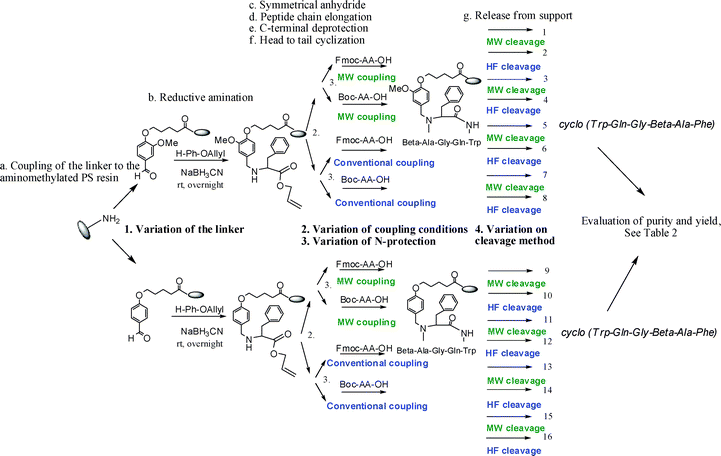 | ||
| Scheme 2 Investigated variations for the synthesis of the model peptide cyclo (Trp-Gln-Gly-β-Ala-Phe). Reagents and conditions: Conventional synthesis (blue): (a) Linker 1 or 2 (3 equiv), HOBt (3 equiv), DIC (3 equiv), r.t., 24 h; (b) NaBH3CN (5 equiv), H-L-Phe-OAll.HCl (5 equiv), DMF–MeOH (9:1), r.t., 18 h; (c) Fmoc- or Boc-β-Ala-OH (5 equiv), DIC (2,5 equiv), CH2Cl2–DMF (9:1), r.t., 2 × 24 h; (d) Fmoc- or Boc AA-OH (3 equiv), HOBt (3 equiv), DIC (3 equiv), r.t., 3 h; (e) Pd(PPh3)4 (5 mol%), PhSiH3 (24 equiv), dry CH2Cl2, 3 h; (f) PyBOP (3 equiv), DIEA (6 equiv), r.t., 2 × 4 h; (g) conditions are given in Table 1. Microwave-assisted synthesis (green): (a) HOBt (3 equiv), DIC (3 equiv), CH2Cl2–DMF (9:1), linker (3 equiv), 60 °C, 20 min; (b) NaBH3CN (5 equiv), H-L-Phe-OAll.HCl (5 equiv), DMF–MeOH (9:1), r.t., 2 × 8 h; (c) Fmoc- or Boc-β-Ala-OH (10 equiv), DIC (5 equiv), CH2Cl2–DMF (9:1), MW, 2 × 40 min; (d) Fmoc- or Boc AA-OH (3 equiv), HOBt (3 equiv), DIC (3 equiv), MW, 60 °C, 20 min; (e) Pd(PPh3)4 (5 mol%), PhSiH3 (24 equiv), dry CH2Cl2, 3 h; (f) PyBOP (3 equiv), DIEA (6 equiv), 2 × 40 min; (g) conditions are given in Table 1. | ||
| Entry | Linker | Coupling conditions a(MW/Conv.) | Boc/Fmocb | Cleavage conditions cHF or MW | Crude yieldd (%) | Purity after cleavagee’c (%) |
|---|---|---|---|---|---|---|
| a MW-coupling: 60 °C, 50 W, 20 min (see methods and materials for detailed conditions). Conventional coupling: r.t., 2 h (see methods and materials for detailed conditions).b Fmoc-Gln-OH was side chain trityl protected.c Cleavage conditions for linker 2: MW, 80 °C, 45 min, 60 W, TFA–TIS (95:5). Cleavage conditions for linker 1: MW, 120 °C, 15 min, 100 W, TFA–DCM–TIS (20:75:5). Cleavage conditions with HF for linker 2 and 1: HF(liq)–p-cresol–p-thiocresol (9:0,5:0,5), 0 °C, 90 min.d Yields are based on the loading of the aminomethylated polystyrene resin (0.2 mmol g−1).e Purity was determined by HPLC with UV detection at 215 nm.f According to LCMS 5–10% of the cyclic peptide was present in the mixture next to many unidentified side compounds. | ||||||
| 1 | 2 | MW | Fmoc | MW | 36 | 75 |
| 2 | 2 | MW | Fmoc | HF | 58 | 65 |
| 3 | 2 | MW | Boc | MW | 40 | 71 |
| 4 | 2 | MW | Boc | HF | 54 | 90 |
| 5 | 2 | Conventional | Fmoc | MW | 39 | 72 |
| 6 | 2 | Conventional | Fmoc | HF | 21 | 51 |
| 7 | 2 | Conventional | Boc | MW | 33 | 78 |
| 8 | 2 | Conventional | Boc | HF | 48 | 53 |
| 9 | 1 | MW | Fmoc | MW | Very lowf | 7 |
| 10 | 1 | MW | Fmoc | HF | 47 | 70 |
| 11 | 1 | MW | Boc | MW | Very lowf | 10 |
| 12 | 1 | MW | Boc | HF | 33 | 69 |
| 13 | 1 | Conventional | Fmoc | MW | Very lowf | 20 |
| 14 | 1 | Conventional | Fmoc | HF | 33 | 90 |
| 15 | 1 | Conventional | Boc | MW | Very lowf | 25 |
| 16 | 1 | Conventional | Boc | HF | 23 | 72 |
Table 2 in contrast shows that the new linker 2 does not need HF for cleavage to obtain good yields and purities (Table 2, entries 1, 3, 5 and 7). Despite the fact that mostly (Table 2, entries 1-8) a slightly higher yield was observed using HF cleavage, it is obvious that cleavage in TFA applying microwave irradiation at a temperature of 80 °C represents a more convenient method than the use of liquid HF. The ease of cleavage under microwave irradiation conditions justifies the loss of yield compared to the hazardous HF conditions.
Microwave irradiation also accelerates the amino acid coupling reaction rates considerably.26–28 Therefore, conventional coupling at room temperature was compared with coupling at elevated temperature under microwave irradiation.26–28 Except for the deprotection of the amino acids and the reductive amination, all coupling steps were done applying microwave irradiation. The completeness of the coupling reactions was followed with our recently developed DESC test29 for the colorimetric detection of resin bound primary and secondary amines.
The microwave-assisted reductive amination, necessary for the coupling of the first amino acid to the resin-bound linker, was already described in literature.19,25 However, when the reductive amination was performed under microwave irradiation at a ceiling temperature of 60 °C for 2 × 40 min we detected at the end of the peptide synthesis, next to the presence of the correct sequence, also a slight amount of deletion peptide, missing the C-terminal Phe. A possible explanation for this phenomenon is an over reduction of the aldehyde to the alcohol by NaCNBH3. During the following amino acid coupling step, β-Ala reacts with this alcohol and further peptide synthesis leads to a deletion sequence lacking the first amino acid Phe. Therefore we decided to run this reaction under conventional conditions. Comparable yields and purities were obtained for the peptides synthesized applying conventional coupling conditions or coupling under microwave irradiation via Boc- or Fmoc-strategy (Table 2, compare entry 1 with 5 and entry 3 with 7). This demonstrates that the application of microwave irradiation can speed up the synthesis without loss of yield and purity compared to conventional coupling conditions.26–28
Finally, the two mainly used N-protective groups for peptide synthesis, Boc and Fmoc were evaluated. Comparing the results of the peptides synthesized via Boc- and Fmoc-strategy, we notice that both protective groups are compatible with the new linker (Table 2, entries 1–8). Orthogonal deprotection of acid labile protective groups, prior to release of the peptide, is possible, as the new linker is stable towards TFA at r.t. (Table 1, entry 7). When Fmoc N-protected amino acids were used, the amide of the Gln side chain was trityl protected. The trityl group was removed before release of the cyclic peptide from the support upon treatment with a TFA–TIS (95:5) mixture at r.t. This did not lead to a decreased yield (Table 2, entries 1 and 5) indicating that orthogonal deprotection prior to release of the peptide is possible with the new BAL 2.
In summary, a new tailor made acid labile linker 2 was developed for the BAL solid phase methodology. The new linker is compatible with Boc- as well as Fmoc-strategy and allows the cleavage of acid labile side chain protective groups at r.t., but is cleavable under acidic conditions (TFA–TIS, 95:5) at elevated temperature (80 °C) upon microwave irradiation. The new linker 2 was thoroughly compared with the existing linker 1, the last requiring very strong acidic conditions (HF) for the release of the peptide from the solid support. Additionally, it was once more demonstrated that the use of microwave irradiation for the coupling reactions reduces the coupling time leading to a seriously decreased total reaction time for the synthesis of cyclic pentapeptides.
Materials and methods
Amino acid derivatives, PyBOP (benzotriazol-1-yl-oxytripyrrolidinophosphonium hexafluorophosphate), DIC (diisopropylcarbodiimide), HOBt (1-hydroxy-benzotriazole) and DIEA (N,N-diisopropylethylamine) were purchased from Iris Biotech or NovaBiochem. Amino acids were Nα-Fmoc or -Boc protected. Gln was only side-chain protected in the Fmoc method, as Fmoc-Gln(Trt). The aminomethylated polystyrene resin (0.2 mmol g−1, 100–125 μm) was obtained from RAPP Polymere. Peptides were synthesized using the conditions described in Scheme 2. Microwave irradiation (2.45 GHz) was performed in a dedicated CEM-Discover monomode microwave apparatus. All experiments were carried out in sealed microwave process vials (10 mL). A temperature control was performed using external infrared sensor. After completion of the reaction, the vial was cooled to 25 °C via air jet cooling before opening. Conventional peptide synthesis was carried out manually in syringes containing a PE frit (MultiSyntech Gmbh, Germany) for filtration.Synthesis of monoalkoxy-BAL 5-(4-formylphenoxy)pentanoic acid 1
:80 mL). After keeping the solution in the freezer for one night white to colorless crystals were obtained (17.42 g, 85%).1H NMR (300 MHz, CDCl3): δ 9.88 (s, 1H), 7.82 (d, J = 8.70, 2H), 6.97 (d, J = 8.60, 2H), 4.07 (t, J = 4.35 Hz, 2H), 2.44 (t, J = 5.19, 2H), 1.86 (m, 4H).
13C NMR (75 MHz, CDCl3): 191.04, 179.16, 164.01, 132.07, 129.81, 114.73, 67.75, 33.52, 28.35, 21.29.
HRMS C12H14O4 calc. 222,0892 found 222.0902.
Synthesis of meta-dialkoxy-BAL 5-(4-formyl-2-methoxyphenoxy)pentanoic acid 2
The procedure followed for the synthesis of compound 2 was identical as for compound 1, except that 4-hydroxy-3-methoxybenzaldehyde (vanillin) was used instead of 4-hydroxybenzaldehyde. Overall yield = 64%.1H NMR (300 MHz, CDCl3): δ 9.83 (s, 1H), 7.41 (m, 2H), 6.94 (d, 6 Hz, 1H), 4.12 (t, J = 4.65 Hz), 3.89 (s, 3H), 2.47 (t, J = 5.52 Hz, 2H), 1.88 (m, 4H).
13C NMR (75 MHz, CDCl3): 191.5, 179.7, 154.4, 150.2, 130.4, 127.24, 111.8, 109.7, 68.9, 56.3, 33.9, 28.6, 21.7.
HRMS C13H16O5 calc. 252,9977 found 252.1002.
Synthesis of cyclic peptide applying microwave irradiation
:1, 3 mL) and preactivated for 10–15 min and added to the resin prepared according to step 1. The mixture was irradiated for 40 min at a ceiling temperature of 60 °C and 60 W maximum power. The mixture was filtered, and the resin was washed with DMF (3×), CH2Cl2 (3×) and MeOH (3×). The coupling and washing steps were repeated once. Completion of the coupling was confirmed by a negative DESC test.29:4, 5 mL) for 2 × 5 min; b) Boc deprotection: the resin was treated at r.t. with TFA–CH2Cl2–TIPS (40:55:5, 5 mL) for 2 × 15 min.The solution was filtered, and the resin was washed with DMF (3×), CH2Cl2 (3×) and MeOH (3×). The DESC test was positive. Fmoc-or Boc-AA-OH (3 equiv), HOBt (3 equiv) DIC (3 equiv) were mixed for 2 min and added to the resin. The mixture was irradiated for 20 min at a ceiling temperature of 60 °C and 50 W maximum power. The mixture was filtered, and the resin was washed with DMF (3×), CH2Cl2 (3×) and MeOH (3×). Completion of the couplings was confirmed by a negative DESC test.29
Synthesis of cyclic peptide using conventional conditions
:2, 3 mL), and the suspension was added to the loaded resin and shaken for 18 h. The solution was filtered, and the resin was washed with DMF (3×), CH2Cl2 (3×) and MeOH (3×).Release from the support
:5) for 1 h at r.t. to remove the side-chain protective trityl group from Gln, in cases where the Fmoc protective group was used. No side chain protective group was used with the Boc-strategy.The resin bound cyclic peptide was suspended in TFA–CH2Cl2–TIPS (20:75:5) and irradiated for 15 min at a ceiling temperature of 120 °C and 100 W maximum power. The resin was filtered and washed with TFA and CH2Cl2 (3×). The combined phases were concentrated, and the crude peptide was precipitated with diethyl ether, filtered, dissolved in acetic acid and lyophilised.
:5) was used for 45 min at a ceiling temperature of 80 °C and 60 W maximum power.:5) to remove the side-chain protective trityl group from Gln, in cases where the Fmoc protection group was used. No side chain protective group was used with the Boc-strategy.The resin bound cyclic peptide was treated for 90 min at 0 °C with liquid HF in the presence of p-cresol and p-thiocresol. After removal of the HF under reduced pressure, the crude peptide was precipitated in anhydrous ether, filtered, dissolved in acetic acid and lyophilised.
Cyclo(Trp-Gln-Gly-β-Ala-Phe)
Analytical HPLC was performed using a GRACE C18 reversed-phase column (0.21 × 15 cm) on an Agilent 6110 instrument, configured with a quaternary pump and a UV-DAD detector. UV detection was at 215 nm and linear gradients of CH3CN and 0,1% aqueous CHOOH were run at 0.2 mL min−1 flow rate from 1:9 to 1:0 over 20 min. Tr= 13.6 min., MS (ESI-MH+) = 590.2 g mol−1.
Acknowledgements
The authors thank the I.W.T. (Institute for the Promotion of Innovation by Science and Technology in Flanders) for financial support (SBO-project IWT 040083). They are grateful to the company Milestone (Via Fatebenefratelli, 1/524010 Sorisole (BG) Italy) for giving the opportunity to evaluate their Multimode microwave instrument in the course of this project.References
- R. B. Merrifield, J. Am. Chem. Soc., 1963, 85(14), 2149–54 CrossRef CAS.
- W. Bannwarth, Methods Princ. Med. Chem., 2006, 26, 33–110 Search PubMed.
- U. Boas, J. Brask and K. J. Jensen, Chem. Rev., 2009, 109(5), 2092–2118 CrossRef CAS.
- M. Royo, W. Van Den Nest, M. del Fresno, A. Frieden, D. Yahalom, M. Rosenblatt, M. Chorev and F. Albericio, Tetrahedron Lett., 2001, 42(42), 7387–7391 CrossRef CAS.
- W. Bauer, U. Briner, W. Doepfner, R. Haller, R. Huguenin, P. Marbach, T. J. Petcher and Pless, Life Sci., 1982, 31(11), 1133–40 CrossRef CAS.
- E. A. Emmel, C. L. Verweij, D. B. Durand, K. M. Higgins, E. Lacy and G. R. Crabtree, Science, 1989, 246(4937), 1617–20 CrossRef CAS.
- K. J. Jensen, J. Alsina, M. F. Songster, J. Vagner, F. Albericio and G. Barany, J. Am. Chem. Soc., 1998, 120(22), 5441–5452 CrossRef CAS.
- J. Jin, T. L. Graybill, M. A. Wang, L. D. Davis and M. L. Moore, J. Comb. Chem., 2001, 3(1), 97–101 CrossRef CAS.
- M. Pittelkow, U. Boas and J. B. Christensen, Org. Lett., 2006, 8(25), 5817–5820 CrossRef CAS.
- U. Boas, J. Brask, J. B. Christensen and K. J. Jensen, J. Comb. Chem., 2002, 4(3), 223–228 CrossRef CAS.
- C. T. Bui, A. M. Bray, F. Ercole, Y. Pham, F. A. Rasoul and N. J. Maeji, Tetrahedron Lett., 1999, 40(17), 3471–3474 CrossRef CAS.
- T. Okayama, A. Burritt and V. J. Hruby, Org. Lett., 2000, 2(13), 1787–1790 CrossRef CAS.
- W. Gu and R. B. Silverman, Org. Lett., 2003, 5(4), 415–418 CrossRef CAS.
- G. T. Bourne, W. D. F. Meutermans and M. L. Smythe, Tetrahedron Lett., 1999, 40(40), 7271–7274 CrossRef CAS.
- G. T. Bourne, W. D. F. Meutermans, P. F. Alewood, R. P. McGeary, M. Scanlon, A. A. Watson and M. L. Smythe, J. Org. Chem., 1999, 64(9), 3095–3101 CrossRef CAS.
- K. G. Estep, C. E. Neipp, L. M. S. Stramiello, M. D. Adam, M. P. Allen, S. Robinson and E. J. Roskamp, J. Org. Chem., 1998, 63(16), 5300–5301 CrossRef CAS.
- U. Boas, J. B. Christensen and K. J. Jensen, J. Comb. Chem., 2004, 6(4), 497–503 CrossRef CAS.
- M. Pittelkow, U. Boas, M. Jessing, K. J. Jensen and J. B. Christensen, Org. Biomol. Chem., 2005, 3(3), 508–514 RSC.
- M. Jessing, M. Brandt, K. J. Jensen, J. B. Christensen and U. Boas, J. Org. Chem., 2006, 71(18), 6734–6741 CrossRef CAS.
- S. Braese, S. Dahmen and M. Pfefferkorn, J. Comb. Chem., 2000, 2(6), 710–715 CrossRef CAS.
- P.-O. Norrby and K. J. Jensen, Int. J. Pept. Res. Ther., 2006, 12(4), 335–339 Search PubMed.
- R. Minkwitz and M. Meldal, QSAR Comb. Sci., 2005, 24(3), 343–353 Search PubMed.
- J. Alsina, K. J. Jensen, F. Albericio and G. Barany, Chem.–Eur. J., 1999, 5(10), 2787–2795 CrossRef CAS.
- G. T. Bourne, S. W. Golding, W. D. F. Meutermans and M. L. Smythe, Lett. Pept. Sci., 2001, 7(6), 311–316.
- M. Brandt, S. Gammeltoft and K. J. Jensen, Int. J. Pept. Res. Ther., 2006, 12(4), 349–357 Search PubMed.
- B. Bacsa, K. Horvati, S. Bosze, F. Andreae and C. O. Kappe, J. Org. Chem., 2008, 73(19), 7532–7542 CrossRef CAS.
- M. Erdelyi and A. Gogoll, Synthesis, 2002,(11), 1592–1596 CAS.
- F. Rizzolo, G. Sabatino, M. Chelli, P. Rovero and A. M. Papini, Int. J. Pept. Res. Ther., 2007, 13(1-2), 203–208 Search PubMed.
- S. Claerhout, D. S. Ermolat'ev and E. V. Van der Eycken, J. Comb. Chem., 2008, 10(4), 580–585 CrossRef CAS.
Footnote |
| † This paper is part of an Organic & Biomolecular Chemistry web theme issue on enabling technologies for organic synthesis. |
| This journal is © The Royal Society of Chemistry 2010 |