Semi-micro solid phase extraction with electrospun polystyrene fiber disks
Samuel
Chigome
a,
Godfred
Darko
a,
Ulrich
Buttner
b and
Nelson
Torto
*a
aDepartment of Chemistry, Rhodes University, P.O. Box 94, Grahamstown, 6140, South Africa. E-mail: N.Torto@ru.ac.za
bDepartment of Electrical and Electronic Engineering, University of Stellenbosch, South Africa
First published on 30th April 2010
Abstract
A technique is described for performing solid phase extraction (SPE) at a semi-microscale. 10 mg of electrospun polystyrene fibers (average diameter 2.7 µm) were packed into a SPE barrel to form a disk (5 mm × 1 mm). The device was employed to evaluate the extraction of four steroids: prednisone, hydrocortisone, cortisone acetate and 19-nortestosterone from water and plasma matrices. The analytes were desorbed from the fibers with 100 µl methanol and monitored by high performance liquid chromatography with a diode array detector (HPLC-DAD). The semi-micro SPE method provided extraction recoveries of 51.14–80.13% in plasma and 66.07–93.43% in water. The breakthrough volumes at 500 ng ml−1 ranged from 200–400 µl for all analytes. At optimal conditions, the four analytes followed an excellent linear relationship in the range 12.5–400 ng ml−1 with coefficients of determination (r2) greater than 0.99 and the limits of detection ranged from 0.75 to 1.29 ng ml−1. Due to its simplicity, it is anticipated that the method will greatly simplify disk solid phase extraction.
Introduction
There is a growing realization that faster, more efficient and miniaturized methods for sample handling are essential. Solid phase extraction is a sample clean-up and preconcentration technique that has been used widely to isolate target analytes due to its simplicity of operation, high selectivity and good reproducibility. A current challenge is to conduct “flow-through” solid phase extraction quickly and efficiently on a smaller scale. The two possible ways of overcoming this challenge are to reduce the sorbent bed mass as well as the particle size. Sorbent packing format (disk or cartridge) is also an important aspect in making SPE more efficient.Disks have a larger surface area per unit bed mass compared to the cartridge format. They have a larger cross-sectional area that results in a reduced pressure drop. Such a configuration enables higher sample flow rates, and consequently shorter sample processing times. Although the bed height of SPE devices is considerably less when disks are used compared to cartridges or packed columns, the plate height realized for disks is much smaller because the extractive particle size in disks is typically 8–12 µm compared to 50 µm or more for packed columns. Cartridges are easily prepared in the laboratory while disks, so far, have been produced mostly in a manufacturing setting.1,2
To date, methods available for SPE disk fabrication involve a complicated multi-step process in which particles are tightly held together within an inert fiber matrix such as polytetrafluoroethylene (PTFE) (90% micro-particles and 10% PTFE by weight). Disks, therefore, are available in only a limited range of sorbent chemistries largely dictated by market forces, which makes them significantly more expensive than cartridges.3
A possible way to simplify and improve efficiency of SPE disk technology would be to explore fiber based sorbents. This could be achieved by fabricating polymeric fibers using electrospinning; a simple technique that uses repulsive electrostatic forces to draw nano- to microfibers from a polymeric solution.4,5 The use of small diameter fibers in the nano- to microscale results in an increase in the specific surface area compared to the currently available micro-particles, this allows a reduction in sorbent bed mass.6
To the best of our knowledge, the use of electrospun polystyrene microfiber disks for SPE at the semi-microscale has not been reported in literature. It is anticipated that this approach will open up a wide range of SPE disk sorbent chemistries that can address past and emerging matrix challenges in biological, biotechnological and environmental samples.
Experimental
Equipment and reagents
The chromatographic system used consisted of a G1312B binary pump and a G1315C diode array detector (Waldbronn, Germany). The HPLC flow rate was 1 ml min−1 and the mobile phase was a methanol–water (58 : 42, v/v) mixture. Separation was achieved on a Zorbax Eclipse Plus C8, 3.5 µm × 4.6 mm × 100 mm, with subsequent detection at 240 nm. The morphology of the fibers was observed by a scanning electron microscope (SEM; Vega Tescan, USA). Except for the HPLC grade methanol, all other reagents were of analytical reagent grade and deionised water was from a millipore water purification system.Synthesis of polystyrene
Polystyrene was synthesized by boiling medium emulsion polymerisation by the following procedure: 20 ml of styrene monomer and 100 ml of deionised water were added to a three necked flask and the stirring speed set at 300 rpm. The mixture was raised to reflux, and after the medium had boiled for 10 min, 0.2 g potassium persulfate powder was added to the solution. The reaction was stopped after 3 h, after which the polymer powder was purified and dried for use.Microfiber fabrication
Polystyrene microfibers were fabricated by electrospinning. Firstly, polystyrene was dissolved in a mixture of dimethylformamide (DMF) and tetrahydrofuran (THF) (3 : 7, v/v) to make a solution of 20 wt%. The solution was loaded into a 10 ml glass syringe with a stainless steel needle that was connected to the anode of a high voltage power supply. A rotating PVC drum wrapped with aluminium foil was used as a collector. Controlled deposition of the micro-fibers in the form of a sheet was achieved by placing a sharp edged copper strip (counter electrode, cathode to converge the electric field lines and thus focus the deposition of the fibers) behind the rotating drum. Electrospinning conditions used were: +15 kV (needle tip), −5 kV (counter electrode), 15 cm (tip to collector distance) and 0.01 ml h−1 (flow rate).Construction of disk SPE device and solid phase extraction procedure
The disk SPE device was prepared manually by packing 10 mg of polystyrene fibers into a 1 ml polypropylene SPE barrel. Fig. 1a shows a SEM image of a polystyrene microfiber disk (5 mm diameter and a height of 1 mm). The polystyrene fibers were forced into a 1 ml SPE barrel sandwiched between polyethylene frits and made firm by a glass rod (5 mm diameter). The polystyrene fiber disk was then separated from the frits and independently forced to sit tightly at the base of the SPE barrel. Fig. 1b shows an expanded view of the polystyrene microfiber disk sitting at the base of the SPE barrel. Solid phase extraction was performed by conditioning the disk with 100 µl methanol and equilibrating with 100 µl water. After loading 100 µl of spiked plasma the sorbent was washed with 100 µl water, and the analytes were eluted with 100 µl methanol. Fig. 1c shows the microfiber disk SPE device on a 10 port vacuum manifold for the SPE procedure reported in this work. The initial experiments were conducted using a water solution of the steroids as the model sample. | ||
| Fig. 1 Electrospun fiber SPE disk device (a) SEM image of the electrospun fiber disk (5 mm × 1 mm); (b) magnification of the electrospun fiber disk at the base of the SPE barrel; (c) SPE barrel (with microfiber disk) connected to a 10 port SPE vacuum manifold. | ||
Results and discussion
The goal of this research was to explore the possibility of using electrospun fibers packed as disk format for solid phase extraction. This work was prompted by the need to simplify the fabrication of disk solid phase extraction devices. The first part of the work involved an investigation of the effect of packing format and sorbent mass on the extraction efficiency. Two packing formats were compared, that is the disk (reported in this work) and that reported by Zhang et al. who packed the electrospun fibers into a micropipette tip (column).7–9Initial recovery studies of the polystyrene microfibers were carried out on water samples spiked with 500 ng ml−1 of each of the four steroids. In order to investigate the effect of sorbent mass on extraction efficiency, 5 mg and 10 mg of polystyrene fibers were packed. As shown in Fig. 2, the extraction recoveries for the SPE disks improved from 66.07–81.43% to 83.76–93.43% when the sorbent mass was increased from 5 mg to 10 mg. The micropipette tip exhibited slightly higher recoveries of 101.07–124.29% compared to the disk format, which can be attributed mainly to the increased sorbent bed height. However, the SPE procedure for the disk format was more simplified as packing the fibers was easier and the extraction was achieved in less than 15 minutes on a vacuum manifold.
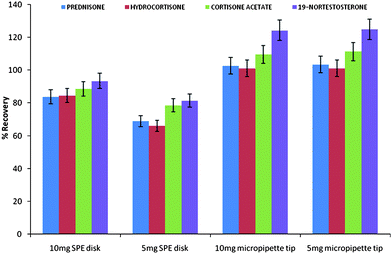 | ||
| Fig. 2 Effect of sorbent mass and packing format on polystyrene microfibers extraction efficiency of four steroids. | ||
Extraction ability of the polystyrene fiber disks was further evaluated by determining the extraction recoveries of the analytes in spiked plasma at three different concentrations (500, 125 and 31.25 ng ml−1). The extraction recoveries ranged from 51.14 to 80.13 over the three concentration ranges. The analytical characteristic data of the semi-micro SPE method coupled with HPLC for the determination of the four target analytes are summarised in Table 1. Good linearity of the four analytes was achieved in the range 12.5–400 ng ml−1 with correlation coefficients greater than 0.99. The detection limits were calculated at a signal to noise ratio of 3 and ranged from 0.75 to 1.29 ng ml−1. The repeatability of the semi-micro method (expressed as the relative standard deviations of the peak areas) was determined for five analyses of the same samples; it ranged from 2.81 to 8.92%. Although there was a decrease in absolute recoveries at 500 ng ml−1, the results still indicated to the suitability of the SPE method for the trace determination of the target analytes. On the basis of the analytical parameters, it was successfully demonstrated that semi-micro SPE using polystyrene microfiber disks was viable.
| Analyte | Linear range/ng ml−1 | Linearity, r2 | Repeatability (% RSD) | LOD/ng ml−1 | LOQ/ng ml−1 | Absolute recovery (plasma) | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| 500 ng ml−1 | 125 ng ml−1 | 31.25 ng ml−1 | 500 ng ml−1 | 125 ng ml−1 | 31.25 ng ml−1 | |||||
| Prednisone | 12.5–400 | 0.9954 | 7.83 | 6.27 | 7.19 | 1.27 | 4.22 | 51.14 | 62.96 | 70.34 |
| Hydrocortisone | 12.5–400 | 0.9965 | 2.81 | 4.16 | 5.66 | 0.75 | 2.51 | 51.85 | 65.13 | 69.45 |
| Cortisone acetate | 12.5–400 | 0.9971 | 8.92 | 8.76 | 3.72 | 0.95 | 3.15 | 54.29 | 70.2 | 78.34 |
| 19-Nortestosterone | 12.5–400 | 0.9958 | 7.11 | 2.7 | 7.44 | 1.29 | 4.29 | 55.43 | 70.59 | 80.13 |
One of the most important characteristic parameters in establishing the suitability of a SPE sorbent for isolating target analytes is the breakthrough volume (VB), as it gives an indication of the sorbent's loading capacity for the target analytes. The knowledge of the equilibrium volume (VE) and the retention volume (VR) is also invaluable in method development for solid phase extraction. Experimental determination of these three key parameters was achieved graphically from the breakthrough curves for all analytes which were obtained by frontal chromatography. Frontal analysis was carried out by loading 500 ng ml−1 aliquots of spiked water samples ranging from 100 µl to 4000 µl and monitoring the eluates by HPLC-DAD. Under ideal conditions, the breakthrough curve has a bilogarithmic shape, however, experimentally the breakthrough curve is plotted by using the line of best fit. Fig. 3 shows the breakthrough curves for the four steroids plotted as the lines of best fit. Due to the difficulties associated with determining the small changes in analyte concentration, the breakthrough volumes were extrapolated as 10% of the outlet maximum concentration. As shown in Table 2, the breakthrough volumes for all analytes ranged from 200 to 400 µl. These values were all well above the loading volume (100 µl), thus confirming that quantitative recovery (100%) could be achieved. Higher values of VB could have been obtained at lower analyte concentrations as evidenced by the high extraction recoveries obtained in the initial experiments (see Table 1) at lower analyte concentrations.
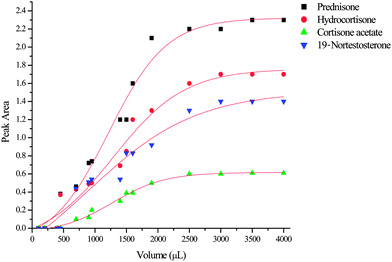 | ||
| Fig. 3 Breakthrough curves determined for the four steroids on the polystyrene microfiber disks. | ||
Determination of the number of theoretical plates (N) is necessary as it gives an indication of the SPE column efficiency. The plate number was determined from the breakthrough curve using the following equation:
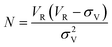
Unlike HPLC columns, for SPE columns the sample enters as a front instead of a narrow plug as no separation of the same class of analytes is intended, therefore it is not necessary to have many theoretical plates as for an HPLC column. However, the retention volume must be large enough in order to permit the handling of larger sample volumes. Typical cartridges provide about 5 to 15 theoretical plates per cm of bed height and particle loaded membrane disks provide about 4 to 9 theoretical plates. As seen from Table 2, the number of theoretical plates (1.47 to 3.25) is smaller compared to conventional disks. This could simply be due to the errors associated with the graphical determination of VR and σv. Although the theoretical plates were lower, the retention volumes were large enough. On average the retention volumes were 14 times the sample volume and thus confirming that the SPE device is suitable for this application. Since this particular SPE sorbent was primarily meant for trace analysis, the results obtained at 500 ng ml−1 demonstrate that it can handle larger sample volumes at lower concentrations. An important conclusion to be drawn from this study is that sampling devices that exhibit very low values of N might still have a capacity that is sufficient for practical purposes, provided that the retention is large enough.
Conclusions
A novel method for solid phase extraction at the semi-microscale using electrospun polystyrene microfiber disks was successfully demonstrated. This simplified approach could be an attractive alternative for SPE disk fabrication. It is therefore anticipated that through this approach, fabrication of SPE disks of a wide range sorbent chemistries will be possible.References
- W. Pipkin, Am. Lab. (Shelton, Conn.), November 1990, 40D Search PubMed.
- E. M. Thurman and K. Snavely, TrAC, Trends Anal. Chem. (Pers. Ed.), 2000, 19, 18 CrossRef CAS.
- J. S. Fritz and J. J. Masso, J. Chromatogr., A, 2001, 909, 79 CrossRef CAS.
- J. Doshi and D. H. Reneker, J. Electrostat., 1995, 35, 151 CrossRef CAS.
- D. Li and Y. Xia, Adv. Mater., 2004, 16, 1151 CrossRef CAS.
- Z. M. Huang, Y. Z. Zhang, M. Kotaki and S. Ramakrishna, Compos. Sci. Technol., 2003, 63, 2223 CrossRef CAS.
- X. J. Kang, L. Q. Chen, Y. Y. Zhang, Y. W. Liu and Z. Z. Gu, J. Sep. Sci., 2008, 31, 3272 CrossRef CAS.
- X. J. Kang, C. Pan, Q. Xu, Y. Yao, Y. Wang, D. Qi and Z. Gu, Anal. Chim. Acta, 2007, 587, 75 CrossRef CAS.
- Y. Zhang, X. Kang, L. Chen, C. Pan, Y. Yao and Z. Z. Gu, Anal. Bioanal. Chem., 2008, 391, 2189 CrossRef CAS.
- K. B. Daszkiewicz and A. Voekl, Talanta, 2009, 80, 614 CrossRef.
- C. F. Poole, A. D. Gunatilleka and R. Sethuraman, J. Chromatogr., A, 2000, 885, 17 CrossRef CAS.
| This journal is © The Royal Society of Chemistry 2010 |