A reagentless signal-on architecture for electronic, real-time copper sensors based on self-cleavage of DNAzymes†
Lidong
Li
*,
Long
Luo
,
Xiaojiao
Mu
,
Tianyu
Sun
and
Lin
Guo
*
School of Chemistry & Environment, Beijing University of Aeronautics & Astronautics, Beijing, 100191, China. E-mail: lilidong@buaa.edu.cn; guolin@buaa.edu.cn; Tel: +086-010-82316498
First published on 28th April 2010
Abstract
Herein, a rapid electrochemical biosensor based on highly specific, metal-induced self-cleaving DNAzymes was designed. This proposed sensing protocol offers reasonable selectivity, amazingly fast speed and operational convenience for copper assays on-line.
The development of specific ion sensors is linked to pressing needs for the rapid detection of toxic metals, such as Cu2+. Currently, flame and graphite furnace atomic absorption spectrometry and spectrophotometric methods are known as the most common techniques used for copper determinations.1 These techniques suffer from their insufficient sensitivity to very low concentration of the metal in many environmental and biological samples. Very recently, scanning electrochemical microscopy (SECM), combined with surface plasmon resonance (SPR), SECM-SPR, was applied for real-time detection of the incorporation of copper ion,2 which requires expensive robotic facilities, and complicated operation process. In recent years, fluorescent and optical fiber sensors have been developed to achieve parts-per-billion detection limits.3 These optical methods, however, suffer from possible drawbacks including unstable chemical and florescence properties, potential false signals arising from contaminating colorants, fluorophores and quenchers, and, frequently, a reliance on cumbersome optical equipment.4 Electrochemical methods, in contrast, benefit from the impressive miniaturization of modern microelectronics, the relative paucity of electroactive contaminants, and the relative stability and environmental insensitivity of electroactive labels and thus are less likely to suffer from these potential drawbacks. Nevertheless, the classical polarographic techniques, which employ a dropping mercury electrode (DME) with a glass capillary, lead to irreproducible dropping characteristics and inaccuracy because of the etching of the glass capillary itself.5 The electrochemical copper detection methods reported to date require complex, multistep protocols involving the reductive deposition of metallic copper followed by anodic stripping voltammetry, and the relatively high detection limit suffers as well.6
Aptamers, short oligonucleotides selected in vitro for specific, high-affinity binding to a broad range of molecular targets, are considered as promising recognition elements for biosensor applications.7 Recently, electrochemical aptamer-based (E-AB) sensors have emerged as a promising and versatile new biosensor platform.8 E-AB signal generation occurs when a binding-induced conformational change significantly alters electron tunneling from a redox-tagged, electrode-bound aptamer to the sensing electrode. E-AB sensors have been widely applied to biological systems by Plaxco and Fan's group.9 Herein, based on E-AB sensing mechanisms, we propose a simple electrochemical copper detection approach based on the highly specific, metal-induced self-cleaving DNAzymes.
Generally, biological catalysis is dominated by enzymes made of protein and, to a lesser extent, by enzymes made of RNA.10 DNA is considerably more resistant than RNA to degradation in aqueous solutions and hence is well-suited to serve as a reservoir for biological information. However, in vitro selection for catalytic polynucleotieds, has been used recently to create several classes of DNA enzymes, including DNAs that cleave RNA, ligate chemically activated DNAs and promote porphyrin metallation. It was found that DNA is more susceptible to scission via depurination followed by elimination or via oxidative mechanisms than by hydrolysis.11 Based on in vitro selection of self-cleaving DNAs, Breaker et al. reported that single-stranded DNA can adopt structures that promote divalent-metal-dependent self-cleavage via an oxidative mechanism, suggesting that efficient DNA enzyme might be made to cleave DNA in a biological context.12 In their findings, an optimized single stranded DNA displays significant cleavage when only Cu2+ is added. Our copper sensing protocol is based on the copper-dependent and highly site-specific cleavage of single stranded DNA.
The copper-dependent self-cleaving DNAzyme (DNA(1)) we have employed is totally composed of 87 nucleotides. Previous studies indicate that the most frequent sites of cleavage are located at two different regions: from 9 to 14 nucleotides and near position 72 of the sequence domain.12 To construct a signal-on architecture for a self-cleaving DNAzyme sensor, a short ferrocene(Fc)-tagged 26 base oligonucleotide DNA(2) was introduced. The DNA(2) is predesigned by partially complementary to DNA(1) from 1 to 8 nucleotides, and from 15 to 22 nucleotides (reversed sequence of DNA(2)). In this design, we leave 9–14 in DNA(2) unbound to DNA(1) because this is a very sensitive region to be cleaved in DNA(1). Meanwhile, to give a stable secondary structure of DNA(2) when releasing from DNA(1), we make the last 4 bases at the 5′ end complementary to the first 4 bases at the 3′ end, in this case, the single strand DNA(2) can bend over itself to give very strong redox signal based on its favorable secondary structure (Fig. S1†). This proposed sensor is also constructed by covalently attaching a thiolated DNA (2) to a gold electrode via well-established self-assembled monolayer chemistry.
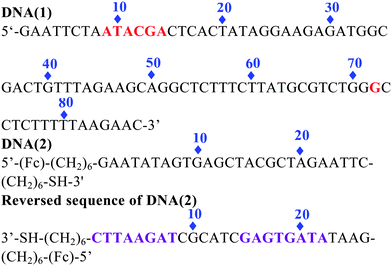
In the absence of Cu2+ (Scheme 1 left), this double stranded DNA complex, which is chemi-absorbed to a gold electrode via a 3′ terminal thiol on the single strand DNA(2), is relatively rigid, presumably preventing the Fc from approaching the electrode to transfer electrons. As we know, the redox reaction of ferrocene is highly distance-dependent, which was predicted by the Marcus electron transfer theory. Therefore, at this conformation, theoretically, almost no Faradic current should be observed. In the presence of Cu2+ (Scheme 1 right), the catalytic DNA (1) strand breaks into 3 fragments via two locations (9–14) and 72. These fragments presumably dissociated from the complex, releasing partially complementary DNA(2). The Faradaic currents will be significantly produced, presumably because the DNA(2) place their redox tags in close proximity to the electrode upon folding into their favorable secondary configurations. Of note, the other cleavage site at 72 in DNA(1) in our sensor protocol is ignored. The fragment cleaved from this site will be dissolved into the aqueous phase without affecting the signal change. In addition, the deposition of the catalytic strand during sensor fabrication must be carefully controlled to produce optimal signal gain and reproducibility. As determined by electrochemical measurements,13 the surface coverage of the self-cleaving DNA(1) was maintained within the range of 6.67 pmol cm−2, an electrode loading level at which sensor gain is maximized.
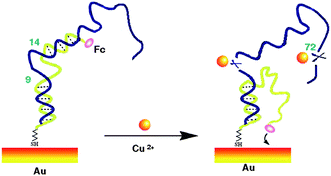 | ||
| Scheme 1 A schematic of the self-cleaving DNAzyme-based electrochemical sensor. | ||
To fabricate the sensor, the mixture of the two partially complementary sequences DNA(1) and DNA(2) (1 μM each) in 1 M NaClO4 containing 1 mM TCEP (TCEP was employed to cleave disulfides) was heated to 90 °C and then slowly cooled to room temperature. Then the cleaned gold electrode was incubated into the above DNA duplex (double stranded DNA) for 18 h. The resulting monolayer-functionalized surface was ready for the copper detection. All the electrochemical measurements were carried out in 10 mM HEPES containing 50 mM NaClO4, 0.5 M NaCl and 0.5 M KCl (pH 7.4) unless otherwise stated. A control experiment was carried out to investigate the effect of HEPES. In the absence of HEPES, the SWV signal of copper is decreased very quickly with time. While, in the presence of HEPES, very stable signals were observed, indicating the stable pH value during the measurement is significantly important. (Fig. S2 and S3†) Meanwhile, in order to reduce the current intensity change due to the operation error, the maximized sensor gain and stable current signal are highly expected. Therefore, another control experiment was done to test the operation methods during the electrochemical measurements. The results show that the maximized and stable current signal can be reached by applying voltage scan consecutive 2 times. The obvious signal intensity change was observed with the different operation method: remain still for 2 min; remain still for 2 min plus apply voltage scan continuously two times; (Fig. S4 and S5†). Scheme S1† illustrates that the Fc tag will be diffused away from the gold electrode surface by gravity whilst remaining still, leading to the decrease of Faradic current. After continuous applying a voltage scan two times, the signal intensity apparently increased due to the electrostatic effect between the negatively charged phosphate backbones of DNA and positive voltage scan, which brings the redox tags into the closest proximity of the electrode surface. After a voltage scan of two times, the signal reached its maximum value and started to level off. All of our electrochemical measurements were carried out by the method of applying a voltage scan two times to reach the optimized signals.
The self-cleaving DNAzyme based sensor is sensitive and specifically responsive to its target ion (Fig. 1a). In the absence of copper, only very tiny Faradaic currents were observed at 0.24 V due to the redox reaction of ferrocene. We presume this residual peak arises because the surface-immobilized Fc-tagged DNA(2) strand is in a conformational equilibrium between a duplex DNA and its favorable bending-over secondary configuration. The baseline descending with time was also investigated during the experiments. The results show that the base line shifts down slowly with measurement time, indicating the charging current of the double layer decreased with time. Of note, in the voltage scan range of SWV, the trend of baseline descending gradually increases from 0.1 V to 0.4 V, which can be ascribed to the charging current increase with increasing voltage (Fig. S6†). Upon increasing Cu2+, we observe a large increase in Faradaic current. The directly measured detection limit of the current sensor architecture is 20 nM, and the signal gain is approximately linear over the range from 20 nM to 600 μM (Fig. 1b), reaching its maximum value around 600 μM, and starts to level off.
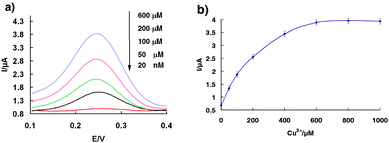 | ||
| Fig. 1 The copper sensor is responsive to its target. (a) Square wave voltammograms of the sensor obtained on-line at various Cu2+ concentrations. (b) Dose-responsive curve for the copper sensor. The error bars represent the standard deviation of four measurements. | ||
As the EPA action level for total copper in drinking water is 1.3 ppb (20.1 μM), and an action level of 0.6 ppb (9.4 μM) has been set for copper in the irrigation water of crops, the detection range around the 10 μM scale is rather important than relatively larger or lower levels. As evidence of the applicability of our sensor for such applications, we have tested its ability to quantify copper in water at parts-per-milllion concentrations (Fig. 2). The signal gain is almost perfectly in a linear relationship over this 10 μM range with R2 = 0.9985, indicating this proposed self-cleaving DNA architecture is an ideal electrochemical sensor for the routine monitoring of copper levels in drinking water and environmental water samples.
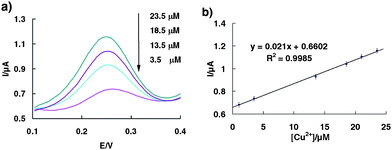 | ||
| Fig. 2 (a) Square wave voltammograms of the sensor obtained on-line at various Cu2+ concentrations from 1 μM to 25 μM. (b) Dose-responsive curve for the copper sensor in the range of 1 μM–25 μM. The error bars represent the standard deviation of four measurements. | ||
Particularly, the sensor equilibrates very rapidly. We observe 98% total signal change within the first half minute and complete saturation in less than 1 min (Fig. S7†). Therefore, it can be served as an on-line sensor for the detection of copper. In our experiments, the fabricated sensor was inserted into the Cu(II) sample solution, measurements were taken in 1 min. We presume this fast response is due to the high catalytic activity of the selected synthetic 87-nucleotide version of DNA(1) with kobs = 0.052 min−1 (10 μM Cu2+, 23 °C) and the fast equilibrium between the duplex configuration and the favorable secondary structure of DNA(2).
The specificity of the sensor was determined by challenging it with the divalent metal ions Mg2+, Zn2+, Ni2+, Mn2+, Co2+, Cu2+, Pb2+, at 1 μM concentrations. Importantly, the SWV signal for the other divalent metals was only as small as the background (in the absence of copper), approximately 10-fold lower than that for 1 μM copper (Fig. 3a). Likewise, the sensor's response to Cu2+ is unaffected by the presence of divalent contaminating ions such as Mg2+, Zn2+, and Ni2+ (Fig. 3b).This excellent selectivity arises not only from the high specificity of the self-cleaving DNA(1) strand, but also from the additional stringency due to the competition between the duplex and the ferrocene tagged DNA(2) strand.
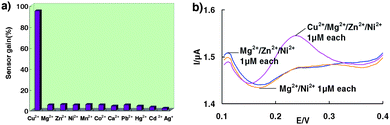 | ||
| Fig. 3 (a) Little signal change is observed when the sensor is challenged with divalent metal ions other than Cu2+, (b) Signals obtained after the sensor is challenged with Mg2+/Ni2+ 1 μM each; Mg2+/Zn2+/Ni2+ 1 μM each and Cu2+/Mg2+/Zn2+/Ni2+ 1 μM each. | ||
To investigate if the method was applicable to real samples, we tested real and spiked lake samples with different Cu2+ ion concentrations. The recoveries of standard addition are 92.8%, 97.6% and 106.2% for Cu2+ ion concentrations of 1.2 μM, 6 μM and 9.6 μM, respectively, when using the proposed electrochemical method. The storage stability of the aptasensor was also checked. The results show that the SWV signal only reduces 6.07% of the original value after one-month storage in a buffer solution.
In summary, the self-cleaving DNA based electronic sensor described here exhibits excellent sensitivity and specificity for its target ion. It also offers reasonable selectivity, amazingly fast speed and operational convenience. The fast response provides a possible approach to be used as an on-line sensor for copper assays. In addition, it is a reagentless sensor since both the recognition element (DNA-cleaving DNAzymes) and the signaling element (ferrocene) have been integrated in a surface-confined configuration. Especially, it offers a novel approach for the signal-on sensor based on DNA-cleaving DNAzymes which has two possible cleavage sites. In our architecture design, the tagged signal strand is only partially complementary to the lower part of DNAzymes, the upper cleavage site in DNAzymes is completely ignored. The good results prove that this short signal strand design makes our sensor even simpler and cheaper. Finally, given the potential for in vitro selection to produce cofactor-dependent DNA-cleaving DNAzymes and given that it appears that DNAzymes can be immobilized without loss of activity, this approach may prove of general utility for electrochemical analyte detection.
This project was financially supported by National Natural Science Foundation of China (20903008 & 50725208) and the National Basic Research Program of China (2010CB934700) as well as the State Key Project of Fundamental Research for Nanoscience and Nanotechnology (2006CB932300).
Notes and references
- (a) B. Welz, Atomic Absorption Spectroscopy, VCH, Amsterdam 1985 Search PubMed; (b) Standard Methods for Examination of Water and Wastewater, 19th ed., American Public Health Association, Washington, DC, 1995 Search PubMed.
- Y. L. Xin, Y. Gao and J. Guo, Biosens. Bioelectron., 2008, 24, 369–375 CrossRef CAS.
- (a) Z. Xu, Y. Xiao, X. Qian, J. Cui and D. Cui, Org. Lett., 2005, 7, 889–892 CrossRef CAS; (b) S. H. Kim, J. S. Kim, S. M. Park and S. K Chang, Org. Lett., 2006, 8, 371–374 CrossRef CAS; (c) L. Zeng, E. Miller, A. Pralle, E. Y. Isacoff and C. J. Chang, J. Am. Chem. Soc., 2006, 128, 10–11 CrossRef CAS; (d) M. Royzen, Z. Dai and J. W. Canary, J. Am. Chem. Soc., 2005, 127, 1612–1613 CrossRef CAS; (e) K. M. K. Swamy, S.-K. Ko, S. K. Kwon, H. N. Lee, C. Mao, J.-M. Kim, K.-H. Lee, J. Kim, I. Shin and J. Yoon, Chem. Commun., 2008, 5915–5917 RSC.
- Y. Xiao, A. A. Rowe and K. W. Plaxco, J. Am. Chem. Soc., 2007, 129, 262–263 CrossRef CAS.
- (a) A. M. Bond and T. A. O'Donnell, Anal. Chem., 1969, 41, 1801–1806 CrossRef CAS; (b) A. M. Bond, T. A. O'Donnell and R. J. Taylor, Anal. Chem., 1972, 44, 464–467 CrossRef CAS.
- C. Truzzi, A. Annibaldi and S. Illuminati, Anal. Bioanal. Chem., 2008, 392, 247–262 CrossRef CAS.
- (a) H. X. Chang, L. H. Tang, Y. Wang, J. H. Jiang and J. H. Li, Anal. Chem., 2010, 82, 2341–2346 CrossRef CAS; (b) Y. Jin, W. Lu, J. Q. Hu, X. Yao and J. H. Li, Electrochem. Commun., 2007, 9, 1086–1090 CrossRef CAS.
- (a) Y. L. Zhang, Y. Wang, H. B. Wang, J. H. Jiang, G. L. Shen, R. Q. Yu and J. H. Li, Anal. Chem., 2009, 81, 1982–1987 CrossRef CAS; (b) Y. Jin, X. Yao, Q. Liu and J. H. Li, Biosens. Bioelectron., 2007, 22, 1126–1130 CrossRef CAS.
- (a) C. H. Fan, K. W. Plaxco and A. J. Heeger, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 9134–9137 CrossRef CAS; (b) B. R. Baker, R. Y. Lai, M. S. Wood, E. H. Doctor, A. J. Heeger and K. W. Plaxco, J. Am. Chem. Soc., 2006, 128, 3138–3139 CrossRef CAS; (c) Y. Xiao, B. D. Piorek, K. W. Plaxco and A. J. Heeger, J. Am. Chem. Soc., 2005, 127, 17990–17991 CrossRef CAS; (d) X. L. Zuo, S. P. Song, J. Zhang, D. Pan, L. H. Wang and C. H. Fan, J. Am. Chem. Soc., 2007, 129, 1042–1043 CrossRef; (e) R. J. White, A. A. Rowe and K. W. Plaxco, Analyst, 2010, 135, 589–594 RSC; (f) J. Zhang, L. H. H. Zhang, F. Boey, S. P. Song and C. H. Fan, Small, 2010, 6, 201–204 CrossRef CAS.
- (a) T. R. Cech, Annu. Rev. Biochem., 1990, 59, 543–568 CrossRef CAS; (b) R. H. Symons, Annu. Rev. Biochem., 1992, 61, 641–671 CrossRef CAS; (c) F. Michel and J. L. Ferat, Annu. Rev. Biochem., 1995, 64, 435–461 CrossRef CAS.
- (a) R. R. Breaker and G. F. Joyce, Trends Biotechnol., 1994, 12, 268–275 CrossRef CAS; (b) R. R. Breaker, Curr. Opin. Biotechnol., 1996, 7, 442–448 CrossRef CAS; (c) R. R. Breaker and G. F. Joyce, Chem. Biol., 1995, 2, 655–660 CrossRef CAS; (d) T. Lindahl, Nature, 1993, 362, 709–715 CrossRef CAS.
- N. Carmi, L. A. Shultz and R. R. Breaker, Chem. Biol., 1996, 3, 1039–1046 CAS.
- H.-Z. Yu, C.-Y. Luo, C. G. Sankar and D. Sen, Anal. Chem., 2003, 75, 3902–3907 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Materials and methods, supplementary Fig. S1–S7 and Scheme S1. See DOI: 10.1039/c0ay00176g |
| This journal is © The Royal Society of Chemistry 2010 |