X-Ray crystallographic studies of three substituted indium(III) phthalocyanines: effect of ring substitution and the axial ligand on molecular geometry and packing
Aurélien
Auger
,
Paul M.
Burnham
,
Isabelle
Chambrier
,
Michael J.
Cook
* and
David L.
Hughes
Wolfson Materials and Catalysis Centre, School of Chemical Sciences and Pharmacy, University of East Anglia, Norwich, UK NR4 7TJ. E-mail: m.cook@uea.ac.uk; Fax: +44 1603 592015; Tel: +44 1603 593135
First published on 10th November 2004
Abstract
Crystal structure analyses of a series of three substituted indium phthalocyanines, viz., chloro[1,4,8,11,15,18,22,25-octakis(hexyl)phthalocyaninato]indium(III), 1, 4-fluorophenyl[1,4,8,11,15,18,22,25-octakis(hexyl)phthalocyaninato]indium(III), 2, and racemic chloro[2-bromo-1,4-dibutoxy-8,11,15,18,22,25-hexakis(hexyl)phthalocyaninato]indium(III), 3, are reported. Despite varying the axial ligand from a chloro group to the more space demanding 4-fluorophenyl group, and introducing somewhat different substituents onto one of the benzenoid rings of one of the derivatives, the three compounds form similar types of columnar stacks. Key components of the stack structure are concave–concave pairs of molecules. The molecules are offset but allow for a significant degree of overlap. Components of these pairs then participate in a lower area convex–convex overlap with adjacent pairs to form an offset column of pairs. However, one of the compounds, 1, shows a different type of global packing to the other two. For this compound, the molecules of one column are tilted with respect to those in an adjacent column and thus exhibit a type of herring bone arrangement; the molecules of the other two compounds pack with their central planes parallel. All three compounds form apparently even and transparent films by the spin coating method. UV-vis spectroscopy shows that the visible region absorption band is shifted to the red relative to the solution phase spectra.
Introduction
Phthalocyanines exhibit a wealth of solid state properties that render them important examples of organic functional materials. Thus specific compounds, or sub-sets, exhibit for example either extrinsic1 or intrinsic semiconductivity,2 photoconductivity3 and non-linear optical behaviour.4 They are in use in technologies as diverse as electrophotography,5,6 laser printing6 and optical data storage,7 and have potential in gas sensors,1,2 charge carrier devices,8 and photovoltaic devices3 including photoelectrochemical cells.A number of the applications utilise phthalocyanines in the solid state, typically in the crystalline form or as thin films. It has been well established that molecular packing can significantly affect semiconduction,1 charge carrier8 and optical absorption properties,9 a dependence that has been much studied for unsubstituted phthalocyanines. The optimisation of the solid state properties of these compounds can become complicated by their rich polymorphology. However, this same polymorphology has also contained the solution to particular problems. Thus the Xerox company's applications patent of 197410 identified the x-polymorph of metal-free phthalocyanine as a superior photoconductive material for electrophotography. Subsequently, attention focused on the polymorphic forms of titanyl phthalocyanine revealing that the so-called Y-form is most effective;11 more recently the V form of hydroxygallium phthalocyanine has attracted research.12 X-Ray crystallography has been used extensively as part of polymorph characterisation.13
Developments in phthalocyanine research over the last 25 years or so have provided a multitude of substituted derivatives.14 These analogues frequently exhibit properties, in particular photophysical properties, that are either modified relative to the unsubstituted parent phthalocyanine or are indeed unique to the compounds themselves, such as liquid crystallinity15 and high solubility in a range of solvents. The latter, among other things, facilitates access to solid state formulations by film deposition methods from the solution phase,16 methods that are appropriate for a variety of advanced technologies. However, X-ray structure determinations of substituted compounds remain few.13 Experience suggests that difficulties are often encountered in obtaining single crystals suitable for such studies. There are, nevertheless, exceptions and these include a number of examples of octa-substituted derivatives where alkyl,17 alkoxy,18 alkylsulfanyl19 and phenyl20 groups are located at the non-peripheral (1,4,8,11,15,18,22,25) positions. These studies have given insight into how such substituents can have a marked effect on both the geometry of the phthalocyanine ring and the molecular packing. However, whereas changes in conformation of the ring induced by substituents can be largely rationalised, understanding variations of molecular packing consequent on changes of metal centre and substituent chains is more challenging. The latter, together with the development of crystal structure/property relationships, provides an intriguing area of study within functional organic materials.
The present paper describes an X-ray crystal study of three substituted indium phthalocyanines, 1, 2 and 3. It has been undertaken to probe, first, the effect of change of the axial ligand on the molecular packing and crystal properties of two 1,4,8,11,15,18,22,25-octahexyl substituted derivatives, 1 and 2. The third compound, the novel chloroindium derivative 3, was investigated to explore the effect of added complexity of the substituents on the geometry and packing of the phthalocyanine. Thus six of the eight hexyl substituents contained in 1 and 2 are retained but two are replaced with two butoxy groups. A bromine atom has also been incorporated because this in the presence of an axial ligand introduces chirality into the molecule. The present work has been conducted on the racemate of 3.
Experimental
Materials
The synthesis and characterisation of both 1 and 2 have been described previously.21Spin-coated films were prepared from solutions in THF (ca. 2.5 mg in 0.1 ml) dropped onto a clean glass slide rotating at 2000 rpm using a spinner (Headway Research Inc.). Spinning was continued for 20 s by which time the solvent had evaporated.
UV-vis spectra of solutions and films were measured using a Hitachi U3000 spectrophotometer.
Chloro[2-bromo-1,4-dibutoxy-8,11,15,18,22,25-hexakis(hexyl)phthalocyaninato]indium(III), 3
X-Ray crystallography
For this study, 1 was recrystallised from a THF–acetone mixture. Single crystals of 2 and 3 were both obtained by recrystallisation from THF–methanol.Crystal structure analyses of compounds 1, 2 and 3 were undertaken. Crystal data and the results of the refinement for each sample are collated in Table 1. Intensity data for 1 were recorded in the Bruker laboratories, courtesy of Dr T. Wagner; crystals of 2 and 3 were measured on equipment at UEA. The procedures of analysis were very similar, and we describe that for complex 3 as typical.
| 1 | 2 | 3 | |
|---|---|---|---|
| Elemental formula | C80H112ClInN8 | C86H116FInN8 | C76H103BrClInN8O2 |
| Formula weight | 1336.1 | 1395.7 | 1390.8 |
| Crystal system | Monoclinic | Triclinic | Triclinic |
| Space group | C2/c (no. 15) | P−1 (no. 2) | P−1 (no. 2) |
| Unit cell dimensions | |||
| a/Å | 29.331(2) | 12.873(3) | 12.694(3) |
| b/Å | 12.1280(10) | 17.910(4) | 17.033(3) |
| c/Å | 41.706(3) | 17.642(4) | 17.996(4) |
| α/° | 90 | 102.92(3) | 108.54(3) |
| β/° | 108.896(2) | 93.73(3) | 103.32(3) |
| γ/° | 90 | 90.50(3) | 92.96(3) |
| Cell volume, V/Å3 | 14036(2) | 3955.0(14) | 3556.8(12) |
| No. of formula units/cell, Z | 8 | 2 | 2 |
| Absorption coefficient/mm−1 | 0.425 | 0.349 | 0.982 |
| Temperature/K | 293(2) | 140(1) | 140(1) |
| Total no. of reflections measured | 76057 | 12642 | 19249 |
| No. of unique reflections | 12398 | 6866 | 11790 |
| R int for equivalents | 0.0485 | 0.104 | 0.094 |
| No. of ‘observed’ reflections (I > 2σI) | 11232 | 3217 | 6567 |
| Final R indices (‘observed’ data) R1, wR2 | 0.058, 0.139 | 0.064, 0.128 | 0.068, 0.171 |
| Final R indices (all data) R1, wR2 | 0.064, 0.143 | 0.158, 0.160 | 0.126, 0.189 |
Crystals of 3 are fine green needles. One was mounted in oil on a glass fibre and fixed in the cold nitrogen stream on a Rigaku R-Axis IIc image-plate diffractometer equipped with a rotating anode X-ray source (Mo-Kα radiation) and graphite monochromator. Using 4° oscillations, 48 exposures of 54 min each were made.
Data were processed using the DENZO/SCALEPACK programs.24 The structure was determined by the direct methods routines in the SHELXS program and refined by full-matrix least-squares methods, on F2’s, in SHELXL.25 There is disorder at the end of one of the butoxy chains and in one of the hexyl chains; there is also an alternative site (5% occupation) for the bromo group—resolution of the corresponding butoxy/hexyl groups has not been attempted. The molecules of the major and minor components of bromo substituents are of opposite enantiomers. The non-hydrogen atoms (except for the carbon atoms in disordered groups, and for the minor Br site) were refined with anisotropic thermal parameters. Hydrogen atoms were included in idealised positions and their Uiso values were set to ride on the Ueq/Uiso values of the parent carbon atoms.
In the refinement process, compounds 1 and 2 were treated similarly except that no hydrogen atoms were included in the disordered hexyl chains.
Scattering factors for neutral atoms were taken from the literature.26 Computer programs used in this analysis have been noted above or in the literature,27 and were run on a Silicon Graphics Indy at the University of East Anglia, or a DEC-AlphaStation 200 4/100 in the Biological Chemistry Department, John Innes Centre, Norwich.
CCDC reference numbers 248916–248918. See http://www.rsc.org/suppdata/jm/b4/b413189b/ for crystallographic data in .cif or other electronic format.
Results and discussion
Compound 1
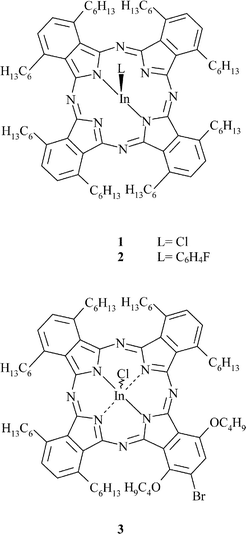
Chloro[1,4,8,11,15,18,22,25-octakis(hexyl)phthalocyaninato]indium(III) 1 crystallises as very fine dark blue needles with the molecular structure shown in Fig. 1. The indium atom is displaced from the central cavity of the macrocycle, lying 0.703(2) Å above the plane of the four central nitrogen atoms N(1,11,21,31). The core of the phthalocyanine ligand is slightly distorted from planarity, although it seems to have no overall systematic shape. Only two of the eight hexyl chains lie within the plane of the ring, those bonded to atoms C(7) and C(34). The chain bonded to atom C(14) is pointing down relative to the In–Cl bond whereas the chains stemming from C(24) and C(37) are facing up. Two of the chains slope slightly away from the ring plane, that of C(17) sloping downwards, and that of C(27) sloping up. The hexyl chain bonded to C(4) veers slightly upwards, towards the side of the C(1) atom, then down at the end. Selected bond dimensions are given in Table 2.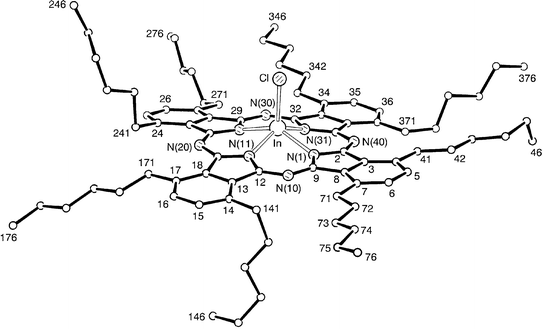 | ||
| Fig. 1 View of a molecule of 1, indicating the atom numbering scheme; atoms labelled ‘n’ represent the carbon atoms C(n). Hydrogen atoms have been omitted for clarity. | ||
| 1 (X = Cl) | 2 (X = PhF) | 3 (X = Cl) | |
|---|---|---|---|
| About the In atom: | |||
| In–N(1) | 2.139(3) | 2.203(9) | 2.150(5) |
| In–N(11) | 2.132(3) | 2.165(9) | 2.147(5) |
| In–N(21) | 2.140(3) | 2.191(9) | 2.134(5) |
| In–N(31) | 2.132(3) | 2.160(9) | 2.161(6) |
| In–X | 2.3528(10) | 2.111(12) | 2.353(2) |
| Mean In–N | 2.136(2) | 2.180(10) | 2.148(6) |
| N(11)–In–N(1) | 83.87(12) | 82.7(5) | 84.4(2) |
| N(1)–In–N(21) | 142.22(12) | 135.3(3) | 142.3(2) |
| N(31)–In–N(1) | 83.50(12) | 80.6(5) | 83.2(2) |
| N(11)–In–N(21) | 83.89(12) | 80.3(4) | 83.4(2) |
| N(11)–In–N(31) | 140.91(12) | 134.9(3) | 140.4(2) |
| N(31)–In–N(21) | 83.86(11) | 82.8(5) | 83.9(2) |
| Mean N–In–N(cis) | 83.78(9) | 81.6(7) | 83.7(3) |
| Mean N–In–N(trans) | 141.6(7) | 135.1(2) | 141.4(9) |
| N(1)–In–X | 107.81(9) | 116.6(3) | 104.99(14) |
| N(11)–In–X | 106.71(9) | 108.1(6) | 109.30(15) |
| N(21)–In–X | 109.94(8) | 108.0(4) | 112.68(14) |
| N(31)–In–X | 112.37(9) | 116.9(6) | 110.18(15) |
| Mean N–In–X | 109.2(12) | 112.(3) | 109.3(16) |
| Deviation of In from central N4 mean-plane | 0.703(2) | 0.833(5) | 0.710(3) |
| Angular deviation of each C6 mean-plane from the central N4 mean-plane: | |||
| C(3–8) | 5.43(11) | 1.5(4) | 7.5(2) |
| C(13–18) | 8.10(12) | 8.3(4) | 5.3(2) |
| C(23–28) | 9.20(12) | 9.5(5) | 3.0(2) |
| C(33–38) | 4.22(12) | 11.9(4) | 13.3(2) |
| Angular deviation of opposite C6 mean-planes: | |||
| C(3–8) vs C(23–28) | 8.33(13) | 9.9(5) | 6.2(2) |
| C(13–18) vs C(33–38) | 12.31(14) | 16.4(5) | 16.2(2) |
There are two principal features of the packing of 1, Fig. 2. The first is that pairs of molecules are packed in a so-called ‘concave–concave’ arrangement with the In–Cl bonds arranged antiparallel and the overlapping sections of the core 3.47 Å apart. This terminology is used by Engel13 who describes the concave side of a Pc molecule as the macrocycle component alone and the convex side as that which contains the macrocycle, the central atom and any ligands co-ordinated to it. The indium atom of one molecule of 1 is displaced considerably from its opposite member; C(39) is close to overlapping C(39′), and N(40) lies over the five-membered ring of N(31′),C(32′,33′,38′,39′). This pair of molecules is stacked less tightly (by overlap of benzenoid rings, 3.61 Å apart, with C(3) closest to the centre of symmetry) with neighbouring pairs, translated one unit cell along the b-axis. There are adjacent, parallel columns which form layers parallel to the a–b plane. The second packing feature is that this plane is bounded, on each side, by parallel planes in which the pairs of molecules have quite a different orientation in the cell. The angle between the normals to the N4 planes, that is the internal nitrogen atoms of the central Pc cavity, is 101.8°. Although this herringbone-like packing is common in unsubstituted M(II) metallated phthalocyanines11 and has been observed also for more complex tin diphthalocyanine28 and unsubstituted iodoindium Pc,29 we believe this to be the first example for a substituted Pc derivative. The role of the hexyl sidechains in the packing cannot be deduced since there are no available X-ray data for the structure of unsubstituted chloroindium Pc. However, a structural analysis of vacuum deposited films of the unsubstituted derivative led to the deduction that the Pc molecules pack closest to each other in a ‘convex–convex’ arrangement,30 thus contrasting to the ‘concave–concave’ packing of 1 in the solid state.
 | ||
| Fig. 2 Packing of the chloroindium phthalocyanine cores of 1, in layers parallel to the a–b plane. | ||
Compound 2
4-Fluorophenyl[1,4,8,11,15,18,22,25-octakis(hexyl)phthalocyaninato]indium(III) 2 (Fig. 3), forms flat blue-green needles upon crystallisation which fall into the triclinic system and have P−1 symmetry. There are two molecules in the unit cell, i.e. a molecule and its inverted relative. The central metal atom is now displaced 0.833(5) Å from the N4 plane, i.e. marginally more than the indium in compound 1, Table 2. This may imply an electronic effect of the fluorophenyl ligand on the indium. Only three of the hexyl chains lie in the same general plane as the Pc, those bonded to atoms C(4), C(7) and C(24). Four of the chains point up to the same side as the fluorophenyl ligand; those bonded to atoms C(17) and C(27) point straight up whereas those bonded to atoms C(34) and C(37) are disordered and generally only point up slightly. The remaining hexyl chain, bonded to C(14), is pointing down. Selected bond lengths and angles are given in Table 2. It can be seen that the values are similar to those obtained for 1 and that differences are related principally to the greater displacement of the In atom from the central N4 mean plane in 2.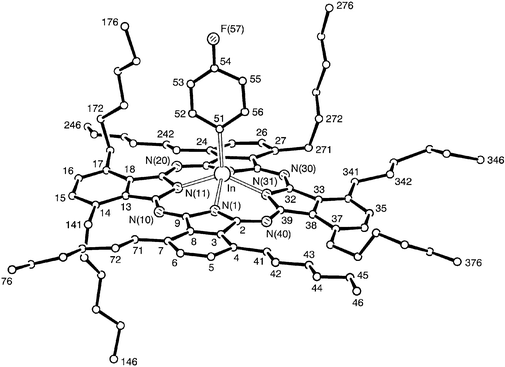 | ||
| Fig. 3 A molecule of 2 indicating the atom numbering scheme. There is disorder in the hexyl side chains at C(17) and C(34); only the major components are shown. Hydrogen atoms have been omitted for clarity. | ||
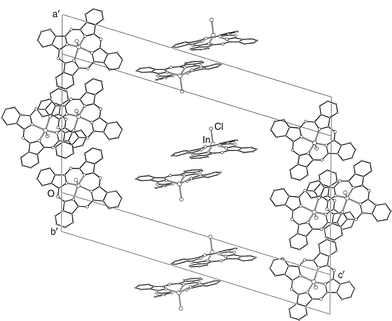
The molecular packing within the crystal of 2 is shown in Fig. 4. As with 1, the molecules are stacked closely, 3.50 Å apart, in pairs about centres of symmetry, with the axial ligands directed outward from the pair, i.e. concave–concave pairing. These pairs are stacked less tightly, by overlap of one phenyl ring pair, on the outer sides, i.e. convex–convex alignment. This is remarkably similar to the arrangements within the sheets in 1. However, there is a significant difference between the packing arrangements of 1 and 2 in that in the case of the latter all the Pc cores lie in parallel planes, so that there are no layers of an alternative orientation in 2.
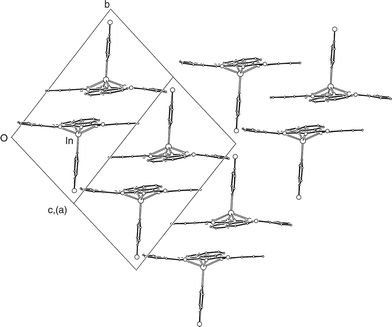 | ||
| Fig. 4 Packing of the molecules of 2, viewed along a cell face-diagonal. Only the phthalocyanine cores and axial ligands are shown. | ||
Compound 3
Chloro[2-bromo-1,4-dibutoxy-8,11,15,18,22,25-hexakis(hexyl)phthalocyaninato]indium(III), 3, was prepared from the metal-free analogue, 2-bromo-1,4-dibutoxy-8,11,15,18,22,25-hexakis(hexyl)phthalocyanine and indium(III) chloride in butanol. The latter phthalocyanine was synthesised by the mixed cyclotetramerisation of 3,6-dihexylphthalonitrile23 and 4-bromo-3,6-dibutoxyphthalonitrile22 under basic conditions.Complex 3, obtained as the racemate, crystallises without spontaneous resolution as fine, green needles in the triclinic system with P−1 symmetry. Two of the hexyl chains and one of the butoxy chains lie within the plane of the ring, those bonded to atoms C(24), C(27) and C(7) respectively, with an all-trans configuration. The butoxy chain at C(4) turns out of plane at O(4). The chain is disordered at its end, but one strand is all-trans. The chain bonded to atom C(14) turns out-of-plane at C(141), then lies all-trans on the same side as the InCl bond, whereas the chain bonded to atom C(34) turns out-of-plane at C(341), then twists with trans–cis–trans angles (in two, disordered arrangements) on the opposite side. The first two carbon atoms in the hexyl chain at C(17) are in-plane, then the chain meanders with trans–cis–cis torsion angles. The hexyl chain at C(37) turns out-of-plane at C(371), then lies all-trans. There are two molecules, related by a centre of symmetry, in the unit cell. The four benzenoid rings are bent away from the mean N4 plane; three deviate to the opposite side from the In and Cl atoms, the concave side, whereas the fourth tilts towards the same side as the In and Cl, the convex side, Fig. 5. The angles between the normals of opposing benzenoid rings are 6.2 and 16.2° respectively, illustrating that, although the Pc core is not planar, the distortion is only minimal. The indium atom is displaced by 0.710 Å from the central cavity of the molecule, a distance comparable to the displacement of the In atom in 1.
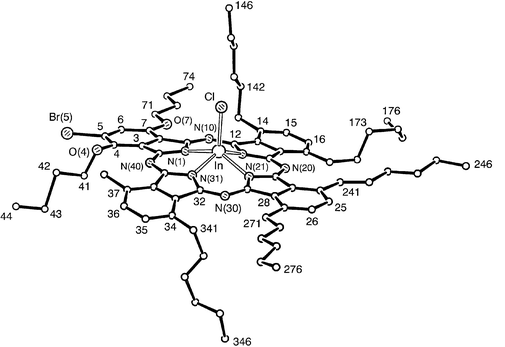 | ||
| Fig. 5 View of a molecule of 3, indicating the atom numbering scheme. Hydrogen atoms and the chain at C(37) have been omitted for clarity. | ||
All the Pc molecules in the crystal of 3 are parallel (by symmetry), in contrast to those of the other chloroindium derivative, 1, and are stacked parallel to the a axis with considerable offset of the In centres between adjacent planes. As with 1 there is significant overlap of nearest neighbour molecules within a concave–concave pair with interplanar distance 3.47 Å, Fig. 6. Both overlapping groups are formed about centres of symmetry. There is also weaker convex–convex overlap of benzenoid rings between molecules of adjacent pairs with 3.63 Å separation. A feature of 3 is that overlap involves benzenoid rings bearing alkyl chains rather than the monobromo-dibutoxy substituted ring. As was also observed for 1 and 2 the consequences of the overlap is that the molecules assemble into stacks. In the case of racemic 3 the two enantiomers alternate up the chain. As reported in the Experimental, and discussed below, there is some packing disorder. However, in the majority ordered component, represented in Fig. 6, all molecules of one enantiomer (labelled In, Cl) have the In–Cl bonds pointing up the page, and that of the other enantiomer [containing In′, Cl′, Br(5′)] all pointing down.
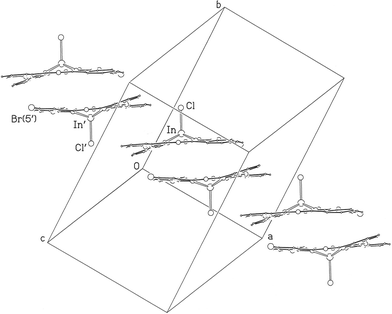 | ||
| Fig. 6 Stacking of molecules of 3 parallel to the a axis with alternation of the pair of enantiomers (ignoring packing disorder, see text). Alkyl chains have been omitted for clarity. | ||
The origin of the crystallographic disorder is shown within Fig. 7 and arises from alternative occupation sites of the bromine. The majority occupation (95%) is at Br(5) but there is 5% occupation at Br(36). In terms of molecular packing, this corresponds to a limited replacement of the expected enantiomer by the ‘wrong’ enantiomer in the ordered packing depicted in Fig. 6. Fig. 7 also provides a ‘plan view’ to illustrate molecular overlap of molecules of 3 and this is discussed below.
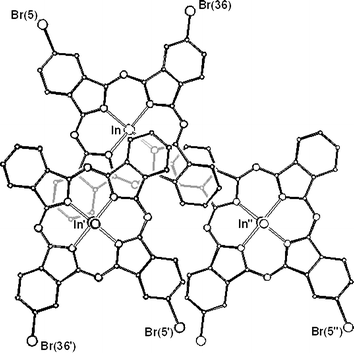 | ||
| Fig. 7 Upper left shows a view of a molecule of 3 containing the indium atom In and with centroid Nx. Disorder arises from alternative sites occupied by the bromine atom as shown. Majority (95%) occupation occurs at Br(5); minority occupation (5%) occurs at Br(36). The figure also shows the molecule overlapping with both its first nearest neighbour, with centroid Nx′ (molecule with In′, lower left) and second nearest neighbour, with centroid Nx″ (molecule containing In″, lower right). Alkyl chains have been omitted for clarity. | ||
Similarities and contrasts
The X-ray structural data of the series of three compounds investigated in this study show differences and similarities in molecular structure and packing. Some of these have been referred to in passing in the discussion above but they merit more in-depth consideration. Table 2 provides comparative data for key molecular parameters and shows that the N–In bond distances, and hence the displacement of the indium from the central mean plane of the four coordinating N atoms, increase in the order 1, 3 and 2. The phthalocyanine ligand is not significantly distorted from planarity in any of the compounds, though compound 3 is exceptional in that one of the benzenoid rings is displaced towards the convex side. As with other non-peripherally substituted phthalocyanines, not all of the alkyl chains can be accommodated simultaneously in the approximate ‘plane’ of the ring.The ‘local’ packing feature that is common to all three compounds is pairing of closest neighbour molecules. The four nitrogen atoms (N4) involved in coordination to the indium atom in a single molecule form the central plane; the N4 mean planes of the two molecules in a pair are parallel. For all three compounds, these paired molecules adopt concave–concave packing so the indium–ligand bonds are anti-parallel. The two molecules constituting a pair are offset, a displacement of one core from the other that allows for a π–π interaction involving a significant component of each of the two molecules of the pair. This packing feature can be seen in Fig. 2 (for compound 1) and more clearly in Fig. 7 (for molecules of 3).
The molecules on the left hand side of Fig. 7 represent a concave–concave pair, the molecule at top left being overlapped with its closest neighbour, lower left. We shall refer to the centroid of the N4 mean plane of the first molecule as Nx, that of the second as Nx′. The third molecule in Fig. 7, with centroid Nx″ (lower right), is a member of a second pair and overlaps to a lesser extent with the first but with convex-convex geometry.
Table 3 collects data for overlapping molecules for all three compounds. Data in the first row for each compound refer to the concave-concave pairs (c-c in the table) in terms of several spatial parameters that relate to the centroids Nx and Nx′ of the two molecules. The parameters are explained in the legend to Table 3 and also shown schematically in Fig. 8. The values for the normal (Norm.) between the parallel planes are closely similar for the two chloroindium derivatives 1 and 3, and are smaller than for 2. However, the Overlap distances, representing closest approach of the overlapping sections, are more closely similar. Thus the closest distance is 3.47 Å for 1 and 3 and 3.50 Å for 2. Values for Offset and α are also in similar ranges. These data point to a favoured overall type of geometry in the concave–concave pairs of all three compounds.
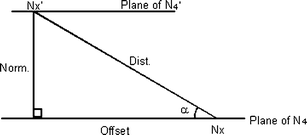 | ||
| Fig. 8 Schematic representation of the parameters defined in Table 3 that refer to the intramolecular geometry of a pair of molecules comprising a molecule with centroid Nx and its first nearest neighbour with centroid Nx′. The same parameters apply also to the relationship of the molecule to its second nearest neighbour (not shown) with centroid Nx″. | ||
| Dist. | Norm. | Offset | Angle, α | Overlap | Pairing | Nx′–Nx–Nx″ angle | |
|---|---|---|---|---|---|---|---|
| a Where: Nx is the centroid of the four central N atoms, Nx′ and Nx″ are the centroids of the symmetry-related N4 groups, Dist. is the Nx⋯Nx′ distance, in Å, Norm. is the perpendicular distance, in Å, between the parallel N4 planes, Offset is the ‘horizontal’ distance, i.e. distance in the N4 planes, in Å, between Nx and the projection of Nx′, Angle, α, is the angle whose sine is Norm./Dist., ‘Overlap’ is the distance, in Å, about a centre of symmetry, between overlapping sections. ‘Pairing’ about the centre of symmetry, viz. concave–concave (c–c) or convex–convex (v–v). Nx′–Nx–Nx″ angle is that subtended at Nx by its nearest neighbours, Nx′ and Nx″. | |||||||
| 1 | |||||||
| Nx⋯Nx′ | 6.875 | 3.717 | 5.784 | 32.73 | 3.47 | c–c | 83.0 |
| Nx⋯Nx″ | 10.862 | 3.936 | 10.124 | 21.25 | 3.61 | v–v | |
| 2 | |||||||
| Nx⋯Nx′ | 7.931 | 4.306 | 6.660 | 32.88 | 3.50 | c–c | 86.3 |
| Nx⋯Nx″ | 10.659 | 3.471 | 10.078 | 19.00 | 3.61 | v–v | |
| 3 | |||||||
| Nx⋯Nx′ | 7.203 | 3.708 | 6.175 | 30.98 | 3.47 | c–c | 88.8 |
| Nx⋯Nx″ | 10.611 | 3.550 | 10.000 | 19.55 | 3.63 | v–v | |
The second row of data for each compound refers to the relative geometry of one molecule of a concave–concave pair and its next nearest neighbour, a member of a second pair. As stated above these experience convex–convex interactions (v–v in Table 3) with a smaller area of overlap. The parameters in Table 3 again relate to the centroids, now Nx and Nx″. The Dist., Offset, α, and Overlap data are particularly consistent through the series of compounds. This similarity of the packing is also manifested in the Nx′–Nx–Nx″ angles for the closest neighbours along the column; for 1, 2 and 3, the angles are 83.0, 86.3 and 88.8°, respectively. The centroids thus form a zigzag pattern along the column in each crystal. It is interesting to note that, qualitatively, the extent of the overlap of a molecule with its first and second nearest neighbours is similar to that reported for one of the polymorphs of unsubstituted titanyl phthalocyanine.31
However, as pointed out earlier, despite the above similarities in the local packing in the series 1, 2 and 3 which give rise to the columnar packing, there is also a significant ‘non-local’ difference in the packing of 1 that is unique to this compound. Thus, unlike the molecules of 2 and 3, not all the N4 planes of 1 are parallel and adjacent columns form a type of herring-bone stack.
Comparisons of solid state spectral band envelopes
The electronic absorption band envelope of phthalocyanines in the solid state is known to be sensitive to molecular packing, and provides a starting point for the study of crystal structure/property relationships. The visible region absorptions exhibited by compounds 1 and 2 in the solution phase are closely similar with the Q-bands absorbing at λmax 726 and 728 nm for 1 and 2 respectively. Compound 3 exhibits a red shift due to the presence of the two butyloxy substituents and absorbs at 741 nm. A convenient procedure for measuring the solid state spectrum of some phthalocyanines is to study ‘burnished films’. These are formed by gently smearing crystals onto a glass substrate. This process was successfully applied to 1 and 3 but proved unsatisfactory for 2. Thus whereas the former pair crystallises as soft crystals, those of 2 are more robust and do not smear. It appears therefore that the axial 4-fluorophenyl ligand of 2 has provided a means of reducing fracture of the crystal. Interestingly the melting points of 1 (112 °C) and 3 (111 °C) are similar and lower than that of 2 (129 °C). An unexpected feature of the mp of 3 is that it is much lower than that of its metal-free precursor (>250 °C). The smeared crystal films of 1 and 3 show a red shift of the solution phase absorption band to 757 and 776 nm respectively. This is expected for nearest neighbour pairs of phthalocyanines that are offset in the solid state. The shift is marginally larger for 3 in terms of wavelength and transition energy and may in part be a manifestation of the different overall molecular packing features of 1 and 3 identified above.All three compounds were spin-coated from THF solutions to form even, transparent films on glass slides. Q-Band absorption data are presented in Table 4. Earlier work from these laboratories has indicated strongly that the molecular packing of phthalocyanine derivatives in a spin coated film can mimic that in the solid state.32 This is indicated for example by the similarity in the visible region absorption pattern shown by a spin coated film and a burnished film of the same compound. Indeed, the two types of film of 1 gave very similar band envelopes with identical λmax values. However, those of 3 were quite different with the spin coated film showing a smaller red shift of the Q-band. This points to a different molecular organisation in the two films. The overall absorption band shape obtained for the spin coated film of compound 2 was similar to those of 1 and 3 but the spectral shift of the Q-band is more similar to that observed for the film of 1.
| Pc | λ max/nm (THF) | λ max/nm (film a)a | λ max/nm (film b)b | Q-Band shift/nmc | ν film − νTHF/cm−1c |
|---|---|---|---|---|---|
| a Film a; smeared films. b Film b; spin-coated films. c Data in parentheses correspond to the smeared films. Other data correspond to the spin-coated films. | |||||
| 1 | 726 | 757 | 757 | +31 (+31) | −564 (−564) |
| 2 | 728 | — | 752 | +24 | −438 |
| 3 | 741 | 776 | 750 | +35 (+9) | −637 (−164) |
Comparative photoconductivity measurements on the film formulations are in hand.
Conclusion
The present study of three substituted indium phthalocyanines, two chloroindium derivatives, 1 and 3, and a 4-fluorophenylindium derivative, 2, is the first of its kind and provides insight into factors that control molecular packing within the series. Despite varying the axial ligand from a chloro group to the more space demanding 4-fluorophenyl group, and introducing somewhat different substituents onto one of the benzenoid rings of one of the chloroindium phthalocyanines, the three compounds form similar types of columnar stacks. Key components of the stack structure are concave–concave pairs of molecules. The molecules are offset but allow for a significant degree of overlap. Components of these pairs then participate in a lower area convex–convex overlap with adjacent pairs to form an offset column of pairs. One of the compounds, 1, however shows a different type of global packing to the other two. For this compound, the molecules of one column are tilted with respect to those in an adjacent column and thus exhibit a type of herring-bone arrangement; the molecules of the other two compounds pack with their central planes parallel.All three compounds show red shifted Q-band absorptions in the solid state relative to those in the solution phase. Their ease of formulation as spin coated films will be exploited in photoconductivity trials.
Acknowledgements
We thank the EPSRC and the University of East Anglia for studentship funding for PMB and AA and the EPSRC mass spectrometry service at Swansea for mass spectra.References
- R. D. Gould, Coord. Chem. Rev., 1996, 156, 237–274 CrossRef CAS.
- M. Bouvet, in The Porphyrin Handbook Vol 19. Applications of Phthalocyanines, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, pp. 37–103 Search PubMed.
- D. Wöhrle, L. Kreienhoop and D. Schlettwein, in Phthalocyanines: Properties and Applications Vol 4, ed. C. C. Leznoff and A. B. P. Lever, VCH, New York, 1996, pp. 79–181 Search PubMed.
- S. R. Flom, in The Porphyrin Handbook Vol 19. Applications of Phthalocyanines, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, pp. 179–190 Search PubMed.
- P. Erk and H. Hengelsberg, in The Porphyrin Handbook Vol 19. Applications of Phthalocyanines, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, pp. 105–149 Search PubMed.
- P. Gregory, J. Porphyrins Phthalocyanines, 2000, 4, 432–437 CrossRef CAS.
- D. Birkett, Chem. Ind., 2000, 178–181 CAS.
- H. Eichhorn, J. Porphyrins Phthalocyanines, 2000, 4, 88–102 CrossRef CAS.
- M. J. Cook, in Spectroscopy of New Materials: Advances in Spectroscopy Vol 22, ed. R. J. H. Clark and R. E. Hester, Wiley, Chichester, 1993, pp. 87–150 Search PubMed.
- Xerox Corporation, US Patent 3,816,118, June 11, 1974.
- Y. Oda, T. Homma and Y. Fujimaki, Electrophotography, 1990, 29, 250–258 Search PubMed; K. Yamasaki, O. Okada, K. Imani, K. Oka, M. Kotani and H. Yamada, J. Phys. Chem. B, 1997, 101, 13–19 CrossRef CAS.
- Fuji Xerox Co. Ltd, US Patent 5,643,703, July 1, 1997.
- M. K. Engel, in The Porphyrin Handbook Vol. 20. Phthalocyanines: Structural Characterisation, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003 Search PubMed.
- N. B. McKeown, in The Porphyrin Handbook Vol 15. Applications of Phthalocyanines, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, pp. 61–124 Search PubMed; M. S. Rodríguez-Morgade, G. de la Torre and T. Torres, in The Porphyrin Handbook Vol 15. Applications of Phthalocyanines, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, pp. 125–262 Search PubMed.
- A. N. Cammidge and R. J. Bushby, in Handbook of Liquid Crystals: Vol 2B. Low molecular weight liquid crystals II, ed. D. Demus, J. Goodby, G. W. Gray, H.-W. Spiess and V. Vill, Wiley-VCH, Weinheim, 1998, pp. 693–748 Search PubMed.
- M. J. Cook and I. Chambrier, in The Porphyrin Handbook Vol 17. Applications of Phthalocyanines, ed. K. M. Kadish, K. M. Smith and R. Guilard, Academic Press, San Diego, 2003, pp. 37–127 Search PubMed.
- I. Chambrier, M. J. Cook, M. Helliwell and A. K. Powell, J. Chem. Soc., Chem. Commun., 1992, 444–445 RSC; I. Chambrier, M. J. Cook and P. T. Wood, J. Chem. Soc., Chem. Commun., 2000, 2133–2134 RSC; M. Helliwell, A. Deacon, K. J. Moon, A. K. Powell and M. J. Cook, Acta Crystallogr., Sect. B, 1997, 53, 231–240 CrossRef; M. J. Chen, J. W. Rathke and J. C. Huffman, Organometallics, 1993, 12, 4673–4677 CrossRef CAS.
- M. J. Cook, J. McMurdo and A. K. Powell, J. Chem. Soc., Chem. Commun., 1993, 903–904 RSC; J. Cai, J. Wang, J. Huang and N. Chen, Chin. Sci. Bull., 2002, 47, 644–646 Search PubMed; J. Wang, J. Huang, J. Cai and N. Chen, Chin. J. Struct. Chem., 2002, 21, 617–620 Search PubMed.
- P. M. Burnham, M. J. Cook, L. A. Gerrard, M. J. Heeney and D. L. Hughes, J. Chem. Soc., Chem. Commun., 2003, 2064–2065 RSC.
- N. Kobayashi, T. Fukuda, K. Ueno and H. Ogino, J. Am. Chem. Soc., 2001, 123, 10740–10741 CrossRef CAS.
- A. Auger, W. J. Blau, P. M. Burnham, I. Chambrier, M. J. Cook, B. Isare, F. Nekelson and S. M. O'Flaherty, J. Mater. Chem., 2003, 13, 1042–1047 RSC.
- M. J. Cook and M. J. Heeney, Chem. Eur. J., 2000, 6, 3958–3967 CrossRef CAS.
- N. B. McKeown, I. Chambrier and M. J. Cook, J. Chem. Soc., Perkin Trans. I, 1990, 1169–1177 RSC.
- Z. Otwinowski and W. Minor, Processing of X-ray diffraction data collected in oscillation mode, Methods in Enzymology, Vol. 276, Macromolecular Crystallography, Part A, ed. C. W. Carter, Jr. and R. M. Sweet, Academic Press, San Diego, 1997, pp. 307–326 Search PubMed.
- G. M. Sheldrick, SHELX-97—Programs for crystal structure determination (SHELXS) and refinement (SHELXL), University of Göttingen, Germany, 1997.
- International Tables for X-ray Crystallography, Vol. C, Kluwer Academic Publishers, Dordrecht 1992, pp. 500, 219 and 193 Search PubMed.
- S. N. Anderson, R. L. Richards and D. L. Hughes, J. Chem. Soc., Dalton Trans., 1986, 245 RSC.
- W. E. Bennett, D. E. Broberg and N. C. Baenziger, Inorg. Chem., 1973, 12, 930–937 CrossRef CAS.
- J. Janczak and R. Kubiak, Inorg. Chim. Acta, 1999, 288, 174–180 CrossRef CAS.
- A. Schmidt, L. K. Chau, A. Back and N. R. Armstrong, in Phthalocyanines: Properties and Applications Vol 4, ed. C. C. Leznoff and A. B. P. Lever, VCH, New York, 1996, pp. 307–342 Search PubMed.
- W. Hiller, J. Strahle, W. Kobel and M. Hanack, Z. Kristallogr., 1982, 159, 173–183 CAS.
- M. J. Cook, D. A. Mayes and R. H. Poynter, J. Mater. Chem., 1995, 5, 2233–2238 RSC.
| This journal is © The Royal Society of Chemistry 2005 |