Magnetic properties of ultrafine cobalt ferrite particles synthesized by hydrolysis in a polyol medium†
Souad
Ammar
a,
Arnaud
Helfen
a,
Noureddine
Jouini
*a,
Fernand
Fiévet
a,
Izio
Rosenman
b,
Françoise
Villain
c,
Philippe
Molinié
d and
Michel
Danot
d
aLaboratoire de Chimie des Matériaux
Divisés et Catalyse, Université Paris 7-Denis
Diderot, 2 Place Jussieu, 75251, Paris, France. E-mail: jouini@ccr.jussieu.fr
bGroupe de Physique du Solide, Université Paris 7-Denis Diderot, 2 Place Jussieu, 75251, Paris, France
cLaboratoire de Chimie Inorganique et
Matériaux Moléculaires, Université Pierre
et Marie Curie, 4 Place Jussieu, 75251, Paris, France
dInstitut Jean Rouxel, 2 Chemin de la Houssinière, 44072, Nantes, France
First published on 12th October 2000
Abstract
Fine CoFe2O4 powders with monodisperse,
almost equi-axial nanometer-sized particles were synthesised in a
polyol medium by forced hydrolysis of ionic Co(II) and Fe(III)
salts at 160![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C. K(Co) XANES and 57Fe Mössbauer
spectroscopy show that the structure of this ferrite is slightly deviated
from an inverse spinel structure: 16% of cobalt atoms are in tetrahedral sites.
The particles are superparamagnetic above 300 K and ferrimagnetic below
this blocking temperature with, at low temperature, strong coercivity, a saturation
magnetisation value close to the bulk value and high reduced remanence. The
saturation magnetisation measured at 5 K is clearly enhanced with respect
to CoFe2O4 nanometer-sized particles previously
prepared by other methods. These magnetic characteristics suggest that these
particles have a high crystallinity which may result from this novel synthesis
route.
°C. K(Co) XANES and 57Fe Mössbauer
spectroscopy show that the structure of this ferrite is slightly deviated
from an inverse spinel structure: 16% of cobalt atoms are in tetrahedral sites.
The particles are superparamagnetic above 300 K and ferrimagnetic below
this blocking temperature with, at low temperature, strong coercivity, a saturation
magnetisation value close to the bulk value and high reduced remanence. The
saturation magnetisation measured at 5 K is clearly enhanced with respect
to CoFe2O4 nanometer-sized particles previously
prepared by other methods. These magnetic characteristics suggest that these
particles have a high crystallinity which may result from this novel synthesis
route.
Introduction
There is increasing interest in magnetic ferrite nanoparticles because of their broad applications in several technological fields including ferrofluids,1 magnetic drug delivery2 and magnetic high-density information storage.3 To fulfil the requirements for use in high-density recording media the particles must have not only suitable magnetic properties (large saturation magnetisation, remanence, moderate coercivity, high blocking temperature) but also reduced size and uniform shape.Among other materials, fine CoFe2O4 ferrite has received renewed attention for its potential use for high-density recording media. This is due to the remarkable properties observed for bulk CoFe2O4: strong anisotropy, high saturation magnetisation and coercivity along with good mechanical hardness and chemical stability.4
A variety of methods have been developed to elaborate CoFe2O4 ultrafine particles: sol–gel method,5 micro-emulsion with oil in water micelles6–8 or reverse micelles,9 aqueous co-precipitation and calcination,10–13 combustion.14 Despite this great number of routes, preparing CoFe2O4 particles suitable for high-density recording is still a challenge. Indeed, the nanomaterials often show a superparamagnetic behaviour at RT and a very low saturation magnetisation compared to the bulk value.
This paper reports on a novel route for the preparation of spinel CoFe2O4
nanoparticles with significantly improved magnetic properties in comparison
with the other synthesis methods. This route involves hydrolysis and inorganic
polymerisation carried out on Co2+ and Fe3+ salts dissolved
in a polyol medium. The polyol acts as a solvent for the precursor salts because
of its high relative permittivity (ε = 32 for 1,2-propanediol),
and allows one to carry out hydrolysis reactions under atmospheric pressure
in a large temperature range up to the boiling point of the polyol (189°C
for 1,2-propanediol). Moreover, polyols appear as crystal growth media
of particular interest for the synthesis of oxide fine particles.15,16
The magnetic characteristics of the particles prepared via this novel route will be discussed and tentatively related to the preparation process with respect to the other preparative routes reported in the literature.
Experimental
Synthesis
The precursor salts, FeCl3 and Co(CH3COO)2·4H2O (Prolabo) in stoichiometric ratio (2 ∶ 1) were added to a given volume (250 mL) of 1,2-propanediol. The total metal concentration was 0.3 mol L−1. Two main parameters were found to play a crucial role: the hydrolysis ratio and the amount of acetate, defined by the water and the acetate ion to metal molar ratios, respectively. CoFe2O4 nanoparticles with a 5.5 nm average diameter were obtained when these two factors were fixed to 9 and 3, respectively, by adding water and sodium acetate. The mixture was then heated to its boiling point (160°C)
with a heating rate of 6°C min−1. The
ferrite phase is formed via a chemical process involving the formation
of an intermediate phase. Accordingly, the mixture had to be refluxed for
at least 5 hours to obtain the final product as a pure solid phase.
After cooling to room temperature, the particles were separated from the supernatant
by centrifugation, washed with ethylene glycol and acetone and then dried
in air at 50°C.
Characterisation
X-Ray diffraction (XRD) patterns were recorded in the range 10–100° (2θ) with a scan step of 0.02° (2θ) for 50 s on a Siemens D5000 diffractometer (Fe Kα radiation). The crystallite size was calculated from line broadening analysis using the Scherrer formula.17 The cell parameter was obtained from XRD data by using the U-fit program.Particle morphology and chemical analysis of metal ions were determined by Transmission Electron Microscopy (TEM) images using a JEOL-100 CX II microscope equipped with an energy dispersive spectrometer (EDX) (electron probe diameter: 200 nm). The mean diameter and standard deviation were inferred from image analysis of ca. 400 particles (Microvision software). Elementary organic analysis of C and H was performed at the Analysis Centre of UPMC University.
Infrared (IR) spectroscopy was conducted on a FTIR Perkin Elmer 1750 apparatus (transmittance mode with at least 20 acquisitions). Spectra were recorded between 4000 and 500 cm−1 with a resolution of 4 cm−1 on pure pelleted powder.
X-Ray absorption near edge spectroscopy (XANES) measurements were performed at LURE, the French synchrotron facility (Orsay), on the XAS 13 beam line of the DCI storage ring using a Si(331) channel cut monochromator. XANES spectra were recorded at the Co K edge (7708.9 eV) in transmission mode using air filled ionisation chambers. The energy calibration was checked by recording simultaneously the spectra of cobalt foil in a third ionisation chamber. Each spectrum was acquired with a 0.3 eV step size and normalised at the middle of the first extended X-ray absorption fine structure (EXAFS) oscillation. The samples were ground and homogeneously dispersed in cellulose pellets enclosed in Kapton.
The 57Fe Mössbauer spectra were recorded using a 57Co/Rh γ-ray source mounted on an electromagnetic drive and using a triangular velocity form. Spectra were obtained in zero magnetic field at 78 K. The Mössbauer spectra were analysed by least-squares fitting to a Lorentzian function. The isomer shifts (IS) were referred to α-Fe at 300 K. The sample (area: 3 cm2) was prepared by dispersion of the compound (100–140 mg) in a specific resin.
Magnetic susceptibility and hysteresis loop measurements were obtained with a super-conducting quantum interference device (SQUID) magnetometer. The DC magnetic susceptibility χ(T) was measured in zero-field cooling (ZFC) and field cooling (FC) modes in the temperature range 4.5–300 K and with an applied field of 100 Oe. The magnetisation curves M(H) were obtained after cooling the sample to the measurement temperature in zero field and then increasing the field from 0 to 50 kOe. In all cases, the powders were moderately compacted into the sampling tubes.
Results
Phase identification, chemical analysis and morphological characteristics
The XRD pattern (Fig. 1) reveals the formation of CoFe2O4 as a single phase. The cell parameter inferred (a = 8.390(2) Å) is close to that of bulk cobalt ferrite (a = 8.395(5) Å).18 EDX analysis shows that the Co/Fe ratio is very close to 0.5 in good agreement with the formula CoFe2O4.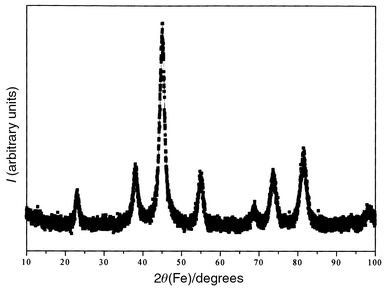 | ||
| Fig. 1 XRD pattern of ultrafine cobalt ferrite powder obtained in 1,2-propanediol. | ||
As already mentioned, the precipitation of the CoFe2O4 oxide occurred through the formation of an intermediate solid phase. Chemical and XRD analyses show that this phase is an iron-rich (FeIII) phase with a cobalt content less than 5 at.% and belongs to the lepidocrocite γ-FeO(OH) structural type. Characterisation of this phase is presently in progress. The ferrite formation process involving an oxo-hydroxide or an hydroxide as an intermediate phase has already been investigated.19–22
The particles are nanosized and almost equiaxial with a mean diameter of 5.5 nm (standard deviation less than 10%) (Fig. 2). Particles can be considered as single crystals since this diameter agrees fairly well with the average crystallite size (7.8 nm) inferred from X-ray line broadening analysis. Indeed, the crystallite size determined from the Scherrer formula is often found to be larger than the diameter measured by TEM. The particle diameter (5.9 nm), inferred from the BET surface area (238 m2 g−1) and the powder density measured by helium pycnometry (4.3 g cm−3), is very consistent with the TEM value confirming the non-porous character of these particles. The density appears lower than the tabulated value for bulk CoFe2O4 (5.3 g cm−3). This is frequently observed for different inorganic materials prepared in polyol media (metal, oxide, hydroxide)23,24 and may be due to the adsorption of organic species. Indeed, a carbon content of 2.5 wt.% is found typically. The main adsorbed organic species are acetate ion and 1,2-propanediol as identified by infrared spectroscopy (Fig. 3). These species appear to be strongly bonded (chemisorbed) to the oxide surface.25,26
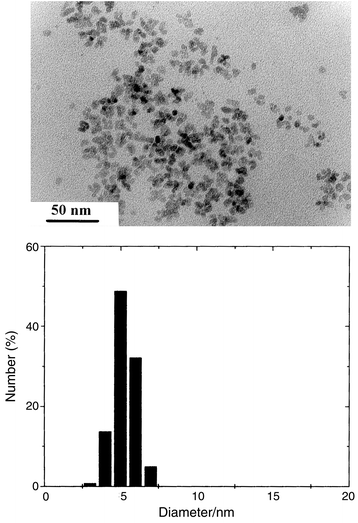 | ||
| Fig. 2 (Upper panel) TEM image of CoFe2O4 particles; (lower panel) histogram of size distribution. | ||
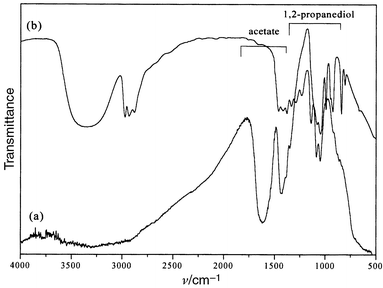 | ||
| Fig. 3 IR spectra of (a) CoFe2O4 powder obtained in 1,2-propanediol; (b) liquid 1,2-propanediol. | ||
X-Ray absorption spectroscopy (XANES)
The Co K-edge XANES spectra of CoFe2O4 powder and three reference compounds, Zn0.5Co0.5O,24 Co(NO3)2·6H2O27 and Co3O4 are shown in Fig. 4. In Zn0.5Co0.5O, CoII is in the tetrahedral sites of a würtzite-like structure. In Co(NO3)2·6H2O, CoII is octahedrally coordinated in a hexa-aquo complex. In Co3O4, CoII is in the tetrahedral sites and CoIII in the octahedral ones of a spinel-like structure.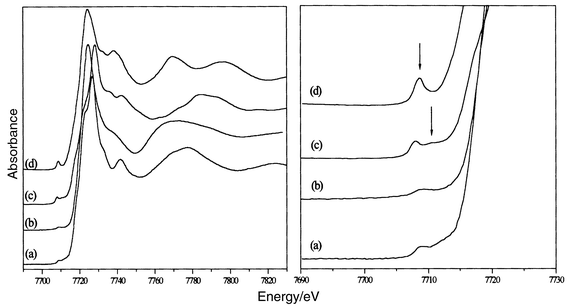 | ||
| Fig. 4 XANES spectra of (a) CoFe2O4; (b) Co(NO3)2·6H2O; (c) Co3O4 and (d) Zn0.5Co0.5O (left); magnified view of the pre-edge region for the same compounds (right). | ||
The energy positions of the main structures observed for CoFe2O4 are in good agreement with the values reported for CoII oxides.28 In particular, all the spectra show a low-intensity pre-edge feature (marked in Fig. 4) which corresponds to 1s → 3d electronic transitions. Its energy position and intensity are related to the oxidation state and the local symmetry, respectively, of Co.29 By comparison to reference compounds, the pre-edge in CoFe2O4 allows us to confirm the oxidation state of cobalt ion as +II and their partial location in tetrahedral sites (Table 1).
| Sample | Energy position/eV | Coordination |
|---|---|---|
| CoFe2O4 (polyol) | 7709.1 ± 0.3 | Co2+: Oh and Td |
| Co3O4 | 7708.1 ± 0.3 | (1/3)Co2+: Td |
| 7710.6 ± 0.3 | (2/3)Co3+: Oh | |
| Co(NO3)2·6H2O | 7709.0 ± 0.3 | Co2+: Oh |
| Zn0.5Co0.5O | 7708.6 ± 0.3 | Co2+: Td |
Mössbauer absorption spectroscopy
The Mössbauer spectrum recorded at 78 K (Fig. 5) is very similar to that observed at lower temperature (4.2 K) for CoFe2O4 nanoparticles with 5 nm average size:6b the spectrum can be fitted as the overlapping of two six-lines patterns with hyperfine fields H = 506 and 535 kOe, corresponding to Fe3+ ions in A (tetrahedral) and B (octahedral) spinel sites respectively. Refinements have been performed according to the model used by Moumen et al.6b with, for the A site, a unique line width (0.22 mm s−1) for the six-line pattern and, for the B site, different widths for the three pairs of lines (0.23, 0.27, and 0.31 mm s−1 with increasing splitting). The Mössbauer parameters for the A site are: isomer shift, IS = 0.38 mm s−1; quadrupole splitting, QS = 0.0 mm s−1; hyperfine magnetic field, H = 506 kOe and relative intensity, Irel = 42%. The corresponding parameters for the B site are IS = 0.48 mm s−1, QS = 0.0 mm s−1, H = 535 kOe and Irel = 58%. Assuming equal recoilless fractions (f) for A and B sites, the A/B population ratio is found to be 0.72, in fair agreement with the value (0.67) obtained by Moumen et al.6b for their 5 nm particles. The cation distribution estimated from our spectrum corresponds to the formula (Co0.16Fe0.84)[Co0.84Fe1.16]O4 where the round and square brackets represent A and B spinel sites respectively.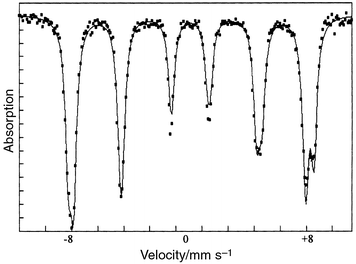 | ||
| Fig. 5 Mössbauer spectrum at 78 K of CoFe2O4 powder. | ||
It is interesting to note that our 78 K hyperfine field values (506 and 535 kOe) are very similar to those reported by Moumen et al.6b (510 and 533 kOe) for particles of equivalent average size. However, in their case, these values are reached only at very low temperature (4.2 K). At 78 K, a noticeable decrease of the thermal field was observed by these authors. This indicates that, for similar particle size, our material is better organised.
Magnetic measurements
A net irreversibility between the curves measured in FC and ZFC modes is observed on DC susceptibility measurements. The ZFC χ(T) curve shows a maximum at 300 K and decreases rapidly to zero at lower temperature, while the FC χ(T) curve increases slightly as the temperature decreases in the temperature range 300–4.5 K. The nanoparticles can therefore be considered as magnetic single domains with a blocking temperature TB around 300 K. Measurements under 200 Oe up to 400 K confirmed this result (Fig. 6). Below this temperature the nanoparticles exhibit ferrimagnetic behaviour with remanence and coercivity which increase when the temperature decreases (Fig. 7). At 5 K the observed values of the squareness ratio and coercivity are 0.67 and 9.3 kOe respectively. Above 300 K, the hysteresis feature vanishes and the nanoparticles have a superparamagnetic behaviour. At 5 K, the saturation magnetisation is very high, 85.1 emu g−1, decreasing to 65 emu g−1 at 300 K owing to thermal fluctuations.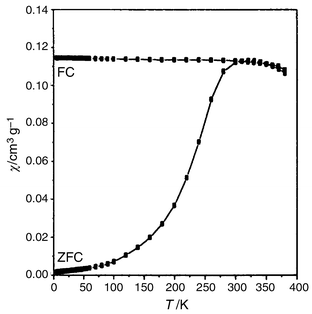 | ||
| Fig. 6 DC susceptibility χ(T) measured in FC and ZFC modes for CoFe2O4 powder from 4.5 to 400 K, under an applied field of 200 Oe. | ||
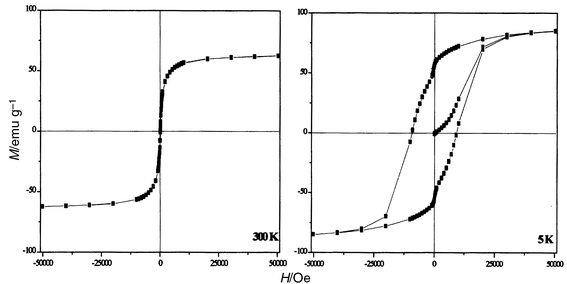 | ||
| Fig. 7 Isothermal hysteresis loops of CoFe2O4 powder; T = 300 K (left) and T = 5 K (right). | ||
It is well known that the blocking temperature and coercivity of nanomaterials depend on several factors: (i) intrinsic factors, which mainly include magnetocrystalline, surface and shape anisotropy and (ii) extrinsic factors, generally related to interactions between particles. This last factor is believed to have a significant influence. In order to appraise this influence, the interparticle interactions were tentatively changed on one hand by dispersing the particles in sugar (≈13 wt.%) and on the other hand by washing the sample in boiling water (residual carbon amount 1.5 wt.%). Magnetic measurements conducted on such samples show no significant changes in blocking temperature and saturation magnetisation. The main change observed concerns the rapid variation of the magnetisation for low magnetic field values when the particles are not sufficiently dispersed. This phenomenon is attributed to interactions between particles being removed by diluting the particles without any significant change in the saturation magnetisation (Fig. 8). This shows that the interactions between particles have been probably lowered by dispersion in sugar. We also conducted a measurement of the temperature dependence of the magnetisation decay. The sample is cooled from RT to 5 K under a magnetic field of 100 Oe. Before the magnetisation is measured, the field is turned off. The remanence decreases when the temperature increases and approaches zero at the blocking temperature (Fig. 9).
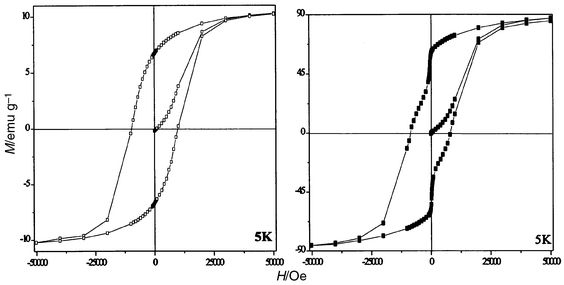 | ||
| Fig. 8 Hysteresis loops measured at 5 K for CoFe2O4 particles: (a) dispersed in sugar, magnetisation data are given per gram of the mixture, and (b) washed in boiling water. | ||
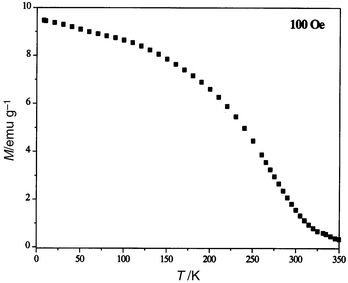 | ||
| Fig. 9 Temperature dependence of the magnetisation decay for 5 nm CoFe2O4 nanoparticles cooled under a magnetic field of 100 Oe. | ||
Altogether these results show that interparticle interactions, although they are certainly present, can be assumed to be rather small and so do not affect significantly the superparamagnetic behaviour of the particles. This may be due to the adsorbed organic species which act as surfactants and help prevent interparticle interactions. A more thorough study of this influence is currently in progress.
Discussion
Table 2 enables us to compare the main magnetic characteristics of the studied nanoparticles with those of similar ones prepared by other synthesis methods.| Ref. | Synthesis method | T B/K | M sat/emu g−1 | H c/kOe | Reduced remanence |
|---|---|---|---|---|---|
| a Magnetic measurements were performed on ferrofluid system. | |||||
| This work | Forced hydrolysis in polyol at 433 K | 300 K (100 Oe) | 65 (300 K) | 9.3 (5 K) | 0.67 |
| 85.1 (5 K) | |||||
| 6 a | Micro-emulsion (oil-in-water micelles at 300 K) | 180 (100 Oe) | 45 (20 K) | 8.8 (20 K) | 0.74 (20 K) |
| 35 (300 K) | |||||
| 9 | Micro-emulsion (reverse micelles at 300 K) | 60 (100 Oe) | 6 (2 K) | 6 (2 K) | 0.37 (2 K) |
| 11 | Calcination at 600 K | 250 K (—) | 13 (300 K) | 13.2 (5 K) | 0.36 |
| 15 (5 K) | |||||
| 12 | Calcination at 600 K | 230–240 (100 Oe) | 13 (5 K) | 14.5 (5 K) | 0.38 |
| 14 | Instantaneous combustion followed by calcination at 673 K | — | <10 (300 K) | 0.05 (300 K) | — |
The coercivity and squareness ratio at 5 K are very close to those observed for nanoparticles obtained by microemulsion (oil in water).6 The squareness value, higher than 0.5, is consistent with randomly oriented equiaxial particles with cubic magnetocrystalline anisotropy.30–32 The particles prepared by calcination show significantly different characteristics: high coercivity and low squareness ratio (0.36) tentatively explained by a uniaxial anisotropy contribution generated by internal strains.10
Examination of Table 2 shows that the nanoparticles prepared here have the highest blocking temperature, close to RT. This value seems to correspond to an intrinsic property of the particles since the interparticle interactions appear to be moderately weak, as previously shown. Thus it may indicate a strong anisotropy effect taking into account the magnetocrystalline and surface anisotropy. The corresponding constant (K) can be inferred from the blocking temperature assuming a random distribution of single domain particles by the relation:33
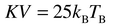 | (1) |
The most interesting result concerns the saturation magnetisation. The measured value (85.1 emu g−1) at 5 K is surprisingly close to that of the bulk (80–93 emu g−1)4 and remains very high (65 emu g−1) at RT. These values are significantly higher than those of CoFe2O4 nanoparticles obtained by either thermal decomposition (calcination11–12 or combustion14) or chimie douce methods such as the micro-emulsion route at RT.6,8,9
This enhancement can partly be explained by the cation distribution. Mössbauer and XANES studies established the presence of cobalt ions in tetrahedral sites leading to an increase of the magnetisation according to the collinear ferrimagnetic Néel model. It is well known that cation distribution in ferrites depends on synthesis factors such as temperature, pressure and nominal composition. The polyol medium appears to be a particular crystal growth medium which favours such an unusual cation distribution.
Nevertheless, the cation distribution alone is unlikely to account for the enhancement of the saturation magnetisation. Indeed, such an interesting increase should be emphasised, because usually it is observed that the saturation magnetisation decreases with particle size. This is believed to be due to spin canting which results from a magnetic ordered core surrounded by a disordered surface layer.34–37 Decreasing the particle size leads to an increase of the relative amount of magnetic cations in the surface layer and favours the spin canting phenomenon. Accordingly the saturation magnetisation of CoFe2O4 nanoparticles reported in the literature never exceeded 50 emu g−1 at 5 K. The high saturation magnetisation observed for nanometer sized CoFe2O4 particles prepared in polyol suggests that they are almost perfectly magnetically ordered single-domains without a significant dead magnetic layer.
Such a result emphasises the crucial influence of the synthesis method on the physical properties of such materials. As previously noticed, high density storage needs particles with uniform size along with improved magnetic characteristics. Examination of Table 2 shows that the synthesis methods reported so far failed to fulfil that demand. Thermal decomposition methods (calcination, combustion) carried out on co-precipitated solid precursors do no provide any control over the morphological characteristics of the particles. Furthermore, decomposition occurred via a release of gas species giving particles with internal pores. This leads to spin canting in the volume of the particle and thus to low saturation magnetisation (less than 15 emu g−1 at 5 K). In contrast, the micro-emulsion route is a powerful way to obtain particles with calibrated shape and size. However, this route proceeds at RT and leads to incompletely ordered materials showing concomitantly a certain disorder in the distribution of the magnetic cations. This leads to saturation magnetisation values lying in a wide range: 6 (2 K)–45 (20 K) emu g−1.
This magnetic disorder may occur not only in the surface, as generally supposed, but also in the whole volume of the particle and leads to the deterioration of magnetic characteristics. Such spin canting in the core has recently been proposed to explain the magnetic properties of nanometer-sized γ-Fe2O3 particles prepared by co-precipitation followed by calcination.38
The new method described here represents significant progress toward the
synthesis of nanoparticles which may be used for magnetic recording. On one
hand, like the micro-emulsion method, this route allows control of the
nucleation and growth steps of the particles. Their nanometer scale and their
narrow size distribution can be explained by a fast nucleation along with
a growth step controlled by the rate of the hydrolysis reaction and/or the
diffusion of the dissolved species toward the surface of the particles. The
adsorbed organic species protect the primary particles against coalescence
and/or aggregation. On the other hand, this crystal growth occurs under thermal
conditions close to hydrothermal ones (160°C under reflux). This
favours good crystallinity and accordingly improves the magnetic order resulting
in a high saturation magnetisation. This high magnetisation shows that the
magnetic disorder is confined in a very thin surface layer. However, this
thin layer, where spin canting due to breaking of the magnetic exchange pathways
(the surface spin having nearest neighbours on one side, none on the other
side even if the first co-ordination shell is completed with adsorbed
species) may exist, appears to be sufficient to enhance anisotropy leading
to a high blocking temperature.
Conclusion
Magnetic properties of finely divided materials are strongly influenced by the shape and size of the particles, their degree of agglomeration and by volume and surface defects. Therefore these properties depend to a large extent on the synthesis process. Among various methods of fabrication of CoFe2O4 nanoparticles, the forced hydrolysis of metal salts in 1,2-propanediol appears as a new and attractive chimie douce route which provides monodisperse particles showing a surprisingly good crystallinity and interesting magnetic characteristics for high density storage applications. They are superparamagnetic above a blocking temperature around 300 K, and exhibit a saturation magnetisation at low temperature close to the bulk value, despite their nanometer size. This high blocking temperature together with high reduced remanence and coercivity measured at low temperature are consistent with a strong magnetic anisotropy.A more exhaustive study of the magnetic properties of this material (interparticle interactions, behaviour at high temperature…) along with High Resolution Electronic Microscopy (HREM) characterisation is currently in progress.
Acknowledgements
We wish to thank P. Gredin for XRD measurements and P. Beaunier for TEM images and EDX analysis. We are grateful to Dr. H. Pascard for fruitful discussions.References
- K. Raj, R. Moskowitz and R. Casciari, J. Magn. Magn. Mater., 1995, 149, 174 CrossRef CAS.
- Scientific and Clinical Applications of Magnetic Carriers, ed. U. Häfelli, W. Schütt, J. Teller and M. Zborowski, Plenum, New York, 1997. Search PubMed.
- M. H. Kryder, MRS Bull., 1996, 21(9), 17 Search PubMed.
- R. Valenzuela, Magnetic ceramics, Cambridge University Press, Cambridge, 1984, p. 212. Search PubMed.
- (a) F. X. Cheng, Z. Y. Peng, C. S. Liao, Z. G. Xu, S. Gao and C. H. Yan, Solid State Commun., 1996, 107, 471 CrossRef CAS; (b) J. G. Lee, J. Y. Park and C. S. Kim, J. Mater. Sci., 1998, 33, 3965 CrossRef CAS.
- (a) N. Moumen, P. Veillet and M. P. Pileni, J. Magn. Magn. Mater., 1995, 149, 67 CrossRef CAS; (b) N. Moumen, P. Bonville and M. P. Pileni, J. Phys. Chem., 1996, 100, 14410 CrossRef CAS; (c) M. P. Pileni, N. Moumen, J. F. Hochepied, P. Bonville and P. Veillet, J. Phys. IV (Fr.), 1997, 7(Supp. C1), 505 Search PubMed; (d) N. Moumen and M. P. Pileni, Chem. Mater., 1996, 8, 1128 CrossRef CAS.
- A. T. Ngo, P. Bonville and M. P. Pileni, Eur. Phys. J. B, 1999, 9, 583 CrossRef CAS.
- V. Pillai and D. O. Shah, J. Magn. Magn. Mater., 1996, 163, 243 CrossRef CAS.
- C. T. Seip, E. E. Carpenter and C. J. O'Connor, IEEE Trans. Magn., 1998, 34(4), 1111 CrossRef CAS.
- K. J. Davis, S. Wells, R. V. Upadhayy, S. W. Charle, K. O. O'Grady, M. El Hilo, T. Meaz and S. Morup, J. Magn. Magn. Mater., 1995, 149, 14 CrossRef.
- M. Grigorova, H. J. Blythe, V. Blaskov, V. Rusanov, V. Petkov, V. Masheva, D. Nihtianova, L. I. M. Martinez, J. S. Munoz and M. Mikhov, J. Magn. Magn. Mater., 1998, 193(1–2), 163 CrossRef CAS.
- V. Blaskov, V. Petkov, V. Rusanov, L. I. M. Martinez, B. Martinez, J. S. Munoz and M. Mikhov, J. Magn. Magn. Mater., 1996, 162, 331 CrossRef CAS.
- E. Uzunova, D. Klissurski, I. Mitov and P. Stefanov, Chem. Mater., 1993, 5, 576 CrossRef CAS.
- C. H. Yan, Z. G. Xu, F. X. Cheng, Z. M. Wang, L. D. Sun, C. S. Liao and J. T. Jia, Solid State Commun., 1999, 111, 287 CrossRef CAS.
- D. Jézéquel, J. Guenot, N. Jouini and F. Fiévet, J. Mater. Res., 1995, 10(1), 77 CrossRef CAS.
- O. Palchik, J. Zhu and A. Gedanken, J. Mater. Chem., 2000, 10, 1251 RSC.
- E. Warren, X-Ray Diffraction, Addison-Wesley, Reading, MA, 1969, p. 264. Search PubMed.
- G. Bate, in Ferromagnetic Materials, ed. E. P. Wohlfarth, North-Holland, Amsterdam, 1980, vol. 2, p. 431. Search PubMed.
- M. Ueda, S. Shimada and M. Inagaki, J. Mater. Chem., 1993, 3, 1199 RSC.
- A. E. Regzzoni, G. A. Urritia, M. A. Blesa and A. J. G. Maroto, J. Inorg. Nucl. Chem., 1981, 43, 1489 Search PubMed.
- P. S. Sidhu, R. J. Gilkes and A. M. Posner, J. Inorg. Nucl. Chem., 1978, 40, 429 Search PubMed.
- E. Matijevic, Pure Appl. Chem., 1980, 52, 1129.
- P. Toneguzzo, Ph.D. Thesis, University Paris 7, France, 1997..
- L. Poul, Ph.D. Thesis, University Paris 6, France, 2000..
- K. Nakamoto, Infrared and Raman Spectra of Inorganic and Coordination Compounds, Wiley Interscience, New York, 1962, p. 232. Search PubMed.
- A. Miyake, Bull. Chem. Soc. Jpn., 1959, 32, 1381 CAS.
- A. Blenzen, C. Lomenech, V. Escax, F. Villain, F. Varret, C. Cartier dit Moulin and M. Verdaguer, J. Am. Chem. Soc., in press. Search PubMed.
- M. Lenglet, A. D'Huysser and J. Dürr, Ann. Chim. Fr., 1988, 13, 505 Search PubMed.
- V. Briois, C. Cartier dit Moulin and M. Verdaguer, L'actualité chimique, 2000, 3, 25 Search PubMed.
- S. W. Charles, R. Chandrasekhar, K. O'Grady and M. Walker, J. Appl. Phys., 1988, 64, 5840 CrossRef CAS.
- T. Lizuka and S. Lida, J. Phys. Soc. Jpn., 1966, 21, 222 Search PubMed.
- A. E. Berkowtiz and E. Kneller, Magnetism and Metallurgy, Academic Press, New York, 1969, vol. 1, ch. 8. Search PubMed.
- J. L. Dormann, Revue Phys. Appl., 1981, 16, 275 Search PubMed.
- R. H. Kodama, A. E. Berkowitz, E. J. McNiff Jr. and S. Foner, Phys. Rev. Lett., 1996, 77, 394 CrossRef CAS.
- B. Martinez, X. Obradors, Ll. Balcells, A. Rouanet and C. Monty, Phys. Rev. Lett., 1998, 80(1), 181 CrossRef CAS.
- R. H. Kodama and A. E. Berkowitz, Phys. Rev. B, 1999, 59, 6321 CrossRef CAS.
- A. Aharoni, Introduction to the theory of ferrimagnetism, Oxford Science Publications, Oxford, 1996, p. 89. Search PubMed.
- M. P. Morales, S. Veintemillas-Verdaguer, M. I. Montero, C. J. Serna, A. Roig, Ll. Casa, B. Martinez and F. Sandiumenge, Chem. Mater., 1999, 11, 3058 CrossRef CAS.
Footnote |
| † Basis of a presentation given at Materials Discussion No. 3, 26–29 September, 2000, University of Cambridge, UK. |
| This journal is © The Royal Society of Chemistry 2001 |