DOI:
10.1039/B003195J
(Paper)
J. Mater. Chem., 2001,
11, 179-185
Magnetic ordering in diluted kagome antiferromagnets†
Received 17th May 2000, Accepted 12th June 2000
First published on 12th October 2000
Abstract
Potassium iron jarosite, KFe3(SO4)2(OH)6,
provides a model kagome Heisenberg antiferromagnet with which to study the
behaviour of this highly frustrated system. Magnetic susceptibility, powder
neutron diffraction and muon spin relaxation measurements have been performed
on samples of protonated and deuterated potassium iron jarosite in which some
of the iron is replaced by aluminium ions to study the effect of diamagnetic
dilution. The two successive magnetic ordering transitions of the pure material
drop in temperature on doping. In all cases the low temperature phase appears
to have long-range ordering of all moments in an array which is coplanar
or nearly coplanar with the crystallographic ab axes, while in the
intermediate phase there is a component of moment along the c-axis
and some degree of dynamic character.
1 Introduction
One of the principal challenges in solid-state science today is to
develop a better understanding of materials with strongly fluctuating ground
states, and particularly those in which fluctuations between states of the
same or similar energy may be coupled to another property such as charge transport
to produce, for example, superconductivity.1
One method of producing such multi-degenerate, or near-degenerate
ground states is by geometrical frustration2,3
which is illustrated for the case of a simple triangular tile in Fig. 1.
In the case where magnetic exchange is antiferromagnetic, it is impossible
to decorate the tile with moments so that they are simultaneously parallel
to both neighbours (Fig. 1(a)); the
compromise arrangements of lowest energy are depicted in Fig. 1(b)
and (c), which differ only in the handedness,
or ‘chirality’ of spins around the triangle. When these tiles
are connected to produce a lattice in two dimensions, the degeneracy will
depend upon the connectivity of the lattice. Fig. 2
depicts the so-called kagome lattice in which the triangular tiles are
connected through vertices, and when this is decorated with antiferromagnetically
coupled moments there are many degenerate ways of arranging them. Theory indicates4–10 that even
when the moments are free to possess components along x, y
or z (where the x–y plane is the plane of the paper
in these Figures), they chose to adopt a co-planar array, and Fig. 2(a) and (b)
depict two degenerate co-planar arrays, respectively called the q = 0
and √3 × √3 configurations on account of the
relative size of the magnetic and nuclear cells.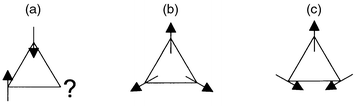 |
| | Fig. 1 Triangular plaquette
of antiferromagnetically coupled moments free to rotate in the plane of the
paper (a) indicates the impossibility of arranging all moments to be simultaneously
antiparallel to their neighbours, while (b) and (c) show degenerate spin configurations
of minimum energy and opposite chirality. | |
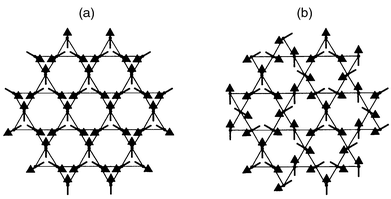 |
| | Fig. 2 Kagome lattices with
(a) q = 0 and (b) √3 × √3
spin arrays. | |
Experimental studies of the nature of the magnetic ground state and excitations
of kagome antiferromagnets have been hindered by difficulties in finding suitable
model compounds.11–21
One system that appears to meet the necessary criteria is the jarosite (see Fig. 3) family of minerals of general formula AFe3(SO4)2(OH)6,
where A+ is usually a univalent cation such as K+ or
H3O+,16–21
Fe3+ may be exchanged for other trivalent ions such as Cr3+22,23 or V3+,23
and the sulfate group may exchanged for CrO42−.17 In all of the iron compounds studied to date, the
material behaves as a kagome array of Heisenberg S = 5/2
spins, coupled with strong antiferromagnetic exchange. In almost all cases,
long-range magnetic order is observed by neutron scattering below a temperature TN
of the order of 55 K, to produce a q = 0 array
in the ab plane and predominantly antiferromagnetic coupling between
these planes to produce a magnetic unit cell that is doubled along the c-axis
relative to the nuclear cell. One exception to this is the hydronium salt
(A+ = H3O+), in which the only
magnetic phase change observed on cooling is a spin-glass like transition
at Tf ≅ 15 K. All the iron jarosites
have essentially the same nuclear structure, but there are at least two reasons
why the hydronium salt may be magnetically so different from the rest of the
family: (i) the hydronium ion may provide an additional efficient pathway
for inter- and intra-layer exchange via H-bonds, and
it also has some degree of orientational disorder that may introduce a disordering
influence;3,19 (ii) the coverage of the
magnetic lattice is generally higher for the hydronium salts relative to the
other members of the family, which have a tendency to lose Fe from the lattice,
compensating for the loss of positive charge by protonation of O–H groups
elsewhere in the structure.
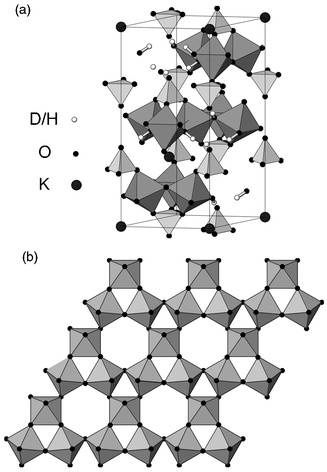 |
| | Fig. 3 (a) Crystal structure
of jarosite, depicting the disposition of FeO6 octahedra and sulfate
tetrahedra and (b) detail of a single layer of Fe–O coordination octahedra
viewed along the c axis with the iron atom at the centre of each
octahedron. | |
We investigated the second of these factors by preparing the diamagnetically-dilute
deuteronium jarosite (D3O)Fe3 − xAly(SO4)2(OD)6 for which the occupation of the magnetic lattice was 89 ± 3%.
This showed a cusp in the dc magnetic susceptibility, χdc,
at 25.5 K, and powder neutron diffraction data taken at 1.4 K
revealed long-range magnetic order;21,24
a similar rise in Tf on diamagnetic doping has also been
observed in (H3O)Fe3 − xGay(SO4)2(OH)6.25 It is tempting therefore to suggest that in this
highly frustrated magnet, diamagnetic dilution pins down a particular spin
configuration from the manifold of ground states, and induces long-range
magnetic order. In this paper we investigate this phenomenon further, preparing
a series of diamagnetically dilute potassium jarosites, KFe3 − xAly(SO4)2(OD)6 to follow the way in which long-range magnetic
order is disturbed in this highly frustrated antiferromagnetic by diamagnetic
dilution of the magnetic lattice.
2 Experimental
Samples of KFe3 − xAly(SO4)2(OD)6
and deuterated analogues were prepared by a hydrothermal technique from anhydrous
or hydrated metal sulfates as described elsewhere.21,23
Preparations were performed at 140 ± 0.5![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) °C
for 24 h. In all cases, the ochre coloured precipitate was washed in
water (or D2O for deuterated samples), and dried in a vacuum oven
at 120°C overnight. The percentage of aluminium and iron in the
product was determined by inductively-coupled plasma atomic emission spectroscopy
using a Thermo Jarrel-Ash instrument on samples that had been digested
in aqua regia for 4 h and diluted with water to give a solution approximately
80 ppm in Fe3+. This revealed that the samples had iron
deficiencies of 5, 8 and 13% and corresponding aluminium levels of 0, 4 and
7% respectively. Thus, it appears more accurate to specify the composition
of the material as KFe3 − xAly(SO4)2(ODz)6 (where charge neutrality is maintained by protonating the OD group
such that z − 1 = 9 − 3x − 3y).
However, the uncertainties in the values of elemental composition, of the
order of 5%, mean that it is not possible to check that the values of x, y
and z are precisely consistent so we will use KFe3 − xAly(SO4)2(OD)6 or KFe3 − xAly(SO4)2(OH)6
as the formulae for the materials through the rest of the work, and we will
refer to the three deuterated materials as the x = 0.15,
0.24 and 0.39 samples in order of decreasing iron content.
°C
for 24 h. In all cases, the ochre coloured precipitate was washed in
water (or D2O for deuterated samples), and dried in a vacuum oven
at 120°C overnight. The percentage of aluminium and iron in the
product was determined by inductively-coupled plasma atomic emission spectroscopy
using a Thermo Jarrel-Ash instrument on samples that had been digested
in aqua regia for 4 h and diluted with water to give a solution approximately
80 ppm in Fe3+. This revealed that the samples had iron
deficiencies of 5, 8 and 13% and corresponding aluminium levels of 0, 4 and
7% respectively. Thus, it appears more accurate to specify the composition
of the material as KFe3 − xAly(SO4)2(ODz)6 (where charge neutrality is maintained by protonating the OD group
such that z − 1 = 9 − 3x − 3y).
However, the uncertainties in the values of elemental composition, of the
order of 5%, mean that it is not possible to check that the values of x, y
and z are precisely consistent so we will use KFe3 − xAly(SO4)2(OD)6 or KFe3 − xAly(SO4)2(OH)6
as the formulae for the materials through the rest of the work, and we will
refer to the three deuterated materials as the x = 0.15,
0.24 and 0.39 samples in order of decreasing iron content.Phase purity was checked by powder X-ray diffraction on a Philips PW1730
diffractometer, with an X'Pert detection system using CuKα radiation.
In all cases, no crystalline impurity phases were detected, nor was there
any evidence through diffuse scattering for significant quantities of amorphous
materials.
χdc was measured for each compound using a Quantum
Design MPMS2 SQUID magnetometer. The dependence of the magnetisation
on an applied field at 5.0 K was found to be linear up to the maximum
operating value of 1.0 T, and data were then taken between 1.8 and
350 K in an applied field of 1.0 T, first after cooling in zero
field and then after cooling in an applied field of 1.0 T.
The nuclear structure and any magnetic correlations were studied by powder
neutron diffraction on the high-flux multidetector instrument D2O at the
Institut Laue Langevin (ILL) in Grenoble. Powder samples whose mass was of
the order of 10 g were loaded in a vanadium can, placed in an ILL ‘Orange’
cryostat, and diffraction patterns taken from 2–160 K at a wavelength
of 2.41(1) Å. The high flux of this instrument allowed a series
of powder diffraction patterns to be measured rapidly as the sample was warmed
up, producing a ‘thermodiffraction’ pattern that allows phase
transitions to be studied. With slightly longer counting times it is possible
to take data of sufficient quality for Rietveld refinement. However, detailed
analysis of the crystallographic structure is limited due to the poor resolution
at high 2θ at this wavelength. Complementary measurements
of any spin freezing process were made by muon spin relaxation (μSR) measurements
on the spectrometer EMU at the ISIS Facility. A protonated sample of KFe3 − xAly(SO4)2(OD)6 was prepared, analysed chemically to
determine x = 0.30 and y = 0.15,
and measurements of χdc made as for the other samples.
Approximately 1 g of sample was loaded into a disc-shaped depression
of 30 mm diameter cut into an aluminium plate; this was covered with
mylar film to retain the sample, and the surrounding aluminium masked by a
silver plate. The sample was then loaded into an Oxford Instruments ‘Variox’
helium cryostat, and the asymmetry in the muon decay measured from 2 to 100 K
in an applied longitudinal field of 50 G (found to be sufficient to
decouple the muon from the nuclear spins) to probe the nature of magnetic
fluctuations above Tf, and the degree of spin freezing
at lower temperatures.
3 Analysis and results
3.1 Magnetic susceptibility
All samples showed strong antiferromagnetic coupling, with Curie–Weiss
behaviour above 150 K, and a maximum in χdc
at a temperature TN of the order of 50 K, falling
as the coverage of the magnetic lattice fell (Fig. 4).
Closer inspection of the maximum in χdc revealed two
peaks in the samples with x = 0.15 and 0.24, and only
one distinct feature in the x = 0.39 sample; values for
the temperatures of the cusps, TN1 and TN2
for higher and lower temperature transitions respectively, are given in Table 1. The value for TN1
for the x = 0.39 sample is in parentheses on account
of the indistinct character of the transition, inferred from a shoulder in
dI/dT from the neutron scattering data, observed to the
high temperature side of the main minimum (see section 3.2). At still lower
temperatures the susceptibility rose on cooling and could be fitted to a Curie–Weiss
expression with a value for the Weiss constant that is considerably smaller
than that at high temperature. This behaviour has been interpreted as arising
from those spins nearer defects in the magnetic lattice 25
which experience on average a weaker exchange field. The data below 25 K
were then fitted to a Curie–Weiss expression to produce the Curie and
Weiss constants C and θ that are given in Table 1. Data taken above 150 K were
then corrected for this contribution and the remaining susceptibility fitted
to a Curie–Weiss expression yielding the Curie and Weiss constants C′
and θ′ given in Table 1.
When C is expressed in emu K−1 mol−1,
it is generally taken to be approximately equal to (μeff)2/8,
where the effective moment μeff2 = g2μB2S(S + 1), while θ is given through the
molecular field approximation by −(4/3k)JS(S + 1),
where J is the exchange constant between a moment and the four nearest
neighbours in the kagome layers.5 Series expansion
calculations5,10 for the specific case
of a Heisenberg kagome antiferromagnet show that although these relations
are correct at relatively high temperatures, that is above T ≅ JS(S + 1)/k,
at lower temperatures C = (μeff)2/9
and θ = −(2/k)JS(S + 1).
For jarosites, this cross-over temperature is approximately 350 K,
so we use the lower-temperature relations to deduce the values of μeff
and J given in Table 1.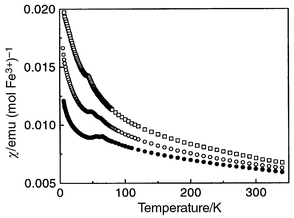 |
| | Fig. 4 Magnetic susceptibility
of KFe3 − xAly(SO4)2(OD)6
as a function of temperature measured after field-cooling in 1.0 T.
Solid circles, open circles and squares refer to data with iron composition x = 0.15,
0.24 and 0.39 respectively. | |
Table 1 Magnetic parameters for aluminium-doped
potassium jarosites with different iron contentsderived from χdc
and powder neutron diffraction data: TN1 and TN2
are the temperatures of the higher and lower temperature cusps in the susceptibility, C
is the gradient of the inverse susceptibility plotted against temperature
for the linear region above 150 K and θ is the intercept
of this line with the temperature axis; μeff and J
are the effective moment and exchange constant derived from C and θrespectively. C′
and θ′ are Curie and Weiss constants extracted from
data taken below 25 K, as described in the text
| Composition x of KFe3 − xAly(SO4)2(OD)6 | TN1, TN2/K | θ/K | C/emu mol−1 K−1 | θ′/K | C′/emu mol−1 K−1 | μeff/μB | J/K |
|---|
| 0.15 | 64.5(0.5), 57.0(0.5) | −663(2) | 5.78(1) | −6.83(12) | 0.0545 | 6.8 | 37.9(1) |
| 0.24 | 60.5(1.0), 45.5(1.0) | −630(5) | 5.69(3) | −12.8(1) | 0.14 | 6.74 | 35.0(3) |
| 0.39 | (53.5(2.0)), 41.5(1.0) | −430(19) | 5.55(3) | — | — | 6.66 | 24.6(1.1) |
3.2 Powder neutron diffraction
All neutron powder diffraction patterns were similar in appearance to those
observed for other pure and dilute jarosites.17,19,21,26
At the lowest temperatures, additional Bragg peaks appeared compared with
data taken above 70 K, and these were attributed to long range magnetic
order (Figs. 5–7). These
peaks could be indexed on a unit cell with the same a parameter as
the nuclear cell, but doubled along the c-axis, as observed for
spin structures in other jarosites.17,19,21,22,26
The evolution of the spin structure on cooling may be illustrated by integrating
the intensity of a prominent magnetic peak and plotting against temperature.
The top panel of Fig. 8 shows how the
intensity I of the (1 1 3/2) reflection for the x = 0.15
sample depends on temperature, while the middle panel shows the derivative
of I with respect to temperature, revealing more clearly a second
transition at lower temperature. The bottom panel reproduces χdc
over this temperature range, mirroring the behaviour of dI/dT.
The precise value of TN derived from the temperature dependence
of the intensity of a magnetic Bragg peak is generally taken as the temperature
at which dI/dT is most negative; at this point the rate
of decay of the magnetic Bragg peak is greatest, and this is compounded by
a maximum in the critical scattering that arises from short-range magnetic
correlations.27 The result of this analysis
applied to all three samples is summarised in Table 1,
and Figs. 9 and 10
reproduce the temperature dependence of I, dI/dT
and χdc for the x = 0.24 and
0.39 samples respectively.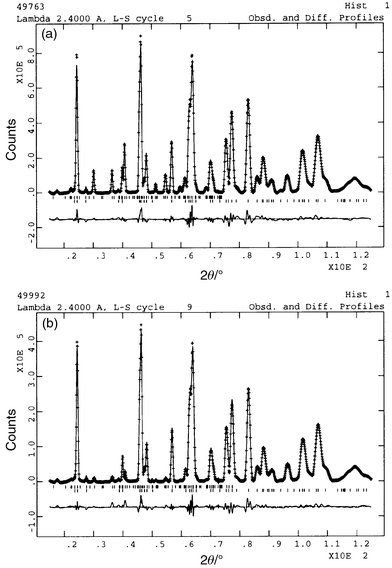 |
| | Fig. 5 Neutron diffraction
data for KFe3 − xAly(SO4)2(OD)6
with x = 0.15 taken at (a) 18.12 K (top) and (b)
61.65 K (bottom). The line through the data represents the profile
calculated after Rietveld refinement, with the difference between calculated
and measured profiles given in the trace below. The upper set of tick marks
indicates reflections for the magnetic scattering, while the lower set is
for the nuclear scattering. | |
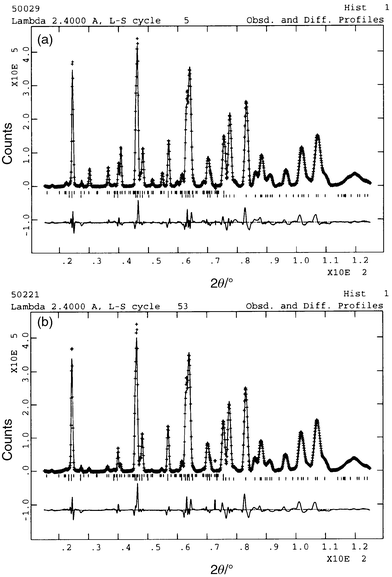 |
| | Fig. 6 Neutron diffraction
data for KFe3 − xAly(SO4)2(OD)6
with x = 0.24 taken at (a) 18.18 K (top) and (b)
57.0 K (bottom). The line through the data represents the profile calculated
after Rietveld refinement, with the difference between calculated and measured
profiles given in the trace below. The upper set of tick marks indicate reflections
for the magnetic scattering, while the lower set is for the nuclear scattering. | |
 |
| | Fig. 7 Neutron diffraction
data for KFe3 − xAly(SO4)2(OD)6
with x = 0.39 taken at (a) 6.03 K (top) and (b)
53.83 K (bottom). The line through the data represents the profile
calculated after Rietveld refinement, with the difference between calculated
and measured profiles given in the trace below. The upper set of tick marks
indicate reflections for the magnetic scattering, while the lower set is for
the nuclear scattering. | |
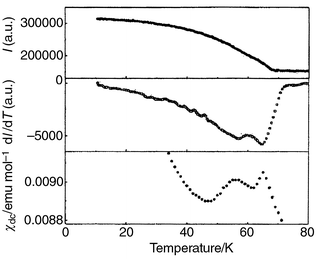 |
| | Fig. 8 Magnetic phase transitions
in KFe3 − xAly(SO4)2(OD)6
with x = 0.15 revealed by the intensity I of
the (1 1 3/2) reflection in the powder neutron diffraction pattern (top),
through dI/dT for the same reflection (middle) and χdc
(bottom). | |
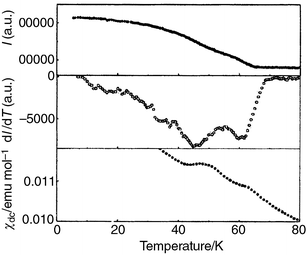 |
| | Fig. 9 Magnetic phase transitions
in KFe3 − xAly(SO4)2(OD)6
with x = 0.24 revealed by the intensity I of
the (1 1 3/2) reflection in the powder neutron diffraction pattern (top),
through dI/dT for the same reflection (middle) and χdc
(bottom). | |
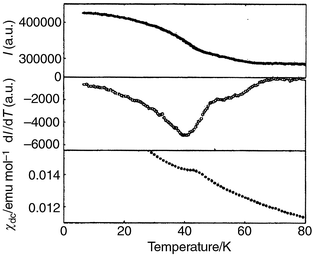 |
| | Fig. 10 Magnetic phase transitions
in KFe3 − xAly(SO4)2(OD)6
with x = 0.39 revealed by the intensity I of
the (1 1 3/2) reflection in the powder neutron diffraction pattern (top),
through dI/dT for the same reflection (middle) and χdc
(bottom). | |
Diffraction patterns were selected for Rietveld refinement of the nuclear
and magnetic structures for each sample at temperatures that appeared to represent
each of the ordered magnetic phases. Thus, for the x = 0.15
sample, patterns were selected at 18.12 K and at 61.65 K; for
the x = 0.24 sample at 18.18 K and 57.0 K;
and for the x = 0.39 sample at 6.03 K and 53.83 K.
This last data set was not taken at an ideal temperature because a judgement
of where best to take a pattern for the intermediate magnetic phase was made
before careful analysis of dI/dT. Initial values for the
various structural parameters were taken from refinements of other AFe(SO4)2(OD)6
compounds, and in particular for the potassium salt,17,26
and the refinement was performed using the program suite GSAS.28
Structural parameters determined in this fashion are given for KFe3 − xAly(SO4)2(OD)6 with x = 0.15, 0.24 and
0.39 and at temperatures of 18.12, 18.18 and 6.03 K in Tables 2,
3 and 4 respectively. The magnetic structure proposed previously
for potassium iron jarosite17 can be described
using a propagation vector (in the nonprimitive hexagonal cell) of k = (0,
0, 3/2). Group theory calculations using the technique of Representational
Analysis29,30 indicate that only three
possible magnetic structures are compatible with this value of k.
A good agreement with the experimental data was achieved only with one of
these values. If the atomic sites are labeled Fe1 = (0.5, 0.5,
0.5), Fe2 = (0.5, 0, 0.5) and Fe3 = (0, 0.5, 0.5)
, this model corresponds to the combination of two basis vectors. With respect
to the hexagonal crystallographic cell these can be written: Ψ1,Fe1 = (1 −1
0), Ψ1,Fe2 = (1 2 0), Ψ1,Fe3 = (−2 −1
0) and Ψ2,Fe1 = Ψ2,Fe2 = Ψ2,Fe3 = (0 0 1)
Table 2 Structural parameters for KFe3 − xAly(SO4)2(OD)6 with x = 0.15 and y = 0
derived from Rietveld refinement of neutron powder diffraction data taken
at 18.12 K in the space group R![[3 with combining macron]](https://www.rsc.org/images/entities/char_0033_0304.gif) m with Z = 3.
Figures reported without errors were not refined. Nuclear cell parameters: a = 7.24402(21) Å, c = 16.99335(74) Å; Rwp = 4.73%, Rp = 3.51%
(at 61.65 K: a = 7.24976(20) Å, c = 17.00999(66) Å)
m with Z = 3.
Figures reported without errors were not refined. Nuclear cell parameters: a = 7.24402(21) Å, c = 16.99335(74) Å; Rwp = 4.73%, Rp = 3.51%
(at 61.65 K: a = 7.24976(20) Å, c = 17.00999(66) Å)
| Atom (Wyckoff site) | x | y | z | Uiso/Å2 | Site symmetry | Fractional occupancy |
|---|
| Fe (9d) | 0.50000 | 0.50000 | 0.50000 | 0.0100(*) | 2/M(110) | 0.95 |
| S (6c) | 0.00000 | 0.00000 | 0.30774(75) | 0.0219(44) | 3M(100) | 1.000 |
| O1 (6c) | 0.00000 | 0.00000 | 0.39068(38) | 0.0058(11) | 3M(100) | 1.000 |
| O2 (18h) | 0.22473(22) | −0.22473(22) | −0.05425(20) | 0.0058(11) | M(110) | 1.000 |
| O3 (18h) | 0.12782(27) | −0.12782(27) | 0.13626(26) | 0.0058(11) | M(110) | 1.000 |
| K (3a) | 0.00000 | 0.00000 | 0.00000 | 0.052(4) | −3M(100) | 1.000 |
| D4 (18h) | 0.19706(22) | −0.19707(23) | 0.11054(18) | 0.0168(18) | M(110) | 1.000 |
Table 3 Structural parameters for KFe3 − xAly(SO4)2(OD)6 with x = 0.24 derived from Rietveld
refinement of neutron powder diffraction data taken at 18.2 K in the
space group Rm with Z = 3.
Figures reported without errors were not refined. Nuclear cell parameters: a = 7.21044(6) Å, c = 16.90761(115) Å; Rwp = 5.96% , Rp = 4.2%
(at 57.0 K: a = 7.21071(7)Å, c = 16.91207(123) Å)
| Atom (Wyckoff site) | x | y | z | Uiso/Å2 | Site symmetry | Fractional occupancy |
|---|
| Fe (9d) | 0.50000 | 0.50000 | 0.50000 | 0.01(*) | 2/M(110) | 0.92 |
| S (6c) | 0.00000 | 0.00000 | 0.30610(115) | 0.0201(67) | 3M(100) | 1.000 |
| O1 (6c) | 0.00000 | 0.00000 | 0.38975(57) | 0.0066(16) | 3M(100) | 1.000 |
| O2 (18h) | 0.22507(32) | −0.22507(32) | −0.05417(30) | 0.0066(16) | M(110) | 1.000 |
| O3 (18h) | 0.12750(40) | −0.12750(40) | 0.13669(40) | 0.0066(16) | M(110) | 1.000 |
| K (3a) | 0.00000 | 0.00000 | 0.00000 | 0.052(4) | −3M(100) | 1.000 |
| D4 (18h) | 0.19653(37) | −0.19653(37) | 0.11046(28) | 0.0220(28) | M(110) | 1.000 |
Table 4 Structural parameters for KFe3 − xAly(SO4)2(OD)6 with x = 0.39 derived from Rietveld
refinement of neutron powder diffraction data taken at 6.03 K in the
space group Rm with Z = 3.
Figures reported without errors were not refined. Nuclear cell parameters: a = 7.21182(23) Å, c = 16.96039(77) Å; Rwp = 4.03% , Rp = 2.85%
(at 53.83 K: a = 7.21321(23) Å, c = 16.96639(77) Å)
| Atom (Wyckoff site) | x | y | z | Uiso/Å2 | Site symmetry | Fractional occupancy |
|---|
| Fe (9d) | 0.50000 | 0.50000 | 0.50000 | 0.0281(*) | 2/M(110) | 0.87 |
| S (6c) | 0.00000 | 0.00000 | 0.30707(73) | 0.0201(46) | 3M(100) | 1.000 |
| O1 (6c) | 0.00000 | 0.00000 | 0.39197(35) | 0.0690(72) | 3M(100) | 1.000 |
| O2 (18h) | 0.22427(20) | −0.22427(20) | −0.05430(19) | 0.0690(72) | M(110) | 1.000 |
| O3 (18h) | 0.12654(30) | −0.12654(30) | 0.13580(30) | 0.0691(72) | M(110) | 1.000 |
| K (3a) | 0.00000 | 0.00000 | 0.00000 | 0.052 (4) | −3M(100) | 1.000 |
| D4 (18h) | 0.19633(21) | −0.19633(21) | 0.11113 (16) | 0.0229 (17) | M(110) | 1.000 |
The first basis vector corresponds to a planar 120° array of moments
and the second to an array with a component along the c-axis. Any
linear combination of these two basis vectors corresponds to a symmetry-allowed
solution, and so in general the possible projections of the magnetic moment, M,
are given by
| | 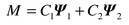 |
(1)
|
Refinement was made directly in terms of these basis vector mixing
coefficients
Cn using a reverse Monte-Carlo
(RMC) algorithm in which only random changes to these coefficients that improved
χ2
were accepted. 400 such cycles were made, and these proved to be sufficient
to find a fit minimum that could not be significantly improved by conventional
least-squares methods. The magnetic structure factor calculations were
performed using the GENLES routine of the GSAS refinement suite.
28
Representational Analysis calculations and the RMC refinement of the basis
vector mixing coefficients were carried out using the package SARA
h.
31Refinements are displayed in Figs. 5–7
in order of decreasing iron coverage. Optimised parameters for the magnetic
structure are given in Table 5.
The site occupancy of the iron atoms was also refined and found to be consistently
2% higher than the values found for chemical analysis. It should also be noted
that it is difficult to make precise and meaningful comparisons between the
ordered magnetic moment refined for the intermediate phases because the temperatures
for which data were taken and refined correspond to different points on the
respective sublattice magnetisation curves i.e. the reduced temperature
(TN1 − T)/TN1
is similar for the x = 0.15 and x = 0.24
samples, but quite different for the x = 0.39 sample.
Table 5 Magnetic moments refined in the space group P1
for KFe3 − xAly(SO4)2(OD)6,
where the components of the moments adopt a q = 0 structure
in the ab plane, and may cant out of this plane by an angle ϕ
| Iron deficiency x | T/K | μeff/μB | Canting angle ϕ/° |
|---|
| 0.15 | 18.12 | 3.942(32) | 4.3 |
| 0.24 | 18.18 | 3.637(43) | 5.7 |
| 0.39 | 6.03 | 2.995(30) | 4.5 |
| 0.15 | 61.65 | 1.779(40) | 14.5 |
| 0.24 | 57.0 | 1.755(82) | 15.5 |
| 0.39 | 53.8 | 1.730(46) | 12.0 |
3.3 Muon spin relaxation
At relatively high temperatures (T > 70 K),
the asymmetry of the decay of the muon polarisation, G(t),
could be fitted well to the expression:| | 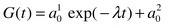 |
(2)
|
The time-independent
term a10 arises from
implantation of some of the muons in the silver part of the sample holder,
while the exponential term is what is expected for muon spin depolarisation
in a paramagnet, where the relaxation rate λ = 1/T1,
and a10 is the initial
asymmetry for this component of the depolarisation. As the sample is cooled
further, a10 was observed
to drop in two stages, first from about 67 K and then from about 47–50 K
as displayed in Fig. 11; the susceptibility
of this sample measured on the SQUID magnetometer is similar to that for the
deuterated x = 0.24 sample (Figs. 4
and 9) with maxima at 47 K, and 65 K. λ
shows relatively little change on cooling, except for a small maximum in the
region of 45 K.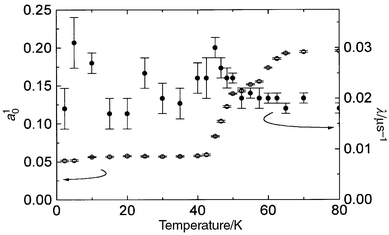 |
| | Fig. 11 Temperature dependence
of the initial asymmetry, (open circles), and the relaxation rate, λ
(closed circles),of the exponential component of decay of muon polarisation
in KFe3 − xAly(SO4)2(OD)6
with x = 0.30 and y = 0.15. | |
If the magnetic moments in the sample freeze in an ordered array, the muon
polarisation will precess at a rate that depends on the magnitude of the internal
field experienced at the implantation site — indeed, previous μSR
studies of potassium iron jarosite32 show
such behaviour below approximately 60 K, with two principal Fourier
components whose frequency saturates at approximately 21 and 38 MHz
on cooling, arising from muons that experience two different static fields
in the sample (though note that the data in that experiment were not taken
with sufficient resolution in temperature to observe two freezing transitions).
However, EMU is fed muon pulses whose width sets an upper limit of 10 MHz
to observe such precession, so we might anticipate the contribution to G(t)
from such a signal will effectively appear as a time-independent term,
centred at 1/3 of the value of a0 for this component;
if the ordered array retains some dynamic character, this signal will also
decay, though previous measurements on potassium jarosite suggest that over
at least the first 0.3 µs of measurements it remains constant
with time.
The onset of spin-freezing may be inferred from the drop in a10 for the exponential component of G(t)
on cooling, with transitions at 67 K, and 45–47 K, consistent
with the susceptibility data. The observation of a weak maximum in λ
at TN2 suggests that a proportion of the moments are still
slowing down and freezing at this temperature — possibly because one
or more components of the moments are not static in the intermediate phase
on the time-scale of the muon experiment. The observation that there is
no such maximum at TN1 might simply arise when muons are
implanted in sites of relatively high symmetry and the ordered array produced
a null field at the muon site. This would be the case, for example, if the
moments froze into triangular antiferromagnetic arrays, and the muons were
implanted at sites equidistant from each moment. It should be noted that a
similar maximum at TN2 in 1/T1 was
observed in 23Na NMR measurements on sodium iron jarosite.26,33
4 Discussion
Diamagnetic dilution of potassium iron jarosite leads to a reduction in
the exchange field experienced by the most strongly coupled moments, and also
in the magnetic freezing temperatures, exactly as might be expected when a
conventional magnet is diluted. The observation of two magnetic transitions
is well documented for a number of other jarosites with A cations K+,
Na+,18,30 Rb+
and Ag+,21 but there has been no
clear assignment of the states involved, and the perturbations that must be
applied to the Hamiltonian for a simple nearest-neighbour kagome Heisenberg
antiferromagnet to cause such behaviour. Further-neighbour exchange is
known to stabilise various forms of magnetic long-range order5,10
and Ising anistropy also causes a symmetry-breaking transition, though
without long-range magnetic order.37,38
Ising kagome antiferromagnets with further-neighbour in-plane interactions
reveal complex magnetic phase diagrams, and successive phase transitions on
cooling,34–38
but the iron jarosites are not expected to represent such models well. The
influence of further-neighbour exchange interactions on XY kagome antiferromagnets
has also been investigated,39 but only up
to in-plane second-neighbour interactions, which induce a single transition
to a long-range ordered state. It therefore appears that there is still
no theoretical work for a Heisenberg kagome antiferromagnet subject to all
the perturbations that are likely to be significant for jarosites, and which
reproduces the magnetic phases observed for much of the family.Our measurements, in combination with observations of ordering processes
in the sodium and potassium salts26,30
shed some light on the nature of the phases between TN1
and TN2 and below TN2, and allow us
to establish the tentative composition-magnetic phase diagram reproduced
in Fig. 12. The low-temperature
phase (C) was determined for polarised neutron scattering measurements on
a single crystal of KFe3(SO4)2(OH)6
to be a coplanar array of moments in the ab plane,40
while the intermediate phase (I) appears to have some degree of order of the
moments along the c-axis, and is likely to retain some fluxional
character according to the muon measurements. It should be noted that our
analysis clearly indicates that phase I has components of the moment both
along cand in the ab plane — attempts to
refine the structure with moments exclusively in one direction or the other
gave very poor fits. The character of the low temperature phase is thus compatible
with theoretical work that predicts that a process of ‘order by disorder’
selects such coplanar states, and within experimental error this coplanarity
is robust towards dilution up to at least the x = 0.39
sample. The character of the intermediate phase is not really treated explicitly
by current theory and one can only speculate that either a temperature-dependent
Ising anisotropy, or the influence of thermal fluctuations, may tip the moments
out of the ab plane before the system becomes paramagnetic.
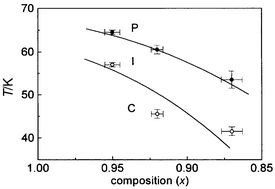 |
| | Fig. 12 Magnetic-compositional
phase diagram for KFe3 – xAly(SO4)2(OD)6
delineating the paramagnetic (P), intermediate (I) and coplanar (C) phases,
where values of x are taken from chemical analysis. | |
The ordered moment in all of the samples at the lowest temperatures is
significantly smaller than would be expected on the basis of the electronic
configuration of Fe3+ in these materials. This may be due in part
to a component of the scattering residing in the diffuse background, as has
been observed in a full three-directional (xyz) polarised neutron
study of KFe3(SO4)2(OD)6.41 However, that study also revealed that the total
integrated static structure factor at 1.5 K was smaller than expected,
suggesting that even at this low temperature there are fluctuations faster
than the time-scale of this neutron experiment (10−11–10−13 s);
this conclusion is compatible with the observation that in the related S = 3/2
system, less than 50% of the sublattice magnetisation is observed in neutron
scattering measurements at T ≈ 0.22
5 Conclusions
It is clear from this and other work on the composition and magnetic properties
of jarosites that it is difficult to produce materials with consistent and
controlled coverage of the magnetic lattice. TN1 and TN2
for potassium iron jarosite are reduced on dilution of the iron sublattice,
along with the average exchange field, which is consistent with the behaviour
anticipated for a conventional magnet. The observation that long-range
order is observed in potassium iron jarosite with at least 95% coverage of
the magnetic lattice suggests that the difference between the jarosites that
show long-range magnetic order, and the hydronium salts which have similar
coverages and show spin-glass order, lies in other factors. Hydronium
salts may provide a different form of further-neighbour inter- and/or
intra-plane exchange, and perhaps also a random element through its structural
disorder, and such influences will be amplified through the sensitivity of
this system to small perturbations in the Hamiltonian. There is clearly a
need to study a wider range of perturbed model systems, exploring the influence
of 2nd and 3rd nearest neighbour in-plane exchange,
as well as interlayer exchange, together with Ising or XY anisotropy, all
of which may be significant in jarosites and lead to the more complex phase
behaviour observed. Finally, detailed single-crystal polarised neutron
studies of the intermediate phase are required to complement data taken in
the low-temperature phase.Acknowledgements
The authors are grateful to the EPSRC for financial support,
and to both The ISIS Facility at the Rutherford Laboratory and also to the
Institut Laue-Langevin for technical support and hospitality. A.H. is
also grateful to the Nuffield Foundation, and D.V. thanks NWO (The Netherlands)
for further financial aid. A.S.W. thanks the TMR project of the EC for his
support, together with the Canadian Institute for Neutron Scattering and to
the Royal Society of Chemistry for further support while some of this work
was being conducted.References
- P. W. Anderson, Science, 1987, 235, 1196 CAS.
- A. P. Ramirez, Ann. Rev. Mater. Sci., 1994, 24, 453 Search PubMed.
- A. P. Ramirez, Handbook of Magnetism, vol. 6 of Magnetic
Materials, ed. K. Buschow, Elsevier, Amsterdam, 2000,
in press. Search PubMed.
- P. Chandra and P. Coleman, Phys.
Rev. Lett., 1991, 66, 100 CrossRef.
- A. B. Harris, C. Kallin and A. J. Berlinsky, Phys. Rev. B, 1992, 45, 2899 CrossRef.
- J. T. Chalker, P. C. W. Holdsworth and E. F. Shender, Phys. Rev.
Lett., 1992, 65, 855 CrossRef CAS.
- A. Chubukov, Phys. Rev. Lett., 1992, 69, 832 CrossRef CAS.
- S. Sachdev, Phys. Rev. B, 1992, 45, 12377 CrossRef.
- E. F. Shender, V. B. Cherepanov, P. C. W. Holdsworth and A. J. Berlinsky, Phys. Rev.
Lett., 1993, 70, 3812 CrossRef.
- J. N. Reimers and A.
J. Berlinsky, Phys. Rev. B, 1993, 48, 9539 CrossRef CAS.
- A.
P. Ramirez, G. P. Espinosa and A. S. Cooper, Phys. Rev. Lett., 1990, 64, 2070 CrossRef CAS.
- C. Broholm, G. Aeppli, G.
P. Espinosa and A. S. Cooper, Phys. Rev. Lett., 1990, 65, 3173 CrossRef CAS.
- B. Martinez, F. Sandiumenge, A. Rouco, A. Labarta, J. Rodríguez-Carvajal, M. Tovar, M. T. Causa, S. Galí and X. Obradors, Phys. Rev. B, 1992, 46, 10786 CrossRef CAS.
- Y. J. Uemura, A. Keren, K. Kojima, L. P. Le, G. M. Luke, W. D. Wu, Y. Ajiro, T. Asano, Y. Kuriyama, M. Mekata, H. Kikuchi and K. Kakurai, Phys. Rev. Lett., 1994, 73, 3306 CrossRef CAS.
- A. P. Ramirez, B. Hessen and M. Winkelmann, Phys. Rev. Lett., 2000, 84, 2957 CrossRef CAS.
- N. Wada, T. Kobayashi, H. Yano, T. Okuno, A. Yamaguchi and K. Awaga, J.
Phys. Soc. Jpn., 1997, 66, 961 Search PubMed.
- M. G. Townsend, G. Longworth and E. Roudaut, Phys. Rev. B, 1986, 33, 49129 CrossRef CAS.
- S. Maegawa, M. Nishiyama, N. Tanaka, A. Oyamada and M. Takano, J. Phys. Soc. Jpn., 1996, 65, 2776 Search PubMed.
- A. S. Wills and A. Harrison, J. Chem.
Soc., Faraday Trans, 1996, 92, 2161 RSC.
- A. S. Wills, A. Harrison, S. A. M. Mentink, T. E. Mason and Z. Tun, Europhys.
Lett., 1998, 42, 325 Search PubMed.
- A. S. Wills, A. Harrison, C. Ritter and R. I. Smith, Phys. Rev. B, 2000, 61, 6157 CrossRef CAS.
- S.-H. Lee, C. Broholm, M. F. Collins, L. Heller, A. P. Ramirez, Ch. Kloc, E. Bucher, R. W. Erwin and N. Lacevic, Phys.
Rev. B., 1997, 56, 8091 CrossRef CAS.
- A. S. Wills, PhD Thesis, The University
of Edinburgh, 1997. .
- A. Harrison, A. S. Wills and C. Ritter, Physica
B, 1998, 241–243, 722.
- S. A. Earle, A. P. Ramirez and R. J. Cava, Physica B, 1999, 262, 199 CrossRef CAS.
- T. Inami, S. Maegawa and M. Takano, J. Magn. Magn. Mater., 1998, 177, 752 CrossRef.
- M. F. Collins, Magnetic Critical Scattering, Oxford University
Press, Oxford, 1989. Search PubMed.
- A.
C. Larson and R. B. Von Dreele, General Structure Analysis
System, 1995. Search PubMed.
- E. F. Bertaut, Acta Crystallogr., Sect. A, 1968, 24, 217 CrossRef CAS.
- E.
F. Bertaut, J. Phys. Colloq., 1971, 1, 462 Search PubMed.
- A. S. Wills, Physica B, 2000, 276–278, 680 CrossRef CAS.
- A. Keren, K. Kojima, L. P. Le, G. M. Luke, W. D. Wu, Y. J. Uemura, M. Takano, H. Dabkowska and M. J. P. Gingras, Phys. Rev. B, 1996, 53, 6451 CrossRef CAS.
- S. Maegawa and M. Nishiyama, personal communication, 1997..
- A. Kuroda and S. Miyashita, J. Phys. Soc. Jpn., 1995, 64, 4509 Search PubMed.
- S. T. Bramwell
and M. J. P. Gingras private
communication, 1994. .
- P. Azaria, H. T. Diep and H. Giacomini, Phys. Rev. Lett., 1987, 59, 1629 CrossRef.
- M. Wolf and K. D. Schotte, J. Phys. A, 1988, 21, 2195 Search PubMed.
- T. Tagaki and M. Mekata, J. Phys.
Soc. Jpn., 1993, 62, 3943 Search PubMed.
- R. S. Gekht and I. N. Bondarenko, JETP, 1998, 86, 1209 CrossRef.
- E. Lelievre-Berna, A. Harrison, G. S. Oakley and D. Visser, Annual Report of the Institut
Laue-Langevin for 1999, 2000, experiment 5-61-37. Search PubMed.
- G. S. Oakley, D. Visser, J. Frunzke, K. H. Andersen, A.
S. Wills and A. Harrison, Physica B, 1999, 267–268, 142 CrossRef CAS.
Footnote |
| † Basis of a presentation given
at Materials Discussion No. 3, 26–29 September, 2000, University
of Cambridge, UK. |
|
| This journal is © The Royal Society of Chemistry 2001 |