
Halogen bonding interaction of chloromethane with several nitrogen donating molecules: addressing the nature of the chlorine surface σ-hole
Abstract
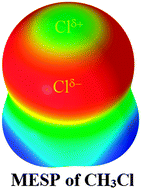
With the goal of understanding the reason for the specific binding affinity of the chlorine in Cl–CH3 in the H2C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O⋯Cl–CH3 complex, we performed molecular electrostatic surface potential (MESP) analysis for isolated H3C–Cl with B3PW91, M06-2X, and MP2(full), all in conjunction with twenty-three Dunning- and Pople-type basis sets of double- and triple-ς valence qualities. The results obtained were benchmarked against the best level of theory employed, CCSD/6-311++G(3d,2p). It was found that the local maximum of the electrostatic potential, Vs,max, on the surface of the chlorine along the outermost extension of the C–Cl bond in Cl–CH3 would vary dramatically from slightly negative to slightly positive values with respect to the basis set sizes and correlation methods employed. Its value, mapped on the 0.001 electrons per bohr3 isodensity surface, is approximately +1.0 kcal mol−1 at the best level. This specific nature of the chlorine's Vs,max is ipso facto more plausible, and is clarified considering the 0.0015 and 0.002 electrons per bohr3 isodensity envelopes, in which cases, Vs,max is apparently small and positive despite the varied basis sets and methods utilized. The thirteen binary complexes investigated using MP2(full)/6-311++G(3d,2p) are thus formed upon the interaction of chlorine's positive σ-hole in Cl–CH3 with the local most negative areas of electrostatic potential, Vs,min, confined on the surface of the nitrogen in the RN series of thirteen monomers, where RN = FCN, ClCN, BrCN, CH3CN, HOCN, HSCN, PCCN, PN, CCl3CN, SiH3CN, NCCN, CNCN, and NaCN. In all instances, the N⋯Cl intermolecular distances of separation evaluated are less than the sum of the chlorine and nitrogen van der Waals radii, 3.48 Å. The N⋯Cl contacts are all stable and have quasi-linear geometries (∠N⋯Cl–C ≅ 175–180°), unraveling the directional nature of the chlorine's positive σ-hole. The effect of substituents –R on the binding energies ΔE of the RN⋯Cl–CH3 complexes was found to be marginal, with values ranging from −0.39 to −1.29 kcal mol−1 with MP2(full) and values from −0.02 to −0.84 kcal mol−1 with CCSD(T). Applications of atoms in molecules and noncovalent interaction reduced-density-gradient methods revealed the N⋯Cl interactions to be of electrostatic origin. The red- and blue-shifts in the vibrational stretching frequencies of the C–Cl bonds estimated with MP2(full) were found to be accompanied with an increase and decrease in the corresponding bonds upon formation of the RN⋯Cl–CH3 complexes, respectively. Natural bond orbital analysis showed that there are several weak interorbital charge transfer interactions persisting between the electron-acceptor and -donor orbitals of the monomers interacting in the RN⋯Cl–CH3 complexes.
O⋯Cl–CH3 complex, we performed molecular electrostatic surface potential (MESP) analysis for isolated H3C–Cl with B3PW91, M06-2X, and MP2(full), all in conjunction with twenty-three Dunning- and Pople-type basis sets of double- and triple-ς valence qualities. The results obtained were benchmarked against the best level of theory employed, CCSD/6-311++G(3d,2p). It was found that the local maximum of the electrostatic potential, Vs,max, on the surface of the chlorine along the outermost extension of the C–Cl bond in Cl–CH3 would vary dramatically from slightly negative to slightly positive values with respect to the basis set sizes and correlation methods employed. Its value, mapped on the 0.001 electrons per bohr3 isodensity surface, is approximately +1.0 kcal mol−1 at the best level. This specific nature of the chlorine's Vs,max is ipso facto more plausible, and is clarified considering the 0.0015 and 0.002 electrons per bohr3 isodensity envelopes, in which cases, Vs,max is apparently small and positive despite the varied basis sets and methods utilized. The thirteen binary complexes investigated using MP2(full)/6-311++G(3d,2p) are thus formed upon the interaction of chlorine's positive σ-hole in Cl–CH3 with the local most negative areas of electrostatic potential, Vs,min, confined on the surface of the nitrogen in the RN series of thirteen monomers, where RN = FCN, ClCN, BrCN, CH3CN, HOCN, HSCN, PCCN, PN, CCl3CN, SiH3CN, NCCN, CNCN, and NaCN. In all instances, the N⋯Cl intermolecular distances of separation evaluated are less than the sum of the chlorine and nitrogen van der Waals radii, 3.48 Å. The N⋯Cl contacts are all stable and have quasi-linear geometries (∠N⋯Cl–C ≅ 175–180°), unraveling the directional nature of the chlorine's positive σ-hole. The effect of substituents –R on the binding energies ΔE of the RN⋯Cl–CH3 complexes was found to be marginal, with values ranging from −0.39 to −1.29 kcal mol−1 with MP2(full) and values from −0.02 to −0.84 kcal mol−1 with CCSD(T). Applications of atoms in molecules and noncovalent interaction reduced-density-gradient methods revealed the N⋯Cl interactions to be of electrostatic origin. The red- and blue-shifts in the vibrational stretching frequencies of the C–Cl bonds estimated with MP2(full) were found to be accompanied with an increase and decrease in the corresponding bonds upon formation of the RN⋯Cl–CH3 complexes, respectively. Natural bond orbital analysis showed that there are several weak interorbital charge transfer interactions persisting between the electron-acceptor and -donor orbitals of the monomers interacting in the RN⋯Cl–CH3 complexes.
 Please wait while we load your content...
Please wait while we load your content...

