
Insights from molecular dynamics simulations for computational protein design
 a
and
Valerie
Daggett
*a
a
and
Valerie
Daggett
*a
Abstract
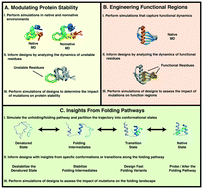
A grand challenge in the field of structural biology is to design and engineer proteins that exhibit targeted functions. Although much success on this front has been achieved, design success rates remain low, an ever-present reminder of our limited understanding of the relationship between amino acid sequences and the structures they adopt. In addition to experimental techniques and rational design strategies, computational methods have been employed to aid in the design and engineering of proteins. Molecular dynamics (MD) is one such method that simulates the motions of proteins according to classical dynamics. Here, we review how insights into protein dynamics derived from MD simulations have influenced the design of proteins. One of the greatest strengths of MD is its capacity to reveal information beyond what is available in the static structures deposited in the Protein Data Bank. In this regard simulations can be used to directly guide protein design by providing atomistic details of the dynamic molecular interactions contributing to protein stability and function. MD simulations can also be used as a virtual screening tool to rank, select, identify, and assess potential designs. MD is uniquely poised to inform protein design efforts where the application requires realistic models of protein dynamics and atomic level descriptions of the relationship between dynamics and function. Here, we review cases where MD simulations were used to modulate protein stability and protein function by providing information regarding the conformation(s), conformational transitions, interactions, and dynamics that govern stability and function. In addition, we discuss cases where conformations from protein folding/unfolding simulations have been exploited for protein design, yielding novel outcomes that could not be obtained from static structures.
 Please wait while we load your content...
Please wait while we load your content...

