
Computational study of C(sp3)–O bond formation at a PdIV centre†
 *a
Alireza
Ariafard,
a
*a
Alireza
Ariafard,
a
Abstract
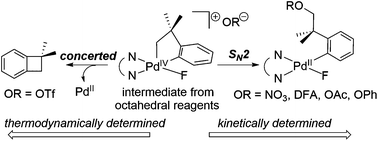
This report describes a computational study of C(sp3)–OR bond formation from PdIV complexes of general structure PdIV(CH2CMe2-o-C6H4-C,C′)(F)(OR)(bpy-N,N′) (bpy = 2,2′-bipyridine). Dissociation of −OR from the different octahedral PdIV starting materials results in a common square-pyramidal PdIV cation. An SN2-type attack by −OR (−OR = phenoxide, acetate, difluoroacetate, and nitrate) then leads to C(sp3)–OR bond formation. In contrast, when −OR = triflate, concerted C(sp3)–C(sp2) bond-forming reductive elimination takes place, and the calculations indicate this outcome is the result of thermodynamic rather than kinetic control. The energy requirements for the dissociation and SN2 steps with different −OR follow opposing trends. The SN2 transition states exhibit “Pd⋯C⋯O” angles in a tight range of 151.5 to 153.0°, resulting from steric interactions between the oxygen atom and the gem-dimethyl group of the ligand. Conformational effects for various OR ligands and isomerisation of the complexes were also examined as components of the solution dynamics in these systems. In all cases, the trends observed computationally agree with those observed experimentally.
 Please wait while we load your content...
Please wait while we load your content...

