
π-Expanded 1,3-diketones – synthesis, optical properties and application in two-photon polymerization†
Rashid
Nazir
ab,
Bjarne
Thorsted
 c,
Evaldas
Balčiūnas
d,
Leszek
Mazur
e,
Irena
Deperasińska
f,
Marek
Samoć
*e,
Jonathan
Brewer
*c,
Maria
Farsari
*d and
Daniel T.
Gryko
*ab
c,
Evaldas
Balčiūnas
d,
Leszek
Mazur
e,
Irena
Deperasińska
f,
Marek
Samoć
*e,
Jonathan
Brewer
*c,
Maria
Farsari
*d and
Daniel T.
Gryko
*ab
aWarsaw University of Technology, Faculty of Chemistry, Noakowskiego 3, 00-664 Warsaw, Poland
bInstitute of Organic Chemistry Polish Academy of Sciences, Kasprzaka 44/52, 01-224 Warsaw, Poland. E-mail: dgryko@icho.edu.pl
cDepartment of Biochemistry and Molecular Biology, University of Southern Denmark, Odense 200-701, Denmark. E-mail: brewer@memphys.sdu.dk
dInstitute of Electronic Structure and Laser (IESL), Foundation for Research and Technology Hellas (FORTH), P.O. Box 1527, 711 10 Heraklion, Crete, Greece. E-mail: mfarsari@iesl.forth.gr
eAdvanced Materials Engineering and Modelling Group, Wroclaw University of Technology, Wybrzeze Wyspianskiego 27, 50-370 Wroclaw, Poland. E-mail: marek.samoc@pwr.edu.pl
fInstitute of Physics, Polish Academy of Sciences, Al. Lotników 32/46, 02-668 Warsaw, Poland
First published on 24th November 2015
Abstract
Four π-expanded α,β-unsaturated 1,3-diketones have been prepared via attaching strong electron-donating and electron-withdrawing groups at positions 9 and 10 of the anthracene scaffold. The strategic incorporation of (C12H25)2N groups at the periphery of these D–π–A molecules resulted in dyes with excellent solubility in most organic solvents. These non-planar diketones possess very broad absorption of light and negligible fluorescence. The two-photon absorption cross-section was measured via Z-scan as well as by two-photon excited fluorescence. The results clearly confirm that, in the case of compounds possessing such small fluorescence quantum yields, the Z-scan was a more reliable approach. Depending on the chemical structure, these compounds exhibited two-photon absorption cross-sections (σ2) up to 2500 GM at 725 nm. For the first time an α,β-unsaturated ketone derived from a proton sponge has been synthesized and was shown to possess optical features distinct from its simpler analogs, such as weak emission. All studied ketones possessed two-photon absorption cross-sections ∼200 GM at wavelength of two-photon polymerization (i.e. 800 nm) and broad fabrication windows have been achieved using all these dyes as photoinitiators.
Introduction
Although the first example of multi-photon photopolymerization (MPP) was reported in 1965 by Pao and Rentzepis,1 it was the pivotal work by the Kawata group in 19972 that brought widespread attention to this technique.3–13 The development of novel monomers14 and photo-initiators for MPP15–25 have been the subject of intense investigation.26–29 Much of the research in this field has followed the pioneering work on classical photoinitiators for polymer synthesis by the Fouassier group.30–39 Several classes of compounds have been investigated in recent years, including α,β-unsaturated ketones,22,23,40 acylphosphine oxides25 and thioxanthones.41 Construction of the scaffolds for tissue engineering and cell growth,42–45 micromechanical devices,46–49 and nanophotonic and metamaterial structures50–52 have been the driving forces towards the rapid development of this technique. Both a large two-photon absorption cross-section and a high yield of photo-initiation are critical for the overall performance of a given photoinitiator. Another important, but often overlooked, factor is the solubility of the photoinitiator in the monomer. Needless to say, liquid photoinitiators are highly sought since solid ones tend to dissolve poorly and slowly in monomers. Ketones are excellent choices, since the carbonyl group is a strong electron-withdrawing moiety which is necessary to obtain large two-photon absorption cross-sections and it is also a known functionality responsible for Norrish type 1 photoinitiators.53–55The α,β-unsaturated ketones possessing electron-donating groups have been the focus of recent studies since they are useful in such diverse areas such as: two-photon excited photodynamic therapy,56,57 two-photon fluorescence microscopy,58 emission in the solid-state59 and photoinitiators of polymerization.18,60 However, only very recently have their photophysical properties been examined in detail.61–63 While benzene is the classical central π-system used in the construction of push–pull dyes,56,64 investigations of larger building blocks such as anthracene, especially anthracene-derived α,β-unsaturated ketones, are virtually unknown. Based on the breakthrough studies by Fouassier and Lalevée, we opted to use 1,3-diketones rather than simple ketones as the electron-withdrawing moiety.35,40,65 1,3-Diketone is a strong electron-withdrawing group.66,67 Its presence results in the decrease in fluorescence quantum yield which is a prerequisite for a compound to be an initiator of photopolymerization. The presence of a 1,3-diketone also broadens the absorption spectrum,35 which, in principle, should also broaden the two-photon absorption spectrum, increasing the chance of achieving MPP using a suitable laser. We reasoned that the combination of D–π–A architecture with 1,3-diketone as acceptor unit and large π-system should lead to notable 2Photon Absorption-cross section (σ2) at the wavelength of interest.
Results and discussion
Design and synthesis
Based on our previous results, based on the efficiency of α,β-unsaturated ketones in MPP,18,19 we designed the next generation of compounds.The crucial part was to maximize the chance that the two-photon absorption cross-section (σ2) would be at the wavelength of interest, i.e. ∼800 nm. At the same time, we sought to introduce more polarized chromophores containing stronger electron acceptors, i.e., 1,3-diketones.
The general structure of these new dyes was based on anthracene as the π-linker connected with an electron donor, dialkylaminobenzene, and an electron acceptor, 1,3-diketone. Two different types of diketones, i.e., indanedione and dimedone (5,5-dimethylcyclohexane-1,3-dione) were chosen as acceptors. Being aware of the work of Blanchard-Desce,68 which clearly showed that linkers with carbon–carbon double bonds or triple bonds had different effects in mediating the charge transfer, we chose to use both these linkers. Since solubility is critical for overall performance of the sensitizer, we opted to use long alkyl chains, which have been proven to ensure proper solubility.18,19
The synthesis started from aldehyde 1, which was prepared from 9,10-dibromoanthracene by means of a metal–halogen exchange reaction (Scheme 1).69 Compound 3 was synthesized via Heck reaction of alkene 2 with 9-bromo-10-formylanthracene (1). Since aldehyde 3 is both sterically hindered and relatively electron-rich, we anticipated that its reactivity in the Knoevenagel condensation would be diminished. In order to achieve the best yield, we performed an optimization study using aldehyde (3) and dimedone (4) as model substrates. The majority of the reported reaction conditions that we utilized (acetic acid in methanol,70L-proline in methanol,71 piperidine in metanol,39 potassium hydroxide in 1,4-dioxane,72 acetic acid/triethylamine in ethanol)73 led to the formation of expected product 6, but only in low yield. The highest yield was achieved by using piperidine and acetic acid in toluene at reflux conditions for 2 h. The same conditions were used to carry out Knoevenagel condensation with ketone 5, leading to dye 7 in 84% yield (Scheme 1). An analogous strategy was employed in the synthesis of ketones 10 and 11 (Scheme 2). Aldehyde 1 was condensed with alkyne 8via Sila-Sonogashira reaction to lead to aldehyde 9. Knoevenagel reaction of aldehyde 9 with ketones 4 and 5 gave corresponding dyes 10 and 11 in 61% and 87% yields, respectively (Scheme 2).
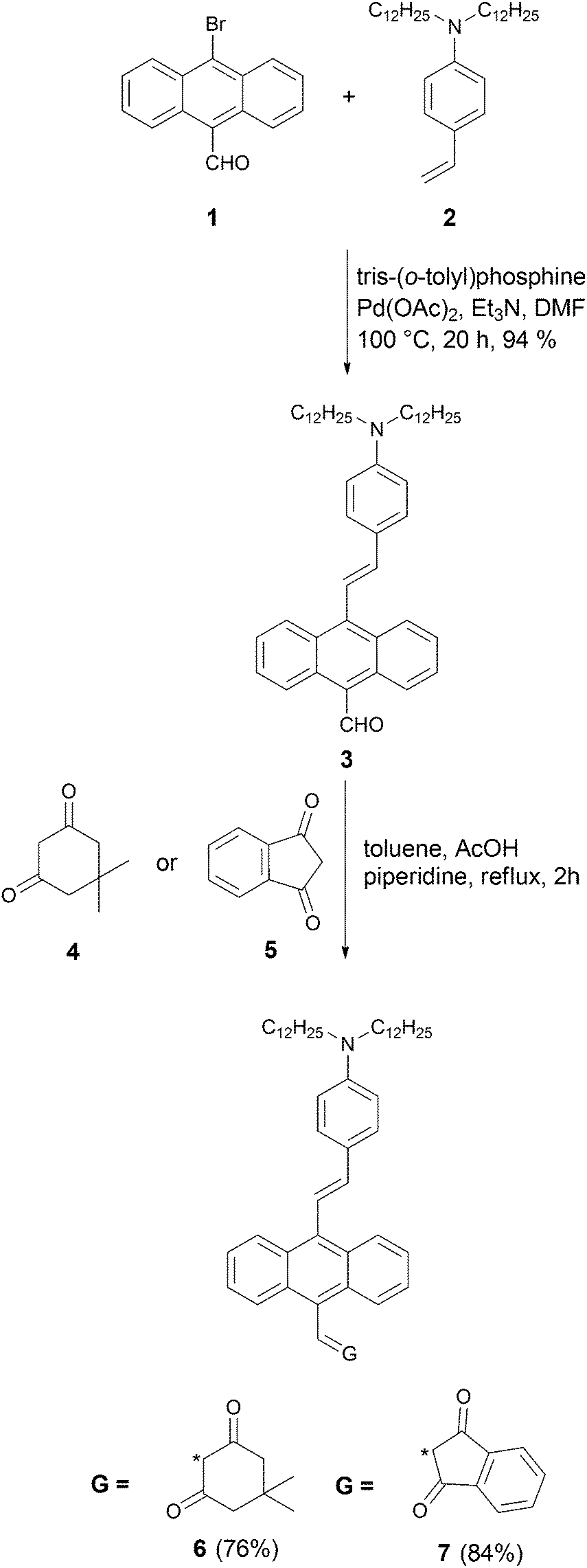 | ||
| Scheme 1 | ||
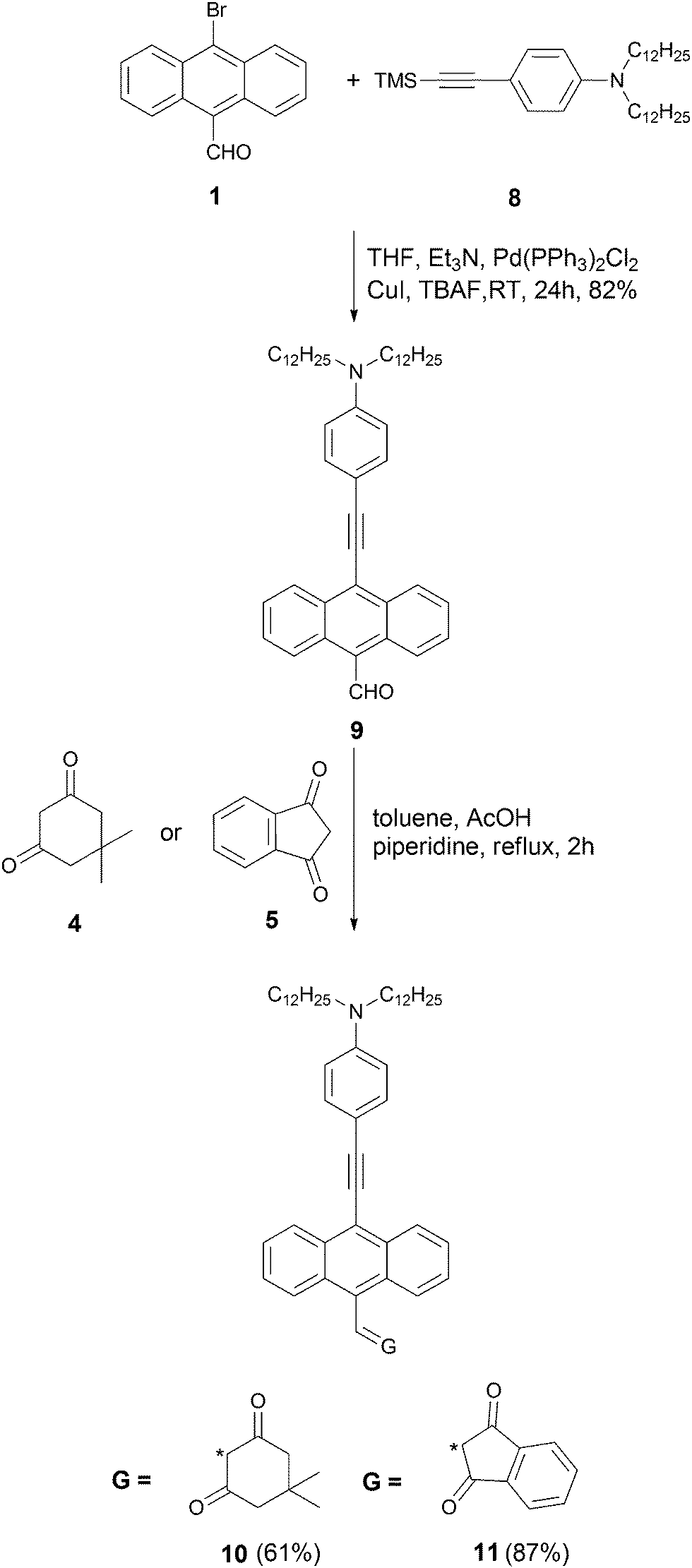 | ||
| Scheme 2 | ||
Finally, we ventured to investigate the influence of a different type of electron-donating moiety, i.e., a proton sponge, on the linear and non-linear optical properties of α,β-unsaturated ketones as well as the photoinitiating ability in MPP. Pozharskii and co-workers developed the synthesis of aldehyde 14via two alternative routes. In the first route they synthesized aldehyde 14 with 7% yield74 and in the second strategy, they synthesized intermediate 13 following oxidation by TiO2 to afford aldehyde 14 in moderate yields.75 The yield of aldehyde 14 improved compared to Pozharskii's procedure via oxidation of 13 using MnO2 (overall 71% yield, Scheme 3). Finally, aldehyde 14 was condensed with cyclopentanone 15 to yield α,β-unsaturated ketone 16 in 61% yield under basic conditions (Scheme 3).
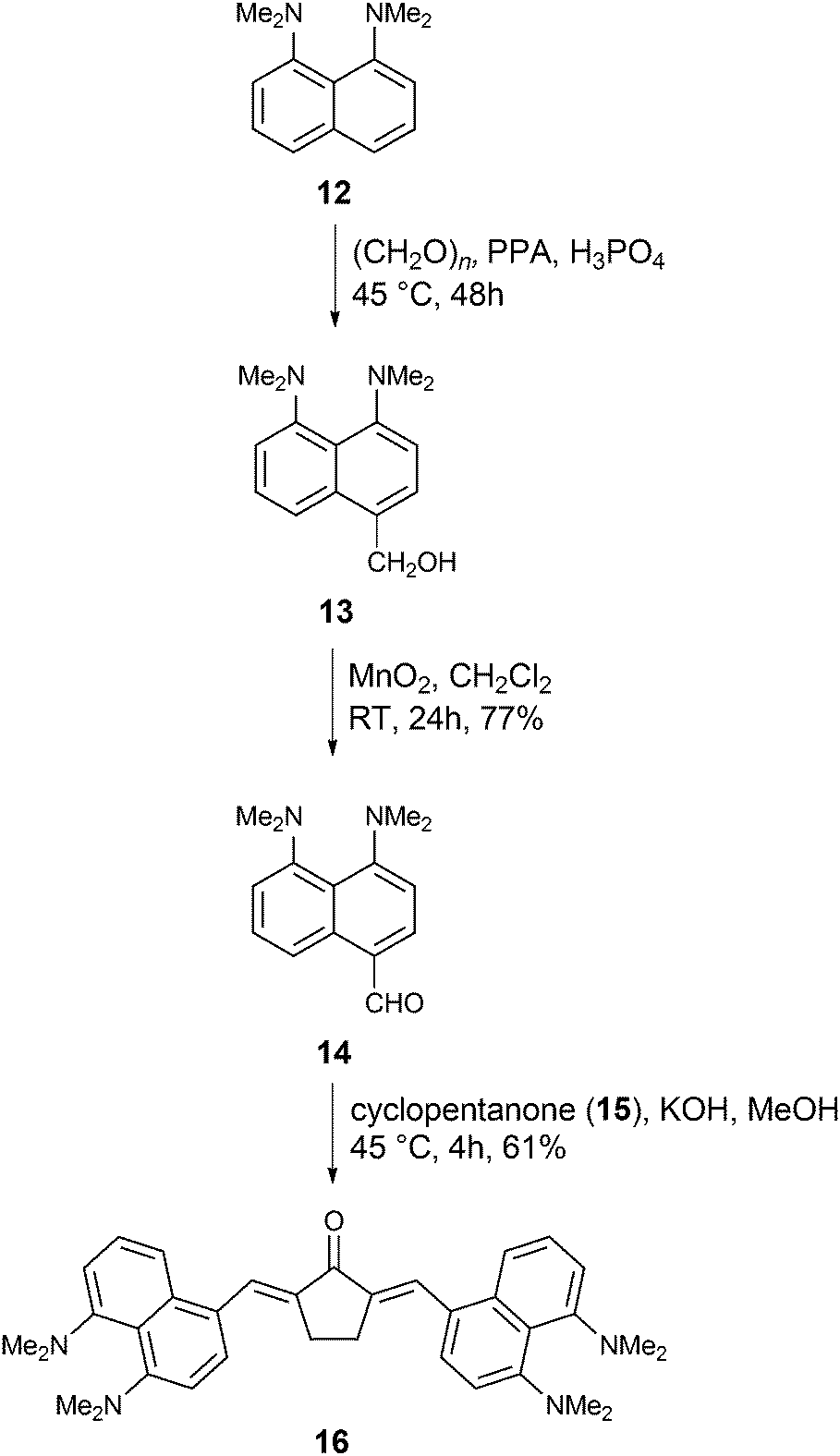 | ||
| Scheme 3 | ||
Linear optical properties
The optical properties of the five ketones were investigated in DCM (Table 1 and Fig. 1, 2). The absorption maxima of ketones 6, 7, 10 and 11 are in the range 500–600 nm (Table 1). Interestingly, in the case of all these compounds, the absorption was very broad but there were no obvious low-energy intramolecular charge transfer (ICT) absorption bands, as observed by Lalevée and co-workers at 620 nm for analogous of 2-(4-(dimethylamino)benzylidene)-indene-1,3-dione.35 The difference in conformation imparted by a larger dihedral angle due to the substitution of a benzene ring with an anthracene ring is most probably responsible for this difference. The deviation from planarity for 1,3-diketones 6, 7, 10 and 11 was also reflected in their low molar absorption coefficients (∼10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000–20000 M−1 cm−1), much lower than that previously reported for α,β-unsaturated-1,3-diketones.35,63
000–20000 M−1 cm−1), much lower than that previously reported for α,β-unsaturated-1,3-diketones.35,63
| Cmpd | λ abs (nm) | ε (M−1 cm−1) | λ em (nm) | Φ fl (%) | Δν (cm−1) | 2λ1PA (nm) | λ maxTPA (nm) | σ max2 (GM) | λ maxTPA (nm) | σ max2 (GM) |
|---|---|---|---|---|---|---|---|---|---|---|
| Z-scan | Z-scan | 2PEF | 2PEF | |||||||
| nd – not determined due to extremely weak emission. | ||||||||||
| 6 | 435 | 7100 | 572 | 0.020 | 2100 | 870 | 725 | 700 | 970 | <8000 |
| 511 | 1020 | |||||||||
| 7 | 395 | 12300 |
629 | 0.010 | 4500 | 790 | 725 | 970 | 710 | <2000 |
| 542 | 1080 | |||||||||
| 10 | 470 | 10700 |
531 | 1.010 | 300 | 940 | 725 | 1000 | 710 | 200 |
| 521 | 1042 | |||||||||
| 11 | 439 | 18500 |
761 | <0.001 | 4700 | 880 | 725 | 2500 | nd | nd |
| 561 | 820 | 1120 | ||||||||
| 16 | 362 | 14900 |
611 | 0.110 | 3500 | 724 | 725 | 600 | 820 | 580 |
| 502 | 1004 | |||||||||
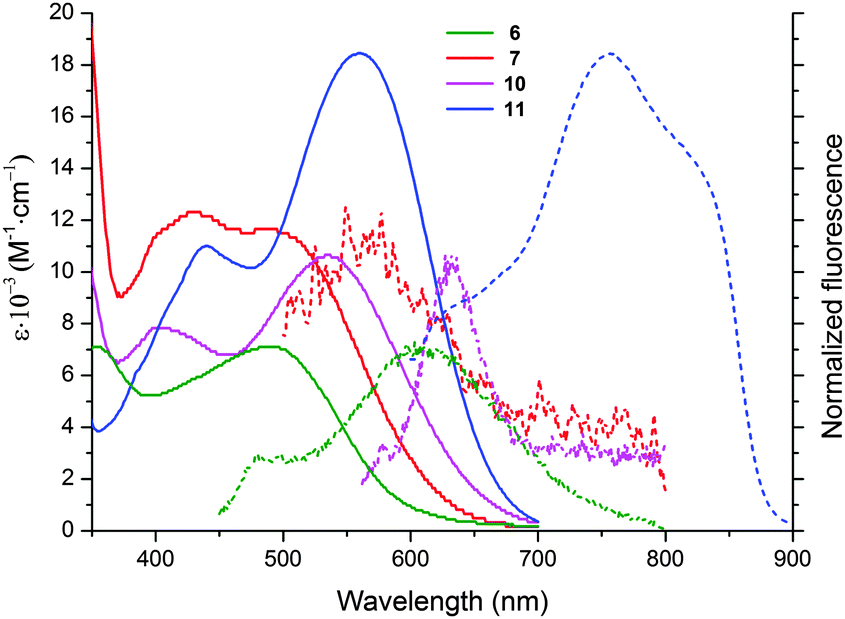 | ||
| Fig. 1 Absorption (solid lines) and emission (dotted lines) spectra of compounds 6 (green), 7 (red), 10 (magenta) and 11 (blue) in DCM. | ||
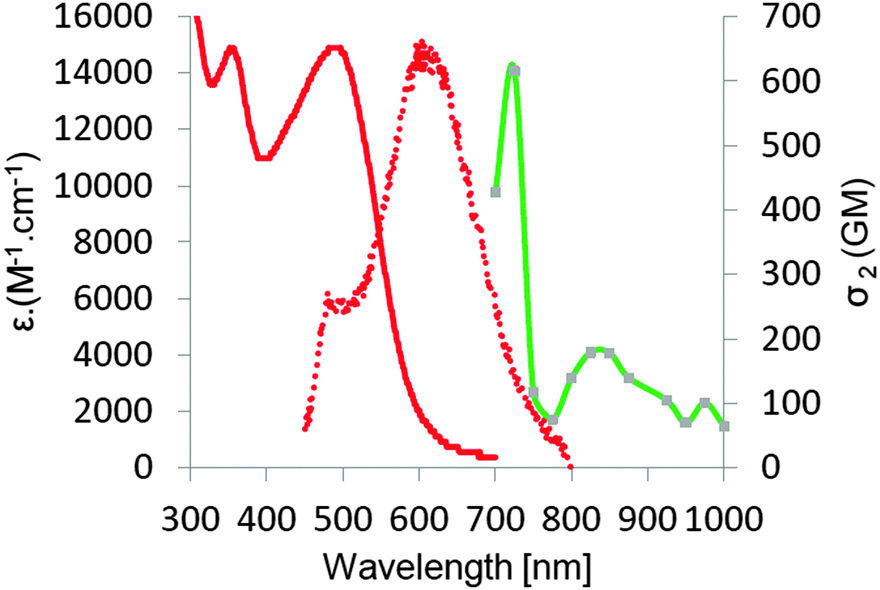 | ||
| Fig. 2 Absorption, emission, and two-photon absorption (green line) spectra of dye 16 in DCM. | ||
For compounds 6/7 and 10/11, one can observe a correlation between the size of the alicyclic ring and the position of the absorption band (Fig. 1 and Table 1). The absorption maxima of 6 and 10 (cyclohexane-1,3-dione ring) were significantly blue-shifted (∼30 nm) relative to 7 and 11 (indene-1,3-dione moiety). The six-membered ring must be even more tilted out-of-plane, as compared to the five-membered ring. The shape of absorption for ketones 6 and 10 is different than that of 7 and 11, namely both bands located in the visible region have similar intensity. Moreover there is almost no bathochromic shift, while proceeding from a more conjugated dye 10 with C–C triple bond as a linker to compound 6 possessing C–C double bond as a linker. The presence of the latter one in dyes 6 and 7 has to cause significant distortion from planarity due to steric interactions between anthracene hydrogens and one of hydrogens attached to C–C double bond. An analogous red-shift while moving from dye 7 to compound 11 is much more noticeable (∼20 nm).
Although all these 1,3-diketones emitted red light, their fluorescence quantum yields were found to be very low (below 0.1%). For ketone 11, we observed broad fluorescence peaks at 760 and 820 nm (Table 1).
Compound 16 possesses different structure and is more related to 2,5-bis(4-(diethylamino)benzylidene)cyclopentanone62 and 2,5-bis((6-(didodecylamino)benzofuran-2-yl)methylene)cyclopentanone.63 Its optical properties are, however, markedly different from those of these two bis(arylidene)cyclopentanones. The comparison of linear optical properties of ketone 16 with 2,5-bis(4-(diethylamino)benzylidene)cyclopentanone clearly showed that there was only small red-shift of absorption (520 nm vs. 480 nm), but there was a strong bathochromic shift of emission (610 nm versus 550 nm). The rationale behind this observation clearly originates from switching of geometry (non-planar in the ground state and planar in the excited state). The fluorescence maximum is comparable with analogous ketone possessing two 6-dialkylaminobenzofurylidene groups as electron donors.63 At the same time, the Φfl values obtained for dye 16 were much lower than any other bis(arylidene)cyclopentanone published so far (0.11% in CHCl3vs. 26% for 2,5-bis(4-diethylamino)benzylidene)cyclopentanone62 and 19% for 2,5-bis((6-(didodecylamino)benzofuran-2-yl)methylene)cyclopentanone.63 The Stokes shift for compound 16 in DCM was comparable to values obtained for 2,5-bis(4-(diethylamino)benzylidene)cyclopentanone and 2,6-bis(4-(diethylamino)benzylidene)cyclohexanone by Wu and co-workers.62
To validate conclusions about the role of steric interaction between anthracene hydrogen and one of the hydrogens attached to C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond, computational calculations with full geometry optimization were carried out using DFT B3LYP/6-31G(d,p) method implemented in the Gaussian09 package.76 In these calculations molecules 6, 7, 10 and 11 were truncated by simple analogues 6′, 7′, 10′ and 11′, wherein the –N(C12H25)2 groups were replaced by –N(CH3)2 groups. Optimization of molecular structures were confirmed by the positive values of all frequencies from their molecular vibrations. In addition, the energies and oscillator strengths for electronic excitations from the ground state S0 to the electronic excited states Si (i = 1–5) have been calculated using TD DFT B3LYP /6-31G(d,p) method. The coordinates of the atoms of the optimized 6′, 7′, 10′ and 11′ molecules and some other results of calculations can be seen in Tables S1 and S2 in the ESI.† According to our calculations the geometries of compounds 6′ and 7′ (with link by CC) differ from the geometries of compounds 10′ and 11′ (with link C
C double bond, computational calculations with full geometry optimization were carried out using DFT B3LYP/6-31G(d,p) method implemented in the Gaussian09 package.76 In these calculations molecules 6, 7, 10 and 11 were truncated by simple analogues 6′, 7′, 10′ and 11′, wherein the –N(C12H25)2 groups were replaced by –N(CH3)2 groups. Optimization of molecular structures were confirmed by the positive values of all frequencies from their molecular vibrations. In addition, the energies and oscillator strengths for electronic excitations from the ground state S0 to the electronic excited states Si (i = 1–5) have been calculated using TD DFT B3LYP /6-31G(d,p) method. The coordinates of the atoms of the optimized 6′, 7′, 10′ and 11′ molecules and some other results of calculations can be seen in Tables S1 and S2 in the ESI.† According to our calculations the geometries of compounds 6′ and 7′ (with link by CC) differ from the geometries of compounds 10′ and 11′ (with link C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) C) by lacking coplanarity between the planes of the anthracene and the dimethylaniline rings (see Fig. 3 and Table S1, ESI†). At an angle of 57 degrees between these two planes in 6′ and 7′, the smallest distances between the H atom of anthracene and H of the CC bridge are 2.152 and 2.139 Å (compared to the co-planar structures whose distance would be approx. 0.97 Å).
C) by lacking coplanarity between the planes of the anthracene and the dimethylaniline rings (see Fig. 3 and Table S1, ESI†). At an angle of 57 degrees between these two planes in 6′ and 7′, the smallest distances between the H atom of anthracene and H of the CC bridge are 2.152 and 2.139 Å (compared to the co-planar structures whose distance would be approx. 0.97 Å).
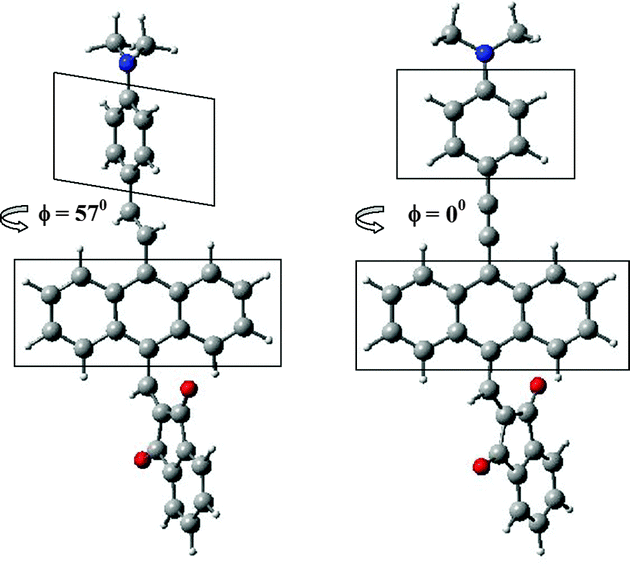 | ||
| Fig. 3 Relative orientation of anthracene and dimethylaniline planes in dependence on linkage between them: (CC (on the left) and CC) for the optimized molecules 7′ and 11′. | ||
The relationship between the electronic spectra and structure of compounds 6′, 7′, 10′ and 11′ can be summarized as follows: according to our calculations (for all four molecules) the excitation S0 → S1 is described as a transition from HOMO to LUMO orbitals. The shapes of these orbitals are shown in Table S2 (ESI†). The HOMO orbitals are composed from the orbitals localized on anthracene and donor, while LUMO orbitals from the orbitals localized on anthracene and acceptor. Therefore: (1) the character of S0 → S1 transition is mixed – locally excited and charge-transfer, (2) the coplanarity or the lack of it between anthracene and donor group has an impact on the spatial shape of the HOMO orbital and (3) in contrast, the type of linkage between the donor and the anthracene has little influence on the shape of the LUMO orbital.
According to our calculations the change in the type of linkage from CC to CC is associated with: (1) a red shift of the absorption band of ∼300 and ∼500 cm−1 in dependence on the type of acceptor group and with the increase of the oscillator strength of 1.3 times. These values are indeed somewhat smaller than those observed experimentally, but describe the same direction of change.
Two-photon absorption
Two-photon absorption cross-sections were measured for compounds 6, 7, 10, 11 and 16 in the range 700–1000 nm using two methods (Table 1). Z-scan measurements were chosen as a primary tool. Z-scan measurements showed, according to expectations, a very strong two-photon response in this family of compounds (Table 1), which originated from the combined effects of a large π-system and the placement of strong electron-donating groups and very strong electron-withdrawing groups within the push–pull systems. In the case of compounds 6, 7, 10 and 11 maxima of 2PA (∼725 nm in all cases) corresponds to no clear transitions at the 1PA spectrum. This wavelength is relatively close to the onset of one-photon absorption, so one cannot exclude some contribution of a sequential process consisting in one-photon absorption followed by excited state absorption. The contribution from a three-photon process, which is two-photon absorption followed by an excited state absorption, is also possible. The values for dyes bearing a carbon–carbon triple bond were higher than for their analogs bearing the ethylene linker. Dyes 7 and 11, bearing stronger electron-withdrawing indene-1,3-dione moiety, possessed higher σ2 than their analogs 6 and 10 bearing six-membered ring.In the two-photon spectra of ketones 6, 7, 10 and 11 both g → c and g → e transitions should be observed since these molecules are not centrosymmetrical.77–79 Strikingly, although indeed two bands are always present in their location it does not strictly correspond to half energies of transitions present in the linear spectra.
The maximum value of 2PA cross-section for ketone 16 was appreciable (600 GM) and identical to what was determined for analogous bis(4-dimethylaminobenzylidene)cyclopentanone.62 The same value can be attributed to interplay between 1,8-bis(dimethylamino)naphthalene being a weaker electron-donor than 4-dimethylaminophenyl, and the overall expansion of the π-system. For bis(4-dimethylaminobenzylidene)cyclopentanone Wu and co-workers confirmed, via fluorescence anisotropy measurements, that the shoulder peak at 425 nm is not of vibronic nature but it comes from an S0–S2 transition, which corresponds to symmetry-allowed transition in 2PA.62 In the case of ketone 16, S0–S2 transition is located at 362 nm, which corresponds to observed maxima at 2PA spectrum at 725 nm. On the other hand, in contrast to what has been observed earlier62 there is no clear S0–S1 transition at 2PA spectrum (which should be visible at ∼1000 nm). Instead, a weak band at 830 nm is observed.
It is important to emphasize that all studied dyes have very similar σ2 (200 ± 50 GM) at wavelength used for two-photon excited polymerization (i.e. 800 nm, Fig. 2 and 4 and ESI†). Analogous dipolar anthracene push–pull dyes bearing the dialkylaminophenylethylene substituent as the electron-donor and 4-cyanophenylethylyne as the electron-acceptor had lower two-photon absorption cross-sections (250 GM).80 In contrast, the analogous quadrupolar compound bearing two diarylaminophenylethynyl substituents displayed a cross-section value of 550 GM.81
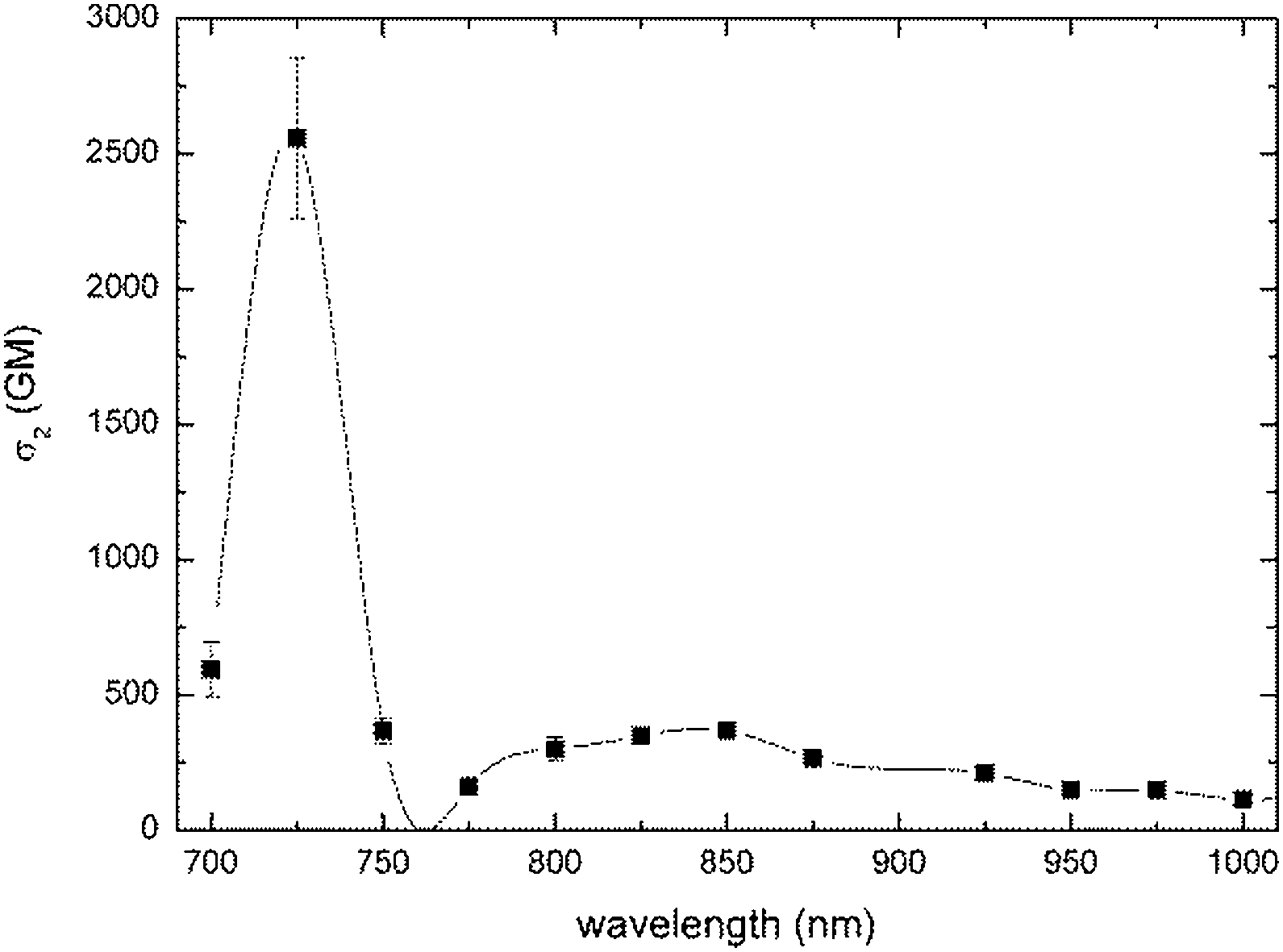 | ||
| Fig. 4 Two-photon absorption spectrum of dye 11 measured by Z-scan technique. | ||
2PA measurements using 2PEF were also performed to investigate how reliable 2PEF can be for compounds possessing such low fluorescence quantum yields. 2PEF is a relative method involving the division of the 2PEF from the dyes with the 2PEF of a reference. Therefore as the fluorescence of the probe goes towards 0, errors become large. Also, the value of the quantum yield, small or large, of the dyes is used for the calculations of the 2PA measurements. Small errors in the absolute values of the quantum yield can lead to large changes of the measured 2PA. In this work it is seen that the 2PEF measurements are reliable for the samples with such low quantum yield. However, it generally overestimates the 2PA cross sections. Therefore the Z scan method is reliable and thus the method of choice.
Polymerization
In order to test whether the investigated photoinitiators were suitable for one-photon polymerization, drop-cast films of each composite were placed under a wide-spectrum UV lamp. All samples fully polymerized after a few minutes of exposure.To evaluate the two-photon initiation potential of the new composites, we studied several parameters related to the response of the composite material with respect to the applied laser power (Fig. 5). (i) The polymerization threshold is the lowest laser power required to have some polymerized structure in the substrate after sample development. This process is characterized by pieces of shapeless polymer, without a well-defined geometry. (ii) The structuring threshold is the lowest laser power required to make complete structures without deformations. (iii) The burning threshold is defined as the laser power at which ablation starts and the material burns. (iv) The fabrication window (FW) is defined as the power difference between the polymerization threshold and the burning threshold. A large FW is desirable, as it allows the use of higher power lasers, allowing fabrication at faster and manufacturing-level scales. As a reference material (R), we used (4,4′-bis(diethylamino) benzophenone), a commercially available photo-initiator used often in MPP.
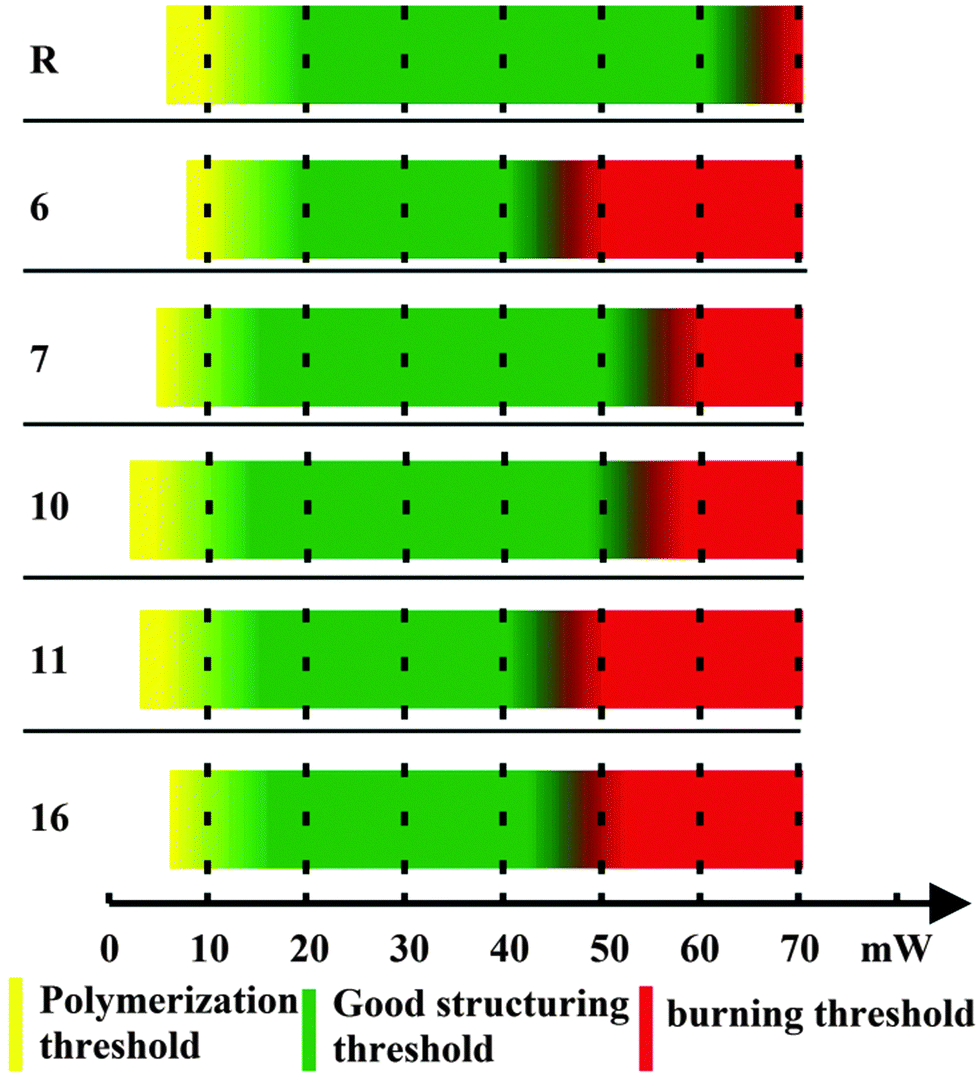 | ||
| Fig. 5 The polymerization (yellow), good structuring (green), and burning (red) thresholds of the five materials prepared in this study. The reference material (R) is 4,4′-bis(diethylamino) benzophenone. | ||
The polymerization parameters were found by structuring the different compounds using a series of increasing laser powers. The results are summarized in Fig. 5. All materials possessed broad and well-defined FWs, comparable to the reference photoinitiator.
Fig. 6a shows an example of 3D free-standing, hollow pyramids fabricated by employing the photopolymer with photoinitiator 10 and increasing laser power (left to right). The three pyramids, prepared with 15, 25 and 40 mW laser power, were complete and well defined. As the average laser power increased the areas outside the pattern started forming and eventually, at laser power of 50 mW, the structures burned and collapsed. Fig. 6b shows in magnification one of the pyramids which was fabricated using 30 mW average laser power. In general, the investigated PIs were comparable to the reference material.
 | ||
| Fig. 6 (a) A SEM micrographs showing an array of micropyramids fabricated using the 10 photoinitiator under conditions of increasing average laser power. (b) A hollow pyramid fabricated using the 10 photoinitiator at 30 mW average laser power. | ||
Experimental section
Materials and synthesis
All commercially available compounds were used as received. All solvents were dried and distilled prior to use. Transformation of oxygen sensitive compounds were performed under argon atmosphere. The reaction progress was monitored by means of thin layer chromatography (TLC) which was performed on aluminium sheets, coated with silica gel 60 F254 (Merck) or aluminium oxide 60 F254 (neutral Merck) with detection by a UV lamp. Product purification was done by means of column chromatography with silica flash P 60 (40–63 μm, SiliCycle) or aluminium oxide 90 (neutral, 70–230 mesh, Merck), dry column vacuum chromatography (DCVC)82 with silica (MN-Kieselgel P/UV254) or aluminum oxide (MNAluminumoxid G). Identity and purity of the prepared compounds were proved by 1H NMR and 13C NMR (Varian 500/200 MHz). High resolution mass spectra (ESI HRMS) were obtained on MaldiSYNAPT G2-S HDMS/GCT Premier, Waters. 9-Bromo-10-anthracenecarboxaldehyde (1),69N,N-didodecyl-4-vinylaniline (2),41 and N,N-didodecyl-4-((trimethylsilyl)ethynyl)aniline (8)83 were obtained by the reported methods.:100 to 5:95) afforded the product as a red viscous oil with 94% yield. Rf = 0.6 (DCM/hexanes 1:2); 1H NMR (CDCl3, 500 MHz) δ 11.5 (s, 1H), 9.02 (d, J = 8.5 Hz, 2H), 8.5 (d, J = 1.5 Hz, 2H), 7.69–7.65 (m, 4H), 7.54–7.49 (m, 4H), 6.85 (d, J = 16.5 Hz, 1H), 6.72 (d, J = 16.5 Hz, 2H), 3.35 (t, 4H), 1.64–1.62 (m, 4H), 1.30–1.22 (m, 36H), 0.89 (t, 6H); 13C NMR (CDCl3, 125 MHz) δ 192.6, 133.5, 133.4 (2 signals), 131.7, 131.2, 129.0, 128.9, 128.1, 128.0, 126.1 (2 signals), 123.7, 111.4 (2 signals), 111.3 (2 signals), 51.0, 31.9, 29.6 (2 signals), 29.5, 29.4 (2 signals), 29.3, 27.2, 27.1, 22.6, 14.1; HRMS (ESI): m/z ([M + H]+) C47H65NO: calculated 659.5066; found: 659.5075.
General procedure for the synthesis of α,β-unsaturated 1,3-diketones 6, 7, 10 and 11
Under the flow of argon the following reagents were added to toluene (5 mL): aldehydes 3 or 9 (0.5 mmol), the corresponding cyclic diketone (0.5 mmol), piperidine (42.5 mg, 0.5 mmol) and glacial acetic acid (30 mg, 0.5 mmol). After refluxing for 2 h, the reaction mixture was cooled, diluted with 50 mL of Et2O and washed with 10 mL saturated NaHCO3 followed by 10 mL of water. The organic phase was dried (MgSO4) and the solvent removed by rotary evaporation.:5 to 1:1) afforded the product as a red gel with 76% yield. Rf = 0.6 (DCM/hexanes 2:1); 1H NMR (CDCl3, 500 MHz) δ 8.92 (s, 1H), 8.44 (d, J = 8.5 Hz, 2H), 7.82 (dd, J1 = 1.5 Hz, J2 = 1.5 Hz, 2H), 7.68 (d, J = 16.5 Hz, 1H), 7.44–7.40 (m, 4H), 6.85 (d, J = 16.5 Hz, 1H), 6.71 (d, J = 16.5 Hz, 2H), 3.33 (t, J = 14.5 Hz, 4H), 2.78 (s, 2H), 2.50 (s, 2H), 1.62 (m, 4H), 1.34-1.27 (m, 36 H), 1.27 (s, 6H), 0.89 (t, J = 13.5 Hz, 6H); 13C NMR (CDCl3, 125 MHz) δ 197.4, 195.5, 149.5, 148.1, 138.1, 136.8, 136.6, 129.2, 128.6, 128.2, 127.9, 127.3, 126.0, 125.3, 124.9, 124.3, 119.1, 111.7, 53.7, 52.9, 51.1, 31.9, 30.0, 29.7, 29.6, 29.5, 29.3, 28.7, 27.3, 27.1, 22.6, 14.1 HRMS (ESI): m/z ([M + H]+) C55H75NO2: calculated 781.5798; found: 781.5823. Anal. calcd for C55H75NO2: C, 84.45; H, 9.66; N, 1.79, found: C, 84.48; H, 9.80; N, 1.69.
:50 to 1:30) afforded the product as a red viscous oil with 84% yield. Rf = 0.6 (acetone/hexanes 1:20); 1H NMR (CDCl3, 500 MHz) δ 8.95 (s, 1H), 8.50 (d, J = 8.5 Hz, 2H), 8.14 (d, J = 7.5 Hz, 1H), 8.03 (d, J = 7.5 Hz, 2H), 7.88–7.78 (m, 4H), 7.73 (d, J = 16.5 Hz, 1H), 7.57 (d, J = 8.5 Hz, 2H), 7.50–7.45 (m, 3H), 6.90 (d, J = 16.5 Hz, 1H), 6.73 (d, J = 8.5 Hz, 2H), 3.35 (t, J = 14.5 Hz, 4H), 1.65 (m, 4H), 1.31–1.27 (m, 36H), 0.90 (t, J = 13.5 Hz, 6H); 13C NMR (CDCl3, 125 MHz) δ 189.6, 187.6, 148.4, 142.8, 142.2, 140.9, 138.7, 135.6, 135.4, 130.2, 129.4, 128.1, 127.4, 126.7, 125.9, 125.3, 124.3, 123.6, 123.5, 119.2, 119.1, 51.3, 32.0, 29.8, 29.8, 29.7, 29.6, 29.5, 27.5, 27.4, 27.3, 22.8, 14.2; HRMS (ESI): m/z ([M + H]+) C56H69NO2: calculated 787.5328; found: 787.5341. Anal. calcd for C56H69NO2: C, 85.34; H, 8.82; N, 1.78, found: C, 85.33; H, 8.74; N, 1.63.
:10 to 1:5) gave the desired compound as a red viscous oil (545 mg, 82%). 1H NMR (CDCl3, 500 MHz) δ 11.48 (s, 1H), 9.00 (d, J = 8.5 Hz, 2H), 8.70 (d, J = 8.5 Hz, 2H), 7.70–7.60 (m, 6H), 6.69 (d, J = 5.5 Hz, 2H), 3.35 (t, J = 14.5 Hz, 4H), 1.64 (m, 4H), 1.34 (m, 36 H), 0.89 (t, J = 14.5 Hz, 6H); 13C NMR (CDCl3, 125 MHz) δ 192.6, 148.7, 134.1, 133.5, 133.4 (3 signals), 131.7, 131.2, 128.9, 128.0, 126.1, 123.7, 111.3, 108.0, 85.0, 51.0, 31.9, 29.65, 29.6 (3 signals), 29.5, 29.4, 29.3, 27.11, 22.6, 14.1; HRMS (ESI): m/z ([M + H]+) C47H63NO: calculated 657.4910; found: 657.4905.
:5 to 1:2) afforded the product as a red viscous oil with 61% yield. Rf = 0.6 (DCM/hexanes 1:1); 1H NMR (CDCl3, 500 MHz) δ 8.87 (s, 1H), 8.72 (d, J = 8.5 Hz, 2H), 7.81 (d, J = 8.5 Hz, 2H), 7.61 (d, J = 7.5 Hz, 1H), 7.56–7.45 (m, 4H), 6.67 (d, J = 6.5 Hz, 2H), 3.33 (t, J = 14.5 Hz, 4H), 2.77 (s, 2H), 2.50 (s, 2H), 1.63(m, 4H), 1.34–1.25 (m, 36 H), 1.17 (s, 6H), 0.89 (t, 6H); 13C NMR (CDCl3, 125 MHz) δ 197.4, 195.5, 148.8, 148.4, 137.0, 134.1, 133.0, 131.4, 129.1, 128.4, 128.0, 127.2, 126.4, 125.9, 125.3, 121.2, 111.3, 84.6, 53.6, 52.8, 51.0, 31.9, 30.0, 29.7, 29.7, 29.6, 29.5, 29.3, 28.7, 27.2, 27.1, 22.6, 14.1.; HRMS (ESI): m/z ([M + H]+) C55H74NO2: calculated 780.5720; found: 780.5726.
:50 to 1:30) afforded the product as a red viscous oil with 87% yield. Rf = 0.6 (acetone/hexanes 1:20); 1H NMR (CDCl3, 500 MHz) δ 8.90 (s, 1H), 8.78 (d, J = 8.5 Hz, 2H), 8.13 (d, J = 7.5 Hz, 2H), 8.02–7.79 (m, 2H), 7.64–7.50 (m, 6H), 6.69 (d, J = 8.5 Hz, 2H), 3.35 (t, J = 14.5 Hz, 4H), 1.65–1.61 (m, 4H), 1.35–1.27 (m, 36 H), 0.90 (t, 6H); 13C NMR (CDCl3, 125 MHz) δ 189.7, 187.7, 142.8, 142.2, 140.9, 138.7, 135.6, 135.3, 130.2, 129.4, 128.1, 127.5, 127.4, 126.7, 126.6, 125.9, 125.3, 124.3, 123.6, 123.5, 119.1, 111.8, 51.3, 32.0, 29.8 (2 signals), 29.7, 29.6, 29.54, 29.52, 27.4, 27.3, 22.8, 14.2.; HRMS (ESI): m/z ([M + H]+) C56H67NO2: calculated 785.5172; found: 785.5201.
:2 to 1:1) afforded a light yellow product in 77% yield. Rf = 0.5 (DCM /hexanes 1:1). 1H NMR (CDCl3, 500 MHz) δ 10.06 (s, 1H), 8.89 (t, J = 8 Hz, 1H), 7.71 (d, J = 8 Hz, 1H), 7.47 (d, J = 16 Hz, 1H), 7.02–6.82 (m, 2H), 2.95 (s, 6H) 2.76 (s, 6H); 13C NMR (CDCl3, 125 MHz) δ 191.1, 156.5, 150.9, 149.8, 138.9, 135.9, 128.8, 125.0, 116.9, 112.6, 108.5, 44.4, 43.3.; HRMS (ESI): m/z ([M + H]+) C15H19N2O: calculated 243.1497; found: 243.1501.
(2E,5E)-2,5-Bis((4,5-bis(dimethylamino)naphthalen-1-yl)methylene)cyclopentanone (16)
To an argon-filled Schlenk tube, the following reagents were added to methanol (10 mL): aldehyde 14 (121 mg, 1 mmol), cyclopentanone 15 (36 μL, 0.44 mmol) and potassium hydroxide (56 mg, 1 mmol). The solution was heated at 45 °C for 4 h and the reaction mixture was cooled. Solvent was concentrated under vacuum and the crude product was purified by DCVC (alumina, acetone/hexanes 5:95) to afford red gel with 61% yield. Rf = 0.6 (dichloromethane/hexanes 1:1) 1H NMR (CDCl3, 500 MHz) δ 8.30 (s, 2H), 7.79 (d, J = 8, 2H), 7.58 (d, J = 8.5 Hz, 2H), 7.41 (t, J = 16.5 Hz, 2H), 6.97–6.89 (m, 4H), 3.03 (s, 4H), 2.87 (s, 12H), 2.80 (s, 12H); 13C NMR (CDCl3, 125 MHz) δ 206.9, 161.8, 156.5, 151.9, 142.1, 136.3, 130.5, 129.8, 126.2, 116.3, 109.5, 108.7, 97.4, 27.3, 27.1, 22.8, HRMS (ESI): m/z ([M + H]+) C35H40N4O: calculated 532.3202; found: 532.3196.
Photophysics
Z-scan measurements
The molecules were dissolved in dichloromethane at a 1.0% (w/w) concentration and the solutions were placed in 1 mm path length optical glass cells. The measurements were carried out in a relative manner, calibrating all the data against Z-scans carried out on a fused silica plate and taking into account the nonlinear signals obtained on a cell containing pure solvent.84 The tunable excitation was achieved by directing the beam from a Quantronix Integra Ti:Sapphire regenerative amplifier (output wavelength 800 nm, pulse duration 130 fs, repetition rate 1 kHz) into an optical parametric amplifier (OPA, a Quantronix Palitra).Two-photon excited fluorescence (2PEF) measurements
Dichloromethane (CHROMASOLV®, for HPLC, ≥99.9%), methanol (CHROMASOLV®, for HPLC, ≥99.9), 5-carboxyfluorescein (99% (HPLC)), Rhodamine B and Rhodamine 6G were purchased from Sigma-Aldrich, Denmark. The samples were dissolved in dichloromethane, Rh B and Rh 6G were dissolved in methanol. The fluorescein was dissolved in a CAPS buffer (N-cyclohexyl-3-aminopropanesulfonic acid) pH 11. Concentrations of the samples and references were determined by a dilution series in a spectrophotometer (Perkin Elmer lambda 35) using quartz cuvettes. Single photon fluorescence emission spectra were measured using a spectrofluorometer (ChronosFD from ISS, Champaign, IL USA). The two photon absorption measurements were performed using a custom built multiphoton excitation spectrofluorometer. In short, the excitation source was a Ti:Sa laser (HPeMaiTai DeepSee, Spectra Physics, Mountain View, CA). The laser power was controlled using motorized halfwave plate together with a polarizer. The laser was focused on the sample using a CFI S Plan Fluor ELWD 60× objective (Nikon). The laser power was monitored by a power meter (PM100D with a S142C head, Thorlabs Sweden AB Goteborg, Sweden). The two photon excited emission was collected through the objective and passed through a Multiphoton-Emitter HC 680/SP (AHF analysentechnik AG, Tuebingen, Germany) to a multimode optical fiber (M200L02S-A, Thorlabs Sweden AB Goteborg, Sweden). The emission was then sent to a monochromator (ARC-SP2155, BFi OPTiLAS, Sweden) and the spectra was then imaged by a cooled CCD camera (PIXIS 400B, Princeton Instruments – New Jersey, USA). A motorized XY stage (Nikon) was used as a sample holder. The laser, laser power, camera and XY stage was controlled using ImageJ72 and custom scripts. Calculations of the two photon cross-sections were done using custom Matlab code (MathWorks, Natick, USA). The absolute 2-photon absorption cross sections were calculated using a relative fluorescence intensity technique as described previously.85 Fluorescein in a CAPS buffer, (pH11), Rh 6G and Rh B in methanol were used to reference the system, using as references well characterized spectra.86 The measurements for the two photon cross-sections were carried out at least five times for the different samples with excitation wavelengths from 700 nm to 1020 nm.To eliminate artifacts, due to photo bleaching or linear absorption, we tested that the fluorescence signal increased as the square of the excitation intensity at the different excitation wavelengths for the different samples.
Photopolymer preparation
For the evaluation of the 1,3-diketones as radical photoinitiators, an organic–inorganic silicate containing zirconium was employed.87 It consisted of methacryloxypropyl trimethoxysilane (MAPTMS), methacrylic acid (MAA), and zirconium n-propoxide (ZPO, 70% in propanol).MAPTMS was hydrolyzed using HCl solution (0.01 M) at a 1:0.75 ratio. ZPO was separately mixed with MAA, at 1:1 ratio. After 45 min, the hydrolyzed MAPTMS was added to the ZPO-MAA mixture. Finally, 31 mM of the investigated photoinitiator was added to each mixture. After stirring for 24 h, the materials were filtered using 0.22 μm syringe filters.
The samples were prepared by depositing a thick droplet onto a glass substrate, which were dried in a vacuum oven at 100 °C for 1 h before being processed by the laser.
3D structure fabrication by MPP
The experimental setup for the 3D structure fabrication by MPP has been described before.88 For the multi-photon polymerization, a Ti:sapphire femtosecond laser was employed (Femtolasers Fusion, 800 nm, 400 mW average power, 75 MHz repetition rate, with integrated dispersive mirrors that pre-compensate the beam delivery and focusing optics to achieve sub-20 fs duration pulses into the sample). The 3D structure was ‘written’ using an x–y galvanometric mirror digital scanner (Scanlabs Hurryscan II), adapted to hold a high numerical aperture focusing microscope objective lens (100×, N.A. = 1.4, Zeiss, Plan Apochromat), and controlled using the SAMLight (SCAPS) software. The laser beam was expanded 5x using a telescope lens to improve focusing. Movement on the z-axis and large-scale movement on the x–y plane was achieved with a three-axes linear encoder stage (PI). The beam on/off was controlled using a mechanical shutter (Uniblitz), while its intensity was controlled by a motorized attenuator (Altechna). The stages, the shutter and the attenuator were computer-controlled via a National Instruments LabVIEW program. A CCD camera was mounted behind a dichroic mirror for the online monitoring of the polymerization.The sample was mounted upside down with the glass slide facing the objective to prevent contact of the material with the oil of the objective, the structures were fabricated layer-by-layer, with the last layer on the surface of the coverslip; this was done to avoid illuminating through the already polymerized region. Scanning speed was always set at 2 mm s−1, and the maximum laser power used was 70 mW, measured before the objective. After the completion of the component building process, the sample was developed in 4-methyl-2-pentanone for 20 min.
Conclusions
We have shown that the detailed design of push–pull dyes can lead to the formation of initiators in multiphoton photopolymerization with desirable properties. The attachment of a strong electron-withdrawing unit at position 9 of anthracene made it possible to maintain absorption maxima of α,β-unsaturated ketones in the green spectral region while broadening the absorption spectrum significantly. Elongation of the π-system did not cause a substantial red-shift of absorption. Indeed, replacing the 4-dimethylaminobenzylidene group with the electron-rich π-extended anthranylidene unit did not bathochromically shift the absorption of studied 1,3-diketones. The steric hindrance imparted by close proximity of two hydrogens at positions 1 and 8 of anthracene and the carbonyl groups forced the molecule to deviate from planarity. Conjugation was thus weakened in comparison to that observed for 2-(4-(dimethylamino)benzylidene)-indene-1,3-dione.35 The conjugation of proton sponge with cyclopentanone led to α,β-unsaturated ketone with slightly bathochromically shifted absorption and significantly red-shifted but very weak emission. Most importantly the maxima of two-photon absorption is 100 nm blue-shifted vs. values for analogous bis(4-dimethylaminobenzylidene)cyclopentanone. The combination of the above mentioned features and superb solubility allowed us to obtain photoinitiators with a fabrication window up to 40 mW, and a polymerization threshold starting as low as 8 mW, under the investigated experimental conditions. Since all studied ketones possessed very similar σ2 at 800 nm, the differences in fabrication window are plausibly related to yield of triplet formation.Acknowledgements
The research leading to these results has received funding from the European Community's Seventh Framework Marie Curie ITN programs TopBio (PITN-GA-2010-264362) and AngioMatTrain (Grant Agreement number: 317304) and from the Ministry of Science and Higher Education of the Republic of Poland (grant MAESTRO-2012/06/A/ST5/00216 and DEC-2013/10/A/ST4/00114). Theoretical calculations were performed at the Interdisciplinary Center of Mathematical and Computer Modeling (ICM) of Warsaw University under the computational grant no. G-32-10.We thank Eli M. Espinoza (US Riverside) for amending the manuscript.References
- Y.-H. Pao and P. M. Rentzepis, Appl. Phys. Lett., 1965, 6, 93–95 CrossRef.
- S. Maruo, O. Nakamura and S. Kawata, Opt. Lett., 1997, 22, 132–134 CrossRef CAS PubMed.
- A. Marino, A. Desii, M. Pellegrino, M. Pellegrini, C. Filippeschi, B. Mazzolai, V. Mattoli and G. Ciofani, ACS Nano, 2014, 8, 11869–11882 CrossRef CAS PubMed.
- O. Kufelt, A. El-Tamer, C. Sehring, S. S. Wolter and B. N. Chichkov, Biomacromolecules, 2014, 15, 650–659 CrossRef CAS PubMed.
- C. M. Marco, A. Gaidukeviciute, R. Kiyan, S. M. Eaton, M. Levi, R. Osellame, B. N. Chichkov and S. A. Turri, Langmuir, 2013, 29, 426–431 CrossRef PubMed.
- H.-B. Sun and S. Kawata, APS, 2004, 170, 169–273 CAS.
- K.-S. Lee, D.-Y. Yang, S. H. Park and R. H. Kim, Polym. Adv. Technol., 2006, 17, 72–82 CrossRef CAS.
- K. D. Belfield, A. R. Morales, B.-S. Kang, J. M. Hales, D. J. Hagan, E. W. V. Stryland, V. M. Chapela and J. Percino, Chem. Mater., 2004, 16, 4634–4641 CrossRef CAS.
- J. M. Hales, D. J. Hagan, E. W. V. Stryland, K. J. Schafer, A. R. Morales, K. D. Belfield, P. Pacher, O. Kwon, E. Zojer and J. L. Bredas, J. Chem. Phys., 2004, 121, 3152–3160 CrossRef CAS PubMed.
- K. D. Belfield, K. J. Schafer, W. Mourad and B. A. Reinhardt, J. Org. Chem., 2000, 65, 4475–4481 CrossRef CAS PubMed.
- C. Martineau, R. Anémian, C. W. I. Andraud, M. Bouriau and P. L. Baldeck, Chem. Phys. Lett., 2002, 362, 291–295 CrossRef CAS.
- G. S. He, L.-S. Tan, Q. Zheng and P. N. Prasad, Chem. Rev., 2008, 108, 1245–1330 CrossRef CAS PubMed.
- M. Wang, X. Ma, J. Yu, X. Jia, D. Han, T. Zhou, J. Yang, J. Nie and T. Wang, Polym. Chem., 2015, 6, 4424–4435 RSC.
- A. I. Ciuciu and P. J. Cywinski, RSC Adv., 2014, 4, 45504–45516 RSC.
- B. H. Cumpston, S. P. Ananthavel, S. Barlow, D. L. Dyer, J. E. Ehrlich, L. L. Erskine, A. A. Heikal, S. M. Kuebler, I. Y. S. Lee, D.-M. Maughon, J. Qin, H. Rockel, M. Rumi, X.-L. Wu, S. R. Marder and J. W. Perry, Nature, 1999, 398, 51–54 CrossRef CAS.
- J. Lalevee, P. Xiao, S. Telitel, M. Lepeltier, F. Dumur, F. M. Savary, D. Gigmes and J. P. Fouassier, Beilstein J. Org. Chem., 2014, 10, 863–876 CrossRef CAS PubMed.
- Z. Q. Li, J. Torgersen, A. Ajami, S. Muhleder, X. H. Qin, W. Husinsky, W. Holnthoner, A. Ovsianikov, J. Stampfl and R. Liska, RSC Adv., 2013, 3, 15939–15946 RSC.
- R. Nazir, F. Bourquard, E. Balčiūnas, S. Smoleń, D. Gray, N. V. Tkachenko, M. Farsari and D. T. Gryko, ChemPhysChem, 2015, 16, 682–690 CrossRef CAS PubMed.
- R. Nazir, P. Danilevicius, A. I. Ciuciu, M. Chatzinikolaidou, D. Gray, L. Flamigni, M. Farsari and D. T. Gryko, Chem. Mater., 2014, 26, 3175–3184 CrossRef CAS.
- G. von Freymann, A. Ledermann, M. Thiel, I. Staude, S. Essig, K. Busch and M. Wegener, Adv. Funct. Mater., 2010, 20, 1038–1052 CrossRef CAS.
- F. Hao, Z. Liu, M. Zhang, J. Liu, S. Zhang, J. Wu, H. Zhou and Y. Tian, Spectrochim. Acta, Part A, 2014, 118, 538–542 CrossRef CAS PubMed.
- Z. Li, N. Pucher, K. Cicha, J. Torgensen, S. C. Ligon, A. Ajami, T. Husinsky, A. Rosspeintner, E. Vauthey, S. Naumov, T. Scherzer, J. Stampfl and R. Liska, Macromolecules, 2013, 46, 352–361 CrossRef CAS.
- N. Pucher, A. Rosspeintner, V. Satzinger, V. Schmidt, G. Gescheidt, J. Stampfl and R. Liska, Macromolecules, 2009, 42, 6519–6528 CrossRef CAS.
- W.-E. Lu, X.-Z. Dong, W.-Q. Chen, Z.-S. Zhao and X.-M. Duan, J. Mater. Chem., 2011, 21, 5650–5659 RSC.
- R. Nazir, P. Danilevicius, D. Gray, M. Farsari and D. T. Gryko, Macromolecules, 2013, 46, 7239–7244 CrossRef CAS.
- K. D. Belfield and K. J. Schafer, Photoinitiated Polymerization, ACS Symposium Series, 2003, vol. 847, ch. 39, pp. 464–481 Search PubMed.
- C. N. LaFratta, J. T. Fourkas, T. Baldacchini and R. A. Farrer, Angew. Chem., Int. Ed., 2007, 46, 6238–6258 CrossRef CAS PubMed.
- K. D. Belfield, K. J. Schafer, Y. Liu, J. Liu, X. Ren and E. W. van Stryland, J. Phys. Org. Chem., 2000, 13, 837–849 CrossRef CAS.
- (a) S. Maruo and J. T. Fourkas, Laser Photonics Rev., 2008, 2, 100–111 CrossRef CAS; (b) W. Wu, R. Tang, Q. Li and Z. Li, Chem. Soc. Rev., 2015, 44, 3997–4022 RSC; (c) J.-F. Xing, M.-L. Zheng and X.-M. Duan, Chem. Soc. Rev., 2015, 44, 5031–5039 RSC.
- S. Telitel, J. Lalevée, N. Blanchard, T. Kavalli, M. A. Tehfe, S. Schweizer, F. Morlet-Savary, B. Graff and J. P. Fouassier, Macromolecules, 2012, 45, 6864–6868 CrossRef CAS.
- M. A. Tehfe, F. Dumur, B. Graff, F. Morlet-Savary, J. P. Fouassier, D. Gigmes and J. Lalevée, Macromolecules, 2012, 45, 8639–8647 CrossRef CAS.
- S. Telitel, S. Schweizer, F. Morlet-Savary, B. Graff, T. Tschamber, N. Blanchard, J. P. Fouassier, M. Lelli, E. Lacôte and J. Lalevée, Macromolecules, 2013, 46, 43–48 CrossRef CAS.
- M. A. Tehfe, F. Dumur, B. Graff, J. L. Clément, D. Gigmes, F. Morlet-Savary, J. P. Fouassier and J. Lalevée, Macromolecules, 2013, 46, 736–746 CrossRef CAS.
- D.-L. Versace, F. Dalmas, J. P. Fouassier and J. Lalevée, ACS Macro Lett., 2013, 2, 341–345 CrossRef CAS.
- M. A. Tehfe, F. Dumur, B. Graff, D. Gigmes, J. P. Fouassier and J. Lalevée, Macromolecules, 2013, 46, 3332–3341 CrossRef CAS.
- M. A. Tehfe, F. Dumur, B. Graff, D. Gigmes, F. Morlet-Savary, J. P. Fouassier, D. Gigmes and J. Lalevée, Macromolecules, 2013, 46, 3761–3770 CrossRef CAS.
- P. Xiao, F. Dumur, T. T. Bui, F. Goubard, B. Graff, F. Morlet-Savary, J. P. Fouassier, D. Gigmes and J. Lalevée, ACS Macro Lett., 2013, 2, 736–740 CrossRef CAS.
- P. Xiao, F. Dumur, D. Thirion, S. Fagour, A. Vacher, X. Sallenave, F. Morlet-Savary, B. Graff, J. P. Fouassier, D. Gigmes and J. Lalevée, Macromolecules, 2013, 46, 6786–6793 CrossRef CAS.
- P. Xiao, F. Dumur, B. Graff, F. Morlet-Savary, L. Vidal, D. Gigmes, J. P. Fouassier and J. Lalevée, Macromolecules, 2014, 47, 26–34 CrossRef CAS.
- K. Belfield, Proceedings of the Radtech Europe Confernce, Vincentz Network, Hannover, 2005, vol. 2, pp. 75–82 Search PubMed.
- S. Dadashi-Silab, C. Aydogan and Y. Yagci, Polym. Chem., 2015, 6, 6595–6615 RSC.
- J. Torgersen, A. Ovsianikov, V. Mironov, N. Pucher, X. H. Qin, Z. Q. Li, K. Cicha, T. Machacek, R. Liska, V. Jantsch and J. Stampfl, J. Biomed. Opt., 2012, 17, 105008 Search PubMed.
- J. Torgersen, X. H. Qin, Z. Q. Li, A. Ovsianikov, R. Liska and J. Stampfl, Adv. Funct. Mater., 2013, 23, 4542–4554 CrossRef CAS.
- M. Chatzinikolaidou, S. Rekstyte, P. Danilevicius, C. Pontikoglou, H. Papadaki, M. Farsari and M. Vamvakaki, Mater. Sci. Eng., C, 2015, 48, 301–309 CrossRef CAS PubMed.
- V. Melissinaki, A. A. Gill, I. Ortega, M. Vamvakaki, A. Ranella, J. W. Haycock, C. Fotakis, M. Farsari and F. Claeyssens, Biofabrication, 2011, 3, 045005 CrossRef CAS PubMed.
- J. K. Gansel, M. Thiel, M. S. Rill, M. Decker, K. Bade, V. Saile, G. von Freymann, S. Linden and M. Wegener, Science, 2009, 325, 1513–1515 CrossRef CAS PubMed.
- Y.-S. Chen, A. Tal and S. M. Kuebler, Chem. Mater., 2007, 19, 3858–3860 CrossRef CAS.
- L. Amato, Y. Gu, N. Bellini, S. M. Eaton, G. Cerullo and R. Osellame, Lab Chip, 2012, 12, 1135–1142 RSC.
- M. H. Olsen, G. M. Hjorto, M. Hansen, O. Met, I. M. Svane and N. B. Larsen, Lab Chip, 2013, 13, 4800–4809 RSC.
- A. Radke, T. Gissibl, T. Klotzbucher, P. V. Braun and H. Giessen, Adv. Mater., 2011, 23, 3018–3021 CrossRef CAS PubMed.
- N. Vasilantonakis, K. Terzaki, I. Sakellari, V. Purlys, D. Gray, C. M. Soukoulis, M. Vamvakaki, M. Kafesaki and M. Farsari, Adv. Mater., 2012, 24, 1101–1105 CrossRef CAS PubMed.
- G. Kenanakis, A. Xomalis, A. Selimis, M. Vamvakaki, M. Farsari, M. Kafesaki, C. M. Soukoulis and E. N. Economou, ACS Photonics, 2015, 2, 287–294 CrossRef CAS.
- B. M. Monroe, W. K. Smothers, D. E. Keys, R. R. Krebs, D. J. Mickish, A. F. Harrington, S. R. Schicker, M. K. Armstrong, D. M. T. Chan and C. I. Weathers, Imaging Sci. J., 1991, 35, 19–25 CAS.
- J. Wu, Y. Zhao, X. Li, M. Shi, F. Wu and X. Fang, New J. Chem., 2006, 30, 1098–1103 RSC.
- S. S. Sarkisov, B. H. Peterson, M. J. Curley, V. N. Nesterov, T. Timofeeva, M. Antipin, E. I. Radovanova, A. Leyderman and P. A. Fleitz, J. Nonlinear Opt. Phys. Mater., 2005, 14, 21–40 CrossRef CAS.
- Y. Zhao, W. Wang, F. Wu, Y. Zhou, N. Huang, Y. Gu, Q. Zou and W. Yang, Org. Biomol. Chem., 2011, 9, 4168–4175 CAS.
- W. Yang, Q. Zou, Y. Zhou, Y. Zhao, N. Huang, Y. Gu and F. Wu, J. Photochem. Photobiol., A, 2011, 222, 228–235 CrossRef CAS.
- D. Kim, H. Moon, S. H. Baik, S. Singha, Y. W. Jun, T. Wang, K. H. Kim, B. S. Park, J. Jung, I. Mook-Jung and K. H. Ahn, J. Am. Chem. Soc., 2015, 137, 6781–6789 CrossRef CAS PubMed.
- X. Cheng, K. Wang, S. Huang, H. Zhang and Y. Wang, Angew. Chem., Int. Ed., 2015, 54, 8369–8373 CrossRef CAS PubMed.
- J. Xue, Y. Zhao, J. Wu and F. Wu, J. Photochem. Photobiol., A, 2008, 195, 261–266 CrossRef CAS.
- K. Yamashita, S. Imahashi and S. Ito, Dyes Pigm., 2008, 76, 748–753 CrossRef CAS.
- Q. Zou, Y. Zhao, N. S. Makarov, J. Campo, H. Yuan, D. C. Fang, J. W. Perry and F. Wu, Phys. Chem. Chem. Phys., 2012, 14, 11743–11752 RSC.
- R. Nazir, T. T. Meiling, P. J. Cywiński and D. T. Gryko, Asian J. Org. Chem., 2015, 4, 929–935 CrossRef CAS.
- B. Strehmel, A. M. Sarker and H. Detert, ChemPhysChem, 2003, 4, 249–259 CrossRef CAS PubMed.
- M. A. Tehfe, F. Dumur, B. Graff, D. Gigmes, F. Morlet-Savary, J. P. Fouassier, D. Gigmes and J. Lalevée, Macromolecules, 2013, 46, 3761–3770 CrossRef CAS.
- T. Kusumoto, D. Kosumi, C. Uragami, H. A. Frank, R. R. Birge, R. J. Cogdell and H. Hashimoto, J. Phys. Chem. A, 2011, 115, 2110–2119 CrossRef CAS PubMed.
- M. Planells and N. Robertson, Eur. J. Org. Chem., 2012, 4947–4953 CrossRef CAS.
- F. Terenziani, C. Katan, E. Badaeva, S. Tretiak and M. Blanchard-Desce, Adv. Mater., 2008, 20, 4641–4678 CrossRef CAS.
- F. Montigny, G. Argouarch and C. Lapinte, Synthesis, 2006, 293–298 Search PubMed.
- H. Cao, H. Zhan, J. Cen, J. Lin, Y. Lin, Q. Zhu, M. Fu and H. Jiang, Org. Lett., 2013, 15, 1080–1083 CrossRef CAS PubMed.
- P. Goswami and B. Das, Tetrahedron Lett., 2009, 50, 897–900 CrossRef CAS.
- R. Abonia, D. Insuasty, J. Castillo, B. Insuasty, J. Quiroga, M. Nogueras and J. Cobo, Eur. J. Med. Chem., 2012, 57, 29–40 CrossRef CAS PubMed.
- G. Audrey, D. Frederic, D. Eddy, M. Fabien, W. Guillaume and M. R. Cedric, Org. Lett., 2010, 12, 2382–2385 CrossRef PubMed.
- N. V. Vistorobskii and A. F. Pozharskii, Zh. Org. Khim., 1989, 25, 2154–2161 CAS.
- N. V. Vistorobskii, O. V. Nogradopa and A. F. Pozharskii, Russ. Chem. Bull., 1997, 46, 334–336 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 09, Revision D.01, Gaussian, Inc., Wallingford CT, 2013 Search PubMed.
- G. Lemercier, C. Martineau, J.-C. Mulatier, I. Wang, O. Stéphan, P. Baldeck and C. Andraud, New J. Chem., 2006, 30, 1606–1613 RSC.
- F. Momicchioli, G. Ponterini and D. Vanossi, Phys. Chem. Chem. Phys., 2014, 16, 15576–15589 RSC.
- K. M. Shafeekh, S. Das, C. Sissa and A. Painelli, J. Phys. Chem. B, 2013, 117, 8536–8546 CrossRef CAS PubMed.
- W. J. Yang, C. H. Kim, M.-Y. Jeong, S. K. Lee, M. J. Piao, S.-J. Jeon and B. R. Cho, Chem. Mater., 2004, 16, 2783–2789 CrossRef CAS.
- B. Strehmel, S. Amthor, J. Schelter and C. Lambert, ChemPhysChem, 2005, 6, 893–896 CrossRef CAS PubMed.
- D. S. Pedersen and C. Rosenbohm, Synthesis, 2001, 2431–2434 CAS.
- R. R. Tykwinski, M. Schreiber, R. P. Carlón, F. Diederich and V. Gramlich, Helv. Chim. Acta, 1996, 79, 2249–2281 CrossRef CAS.
- M. Samoc, A. Samoc, G. T. Dalton, M. P. Cifuentes, M. G. Humphrey and P. A. Fleitz, Two-photon absorption spectra and dispersion of the complex cubic hyperpolarizability γ in organic and organometallic chromophores, in Multiphoton Processes in Organics and Their Application, ed. I. Rau and F. Kajzar, Old City Publishing, Philadelphia, 2011, ch. 7, pp. 341–355 Search PubMed.
- C. A. Schneider, W. S. Rasband and K. W. Eliceiri, Nat. Methods, 2012, 9, 671–675 CrossRef CAS PubMed.
- N. S. Makarov, M. Drobizhev and A. Rebane, Opt. Express, 2008, 16, 4029–4047 CrossRef CAS PubMed.
- A. Ovsianikov, J. Viertl, B. Chichkov, M. Oubaha, B. MacCraith, I. Sakellari, A. Giakoumaki, D. Gray, M. Vamvakaki, M. Farsari and C. Fotakis, ACS Nano, 2008, 2, 2257–2262 CrossRef CAS PubMed.
- I. Sakellari, E. Kabouraki, D. Gray, V. Purlys, C. Fotakis, A. Pikulin, N. Bityurin, M. Vamvakaki and M. Farsari, ACS Nano, 2012, 6, 2302–2311 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Details of optical and computational studies and 1H NMR and 13C NMR spectra of all new compounds. See DOI: 10.1039/c5tc03334a |
| This journal is © The Royal Society of Chemistry 2016 |