
Catalytic role of solid acid catalysts in glycerol acetylation for the production of bio-additives: a review
Abstract
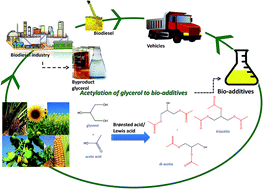
Bio-additives obtained from the acetylation of biodiesel-derived glycerol have been extensively synthesized because of their nature as value-added products and their contribution to environmental sustainability. Glycerol acetylation with acetic acid produces commercially important fuel additives. Considering that the recovery of individual monoacetin, diacetin (DA), and triacetin (TA) is complicated, many endeavours have enhanced the selectivity and total conversion of glycerol using acetic acid during catalytic acetylation. In this work, we extensively review the catalytic activity of different heterogeneous acid catalysts and their important roles in glycerol acetylation and product selectivity. In addition, the most influential operating conditions to attain high yields of combined DA and TA are achieved by closely examining the process. This review also highlights the prospective market, research gaps, and future direction of catalytic glycerol acetylation.
 Please wait while we load your content...
Please wait while we load your content...

