
Citrus hystrix-derived 3,7-dimethyloct-6-enal and 3,7-dimethyloct-6-enyl acetate ameliorate acetylcholine deficits
Shreesh Raj Sammia,
Durga Prasad Mishrab,
Shalini Trivedia,
Shachi S. Smitaa,
Abhishek Nagarc,
Sudeep Tandonc and
Rakesh Pandey*a
aDepartment of Microbial Technology and Nematology, CSIR-Central Institute of Medicinal and Aromatic Plants, Lucknow, 226015, India. E-mail: r.pandeycimap@gmail.com
bDivision of Endocrinology, CSIR-Central Drug Research Institute, Lucknow, 226031, India
cDepartment of Process Chemistry, CSIR-Central Institute of Medicinal and Aromatic Plants, Near Kukrail Picnic Spot, Lucknow, 226015, India
First published on 5th July 2016
Abstract
Cholinergic neurotransmission is an affliction in a plethora of neurodegenerative disorders such as Alzheimer’s, Huntington’s and Parkinson’s diseases and some psychiatric disorders like schizophrenia. The current management of many of these diseases relies heavily on treatments designed to elevate neurotransmission either through curtailing Ache activity or positive modulation of Ach receptors. The present study elaborates for the first time the potential and underlying mechanism of Kaffir lime oil (KLO) and its active constituents, citronellal (3,7-dimethyloct-6-enal) and citronellyl acetate (3,7-dimethyloct-6-enyl acetate), extracted from Citrus hystrix as potential therapeutic agents in the augmentation of cholinergic response in Caenorhabditis elegans. We observed significant elevation of synaptic Ach levels evident through aldicarb assay, being orchestrated through mitigation of Ache activity upon treatment with KLO, recorded at genomic and biochemical levels, along with elevated genomic expression of choline transporter. The active components citronellal and citronellyl acetate were also found to lower Ache activity at biochemical and transcriptomic levels, besides also executing their function through modulation of choline transporter and choline acetyltransferase at genomic levels. Paradoxically, other constituents beta caryophellene and linalool were found to show adverse effects at lower doses. Notably, all of the components of KLO and KLO itself were devoid of any interaction-based Ache inhibitory activity as determined through in vitro assay. Our study has identified and validated KLO and active components for their significant pharmaco-medical importance, presenting novel leads and a new line of drug action compared to the conventional Ache inhibition-based therapeutic approach.
1. Introduction
Acetylcholine (Ach) was the first neurotransmitter identified by Sir Henry Hallet Dale in the year 1914. Ach finds a significant role at neuromuscular junctions and in the autonomic and central nervous systems. Ach synthesis, release and reception are known to be affected in a plethora of neurotoxicological and neurodegenerative conditions. For instance, botulinum toxin is known to inflict damage by disrupting exocytosis of Ach in neuromuscular junctions,1 while atropine antagonizes the binding of Ach onto the muscarinic Ach receptors.2 In the context of neurodegenerative disorders, Ach is associated with Alzheimer’s3,4 Parkinson’s5,6 and Huntington’s diseases.7 Recent scientific advances also advocate the cholinergic system as an important target for therapy in schizophrenia.8 Amongst these disorders the cholinergic system is most severely afflicted in Alzheimer’s disease (AD) where manifestations pertaining to memory deficits arise due to the death of cholinergic neurons.9 In the presence of several debatable theories regarding AD onset, the cholinergic hypothesis is one of the oldest hypothesis and forms the basis of available and acceptable therapeutic interventions.3 The current management of AD is limited to symptomatic relief with the use of acetylcholine-esterase inhibitors such as donepezil, tacrine, rivastigmine and galantamine.10 On similar lines to the current therapies for disorders ensuing due to a dysfunctional cholinergic system, we investigated the efficacy of lower doses of Kaffir lime oil (KLO) and its constituents for their effect on cholinergic function. Kaffir lime (Citrus hystrix D.C.) is a fruit found in tropical parts of the Asian continent. KLO is extracted from the leaves of C. hystrix and finds use in Cambodian and Thai cuisine. Although the plant is known to possess some medicinal properties as well, the information regarding therapeutic use in alleviation of cholinergic dysfunction is totally obscure. Using a simple model organism Caenorhabditis elegans we evaluated the efficacy of KLO as a drug candidate in AD. The studies were extended towards investigating the underlying mechanism of KLO action and identification of the components responsible, presenting a novel scientific insight.2. Materials and methods
2.1. Culture and maintenance of strains
C. elegans strains, Bristol N2 (wild type), were procured from Caenorhabditis Genetics Center, (University of Minnesota, MN, USA) and grown on nematode growth medium (NGM) prepared by adding 50 mM sodium chloride (Hi Media Laboratories, India), 2.5 mg L−1 peptone (Hi Media Laboratories, India) and 17 mg L−1 agar (Hi Media Laboratories, India) in 975 mL double distilled water q.s. and autoclaved (Tomy High Pressure Steam sterilizer ES-215) for 40 minutes at 15 lb per inch2. 1 mL of 5 mg mL−1 cholesterol (Hi Media Laboratories, India) solution, 1 mM calcium chloride (Merk, Germany) (autoclaved), 1 mM magnesium sulphate (Merk, Germany) (autoclaved) and 25 mM potassium dihydrogen phosphate (Merk, Germany) (autoclaved) were added after the medium was cooled to 60 °C. The media was poured in Petri dishes. Escherichia coli OP50 was used as a food source. All experiments were conducted at 22 °C.2.2. Extraction of Kaffir lime oil
The oil content of the plants was determined by hydro-distillation of a C. hystrux plant using a Clevenger apparatus.11 Aerial parts comprising leaves of each replicate were cut into 2–3 cm long pieces and placed with 400 mL of water, in 1 litre capacity round bottom flasks of the Clevenger apparatus. Distillation was conducted at a 60 °C heating mantle temperature for 3 hours. The Kaffir lime oil (KLO) was collected, dehydrated (by anhydrous sodium sulphate), measured and kept in a cool and dark place prior to analysis. For gas chromatographic analysis, a Perkin Elmer gas chromatograph model Auto System XL GC equipped with a FID was used. Capillary Column EQUITY-5 (60 m × 0.32 mm i.d.) was used. The injector temperature and detector temperature was kept at 290 °C and 300 °C respectively. 0.04 μL neat oil was injected with hydrogen at 10 psi pressure used as carrier gas. The initial temperature in the oven program was 70 °C for 2.00 min with a ramp of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3.0 deg min−1 to 250 deg held for 2.00 min, then a ramp of 2:6.0 deg min−1 to 290 deg held for 5.00 min. In order to determine the percentage purity of the individual components of KLO, GC FID was conducted through the procedure described above using a Nucon gas chromatograph.
:3.0 deg min−1 to 250 deg held for 2.00 min, then a ramp of 2:6.0 deg min−1 to 290 deg held for 5.00 min. In order to determine the percentage purity of the individual components of KLO, GC FID was conducted through the procedure described above using a Nucon gas chromatograph.
2.3. Treatment of KLO and its components
A 1000 ppm stock solution of KLO and its components were prepared in 10% dimethyl sulphoxide (DMSO). Citronellal (Ci) and linalool (Li) were isolated from KLO while beta cryophyllene (BCP) and cetronellyl acetate (CA) was purchased from Sigma and Bush Boake Allen respectively. Individual components of Kaffir lime oil were isolated through a glass fractional column under high vacuum and low temperature. Enrichment of the components was done using column chromatography. Gas chromatography was used to ascertain the purity of all the components. The stock solution was accordingly diluted using E. coli OP50 culture (O.D. ∼ 0.3) for various concentrations i.e. 10, 20, 40, 60, 80 and 100 ppm. A total of 20 μL culture mixed with respective concentrations was seeded on 35 mm NGM plates. Plates were incubated overnight at 22 °C and used the next day.2.4. Toxicity assay
The toxicity assay was performed at 22 °C with different doses of KLO and its components (10, 20, 50, 100 and 500 ppm) to assess the lethal effect. The adult age synchronized wild type (N2) worms were transferred to a 24-well plate containing respective doses of the treatment with 1% DMSO serving as control. Percentage survival was observed every hour up to 24 h.2.5. Aldicarb assay
The aldicarb assay was done as described by Mahoney et al. 2006.12 Briefly the age-synchronized N2 embryos were added to treatment plates. Adult worms were washed using M9 buffer. A fixed number of worms (approx. 40) were transferred to 1 mM aldicarb (Sigma) NGM plates (prepared by mixing aldicarb with molten NGM just before pouring onto 24-well plates) and scored for paralysis every 30 minutes. Any worms lost or damaged were disregarded from the study. The percentage of worms paralyzed was calculated for the time point when approximately 50% of the worms were paralyzed in the control.2.6. Levamisole assay
The levamisole assay was done as described by Qian et al. 2008 13 with slight modifications. Briefly the age-synchronized N2 embryos were added to treatment plates. Adult worms were washed from the treatment plates. Approximately 40 worms were transferred to each well of 96-well plates. An equal volume of 50 μM levamisole (Sigma Aldrich U.S.A.) solution (dissolved in M9 buffer) was added to each well and the worms were scored for paralysis. The percentage of worms paralyzed was calculated for the time point when approximately 50% of the worms were paralyzed in control.2.7. Relative quantification of Ache levels
Ache levels were determined using Amplex Red Ach/Ache estimation kit (Life Technologies Co. U.S.A., Cat. no. A12217) according to the manufacturer’s protocol. Briefly, the age-synchronized embryos were added to treatment plates. Adult worms were washed and sonicated in 1× reaction buffer. The worm suspension was then centrifuged at 7000 rpm for 7 minutes. 100 μL of supernatant was added to 100 μL of reaction mixture which was prepared by adding 200 μL of 20 mM Amplex red solution, 100 μL of 200 U mL−1 HRP solution, 10 μL of 100 mM Ach solution and 100 μL of 20 U mL−1 choline oxidase solution in 10 mL q.s. of 1× reaction buffer in black well plates. After 30 minutes incubation at room temperature, fluorescence was measured using a fluorimeter (BMG Polarstar Omega) with excitation at 544 nm and emission at 590 nm. The relative fluorescence thus obtained was normalized using the protein content of the sample (calculated using the Bradford method and plotted against BSA standard ranging from 1 μg to 10 μg) to give relative Ache activity.2.8. Ache inhibition assay
The Ache inhibition assay was done using Amplex Red Ach/Ache estimation kit (Life Technologies Co. U.S.A., Cat. no. A12217). Briefly, different concentrations of KLO and donepezil hydrochloride (Intas Pharma. India) were subjected to estimation by addition to the reaction mixture (prepared by adding 200 μL of 20 mM Amplex red solution, 100 μL of 200 U mL−1 HRP solution, 10 μL of 100 mM Ach solution and 100 μL of 20 U mL−1 choline oxidase solution in 10 mL q.s. of 1× reaction buffer) in black well plates. The fluorescence was read every 5 minutes for 45 minutes using a 96-well plate fluorimeter (BMG Polarstar Omega) with excitation at 544 nm and emission at 590 nm. 10% DMSO was used as a control for KLO and its components while distilled water was used as a control for donepezil hydrochloride (dpz).2.9. RNA isolation, cDNA synthesis, and quantitative real-time PCR
Total RNA was extracted from adult age-synchronized animals using the RNAzol reagent (Molecular Research Centre) according to the manufacturer’s instructions. Briefly, worms were washed off treatment plates, crushed in RNAzol (Molecular Research Centre Inc., Cat. no. RN190) and vortexed for 10 minutes, followed by incubation at room temperature for 10 minutes. The vials were centrifuged at 14000 rpm for 10 minutes at 4 °C. The supernatant was collected and mixed with 100 μL of nuclease-free water (Hi-media). The vials were vortexed for 15 s and kept at room temperature for 15 minutes followed by centrifugation at 12000 rpm at 4 °C. RNA was precipitated from the supernatant by adding equal amount of isopropanol. The vials were centrifuged at 12000 rpm for 10 minutes and the pellet containing RNA was washed twice using 75% chilled ethanol at 6000 rpm for 50 minutes at 4 °C. The pellet was dried and dissolved in nuclease-free water followed by quantification using a nanodrop spectrophotometer.
cDNA synthesis was carried out from 1 μg of total C. elegans RNA in a 96-well thermal cycler using a High capacity cDNA synthesis Kit (Thermo Scientific) according to the manufacturer’s protocol. qRT-PCR studies were done using a Light Cycler 480 machine (Roche Diagnostics, Germany). Differential expression was calculated by the 2−ΔΔCT method. Gpd-1 was used as an internal control and used to normalize the ratios between samples.14 Primers were procured from Integrated DNA technologies, with details as shown in Table 1 (orientation of oligonucleotides: 5′ to 3′).
| Gene name | Primer sequence 5′ to 3′ | |
|---|---|---|
| 1 | gpd-1 F | AAA GTC ATT CCG GAG CTG AAC GGA |
| gpd-1 R | AGC GGC CTT GAC TAC CTT CTT GAT | |
| 2 | ace-1 F | CTG CTA CCA CGT CAG ATA C |
| ace-1 R | TGA GCC AGA GCC ATT TC | |
| 3 | ace-2 F | CTG TAG CTC ACC GGA TAT G |
| ace-2 R | GTC GCC CTG GAA GAA AT | |
| 4 | cho-1 F | GTC GCC CTG GAA GAA AT |
| cho-1 R | CCA CCG GTG AAT GTG TAG | |
| 5 | cha-1 F | GGG AAA GGG AAG AAA CGA |
| cha-1 R | GGA CCA CTG CAC CAT AC | |
| 6 | unc-17 F | ACC ATC ACA ACC TGG ATG TCC GAA |
| unc-17 R | TCC ATA GCC AAC CCA ACC ATA GCA | |
| 7 | unc-29 F | CGA GGA CCA AGA ACT CAT C |
| unc-29 R | ACT GTC CAA CTC CTG GTA | |
| 8 | unc-38 F | CGC TGA CAG CAA CTA CA |
| unc-38 R | CAG GAG CCG AAC TTC AA | |
| 9 | unc-50 F | CAT CCC AGT CAC CGA TTT |
| unc-50 R | TGA GAG CTT GGC GAA TG |
2.10. Statistical analysis
Statistical analysis and graphical representation of data was done using SPSS (Statistical Package for Social Sciences) version 15. Analysis of variance and independent t test was used to calculate statistical significance wherever applicable.3. Results
3.1. Studies on Kaffir lime oil
 | ||
| Fig. 1 Kaffir lime oil enhances cholinergic transmission by elevating synaptic Ach levels: (a) KLO treatment exhibited significantly increased synaptic Ach levels within the dose range 20 to 100 ppm as indicated by the aldicarb assay; (b) representative images of paralyzed and non-paralyzed worms upon exposure to aldicarb; black and white arrows indicate paralyzed and non paralyzed worms respectively; (c) KLO treatment was devoid of any significant effect on nAchR activity, evident from the levamisole assay conducted at doses 10, 20, 40, 60, 80 and 100 ppm; (d) representative images of paralyzed and non-paralyzed worms upon exposure to levamisole; black and white arrows indicate paralyzed and non paralyzed worms, respectively (*p value < 0.05, **p value < 0.005, ***p value < 0.005.); (e) toxicity assay for different doses of KLO indicated it to be safe within the dose range 10 to 500 ppm. | ||
 | ||
| Fig. 2 KLO lacks inherent Ache inhibitory activity and augments cholinergic transmission by down-modulation of ace-1 & ace-2 and upregulation of cho-1, unc-29 & unc-50: (a) 20 and 100 ppm KLO was devoid of any inherent Ache inhibitory activity in vitro; (b) KLO exhibited reduction in gross Ache activity at 20 ppm concentration in vivo; (c) effect of KLO on synaptic Ach levels was also compared with the known Ache inhibitor, donepezil hydrochloride (dpz), which exhibited increased Ach levels only at 1000 ppm and was devoid of any significant effect at 10 and 20 ppm with respect to the control (employing aldicarb assay); (d) 20 ppm KLO resulted in upregulation of cho-1 (high affinity choline transporter), unc-29 (acetylcholine transporter) and unc-50 (regulator of nicotinic acetylcholine receptor), while downregulating ace-1, ace-2 (genes coding for Ache) and cha-1 (choline acetyl transferase); (e) representative GC chromatogram of KLO; citronellal (3,7-dimethyloct-6-enal), beta caryophyllene (4,11,11-trimethyl-8-methylidenebicyclo[7.2.0]undec-4-ene), linalool (2-[(2S,5S)-5-ethenyl-5-methyloxolan-2-yl]propan-2-ol) and citronellyl acetate (3,7-dimethyloct-6-enyl acetate) were found as major components of Kaffir lime oil being 59.93%, 13.23%, 11.2% and 10.24%, respectively (*p value < 0.05, **p value < 0.005, ***p value < 0.005). | ||
226 ± 1038.10), we observed that worms treated with 20 ppm KLO (33233.81 ± 131.70, p = 0.006) exhibited significant lowering of Ache activity. However, the alteration in worms treated with 10 ppm KLO (51098.60 ± 7531.85, p = 0.587) was found to be statistically insignificant (Fig. 2b). The above results implied that KLO negatively modulated Ache activity in vivo.20 ppm KLO showed significant upregulation in the expression of cho-1 (5.47 ± 1.04, p = 0.026), unc-29 (2.16 ± 0.03, p = 0.001), unc-38 (1.66 ± 0.13, p = 0.025) and unc-50 (3.02 ± 0.08, p = 0.001) along with downregulation in the expression of ace-1 (−2.51 ± 0.42, p = 0.004) and ace-2 (−1.91 ± 0.12, p = 0.006). The expression of unc-17 (1.01 ± 0.01, p = 0.996) was close to normal as shown in Fig. 2d. qPCR studies indicated that KLO negatively modulates the expression of ace-1 and ace-2, while upregulating cho-1, unc-29, unc-38 and unc-50.
3.2. Studies on components of Kaffir lime oil
After identifying the individual components of Kaffir lime oil as citronellal (Ci), citronellyl acetate (CA), linalool (Li) and beta caryophyllene (BCP), we expanded our studies towards identification of the active component of KLO. The isolated/procured individual components of KLO were assessed for their purity using GC-FID. As indicated by the gas chromatography, the percentage purity of Ci, CA, BCP and Li were found to be 97.35%, 94.41%, 94.69% and 98.26%, respectively. The gas chromatography for Ci, CA, BCP and Li is shown in Fig. 3 along with their respective structures. | ||
| Fig. 3 Gas chromatography of individual components of KLO: (a) gas chromatograph of citronellal (percentage purity: 97.35%); (b) gas chromatograph of citronellyl acetate (percentage purity: 94.41%); (c) gas chromatograph of beta caryophyllene (percentage purity: 94.69%); (d) gas chromatograph of citronellal (percentage purity: 98.26%). | ||
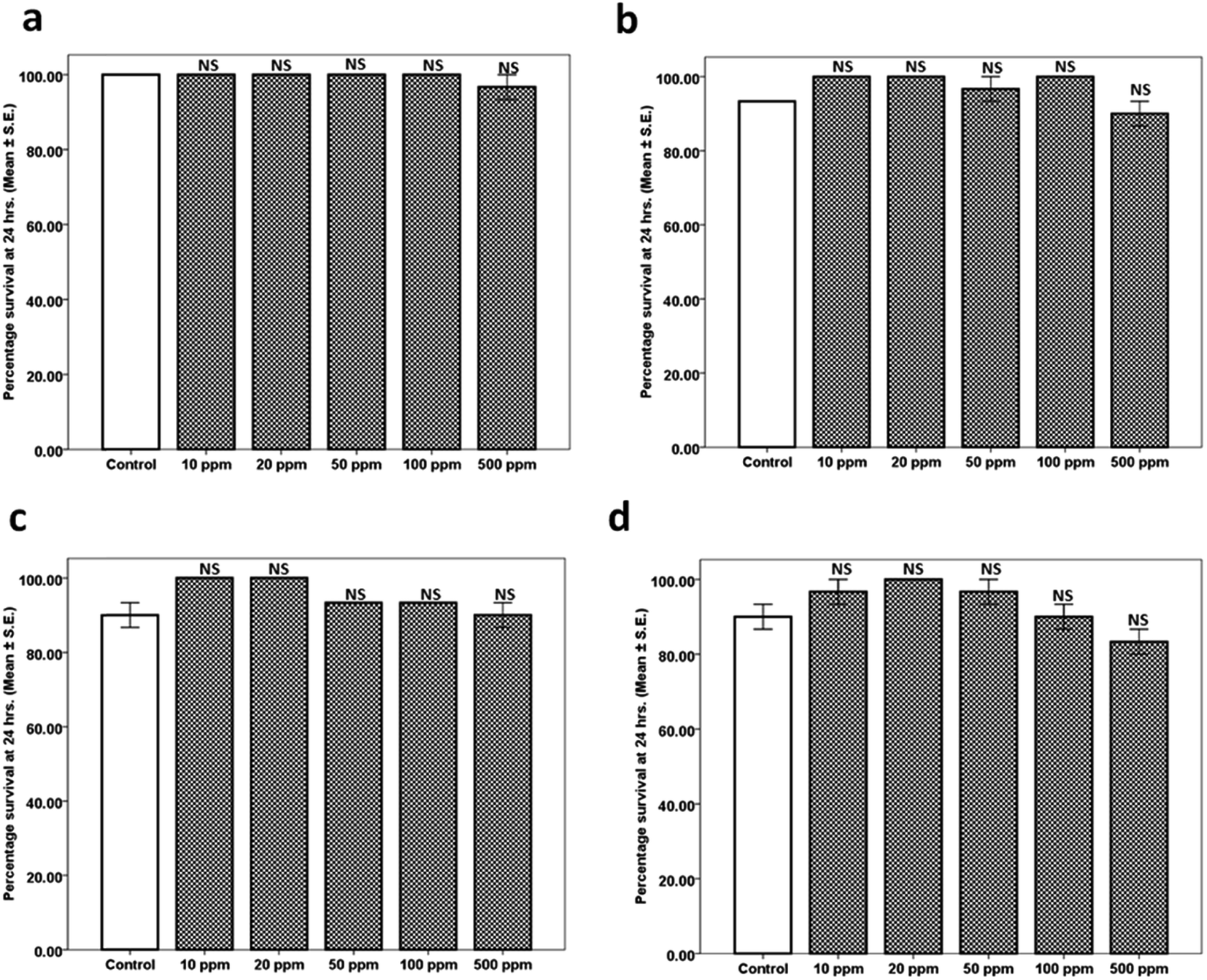 | ||
| Fig. 4 Toxicity assay for different concentrations of Ci, CA, BCP and Li: (a) Ci within the dose range 10, 20, 50, 100 and 500 ppm was devoid of any significant toxicity; (b) CA within the dose range 10, 20, 50, 100 and 500 ppm was devoid of any significant toxicity; (c) BCP within the dose range 10, 20, 50, 100 and 500 ppm was devoid of any significant toxicity; (d) Li within the dose range 10, 20, 50, 100 and 500 ppm was devoid of any significant toxicity. | ||
 | ||
| Fig. 5 Effect of citronellal and citronellyl acetate on synaptic Ach levels and nicotinic Ach receptor activity: (a) Ci exhibited significant augmentation of synaptic Ach levels within the dose range 10 to 100 ppm being highest at 40 ppm; (b) CA exhibited significant increase in synaptic Ach levels in a dose dependent manner at doses 10, 20, 40, 60, 80 and 100 ppm; (c) Ci was devoid of any alteration in nAchR activity at doses 10, 20, 40, 60 and 80 ppm whereas significantly increased nAchR activity was observed at 100 ppm; (d) CA did not show any alteration in nAchR activity (*p value < 0.05, **p value < 0.005, ***p value < 0.005). | ||
The next question that we addressed was to determine if Ci and CA exhibit any modulating effect on nAchR. Hence, the levamisole assay was performed for both Ci and CA in the dose range 10 to 100 ppm, where we observed that neither CA (Fig. 5d) nor Ci (Fig. 5c) possess any nAchR modulating effect, except for the treatment group of Ci at 100 ppm (63.62 ± 0.66, p = 0.007) which exhibited marginal yet significant increase in nAchR activity compared to its respective control (46.57 ± 1.57). The above results indicate that both Ci and CA are responsible for the beneficial effect of KLO which, however, is independent of nAchR activity. The aldicarb and levamisole assays collectively confirmed that Ci and CA elevate Ach levels, and the observed augmented neurotransmission is independent of nAchR participation.
 | ||
| Fig. 6 Effect of beta caryophyllene and linalool on synaptic Ach levels and nicotinic Ach receptor activity: (a) BCP exhibited significant curtailing of synaptic Ach levels at 10 and 20 ppm, whilst showing increased levels at 60, 80 and 100 ppm; (b) Li exhibited a significant decrease in synaptic Ach levels at 20, 40, 60 and 80 ppm; (c) BCP showed lowered nAchR activity at 10, 20 and 60 ppm; it lacked any significant alteration at 60, 80 and 100 ppm; (d) Li showed significant increase in nAchR activity at 100 ppm only (*p value < 0.05, **p value < 0.005, ***p value < 0.005). | ||
The next aim was to determine whether the results observed in the case of BCP and Li are dependent on nAchR. Hence, we performed the levamisole assay for BCP and Li where we observed that BCP showed significant lowering of nAchR activity (evident as percentage of worms paralyzed) at 10 ppm (24.59 ± 1.87, p = 0.006) and 20 ppm (25.30 ± 1.96, p = 0.007) compared to that of its respective control (51.66 ± 1.66). The difference in worms treated with BCP 40, 60, 80 and 100 ppm was found to be statistically insignificant (Fig. 6c). In the case of linalool, the variation in nAchR activity for the worms treated with 10, 20, 40, 60 and 80 ppm was statistically insignificant, except for 100 ppm (80.90 ± 0.90, p = 0.005) which showed elevated nAchR activity in comparison to the control (51.66 ± 1.66) (Fig. 6d). The studies for BCP and Li indicated that both BCP and Li exhibit down-modulation of neurotransmission at lower doses, being complemented through lowered nAchR activity only in the case of BCP.
450.27 ± 35.27, p = 0.030), Ci at 40 ppm (30200.23 ± 385.78, p = 0.016) and CA at 40 ppm (31167.10 ± 312.85, p = 0.021), compared to that of the control (46433.20 ± 272.73). The variation in Ache activity of the worms treated with Ca at 20 ppm was, however, statistically insignificant as shown in Fig. 7a, implying that Ci and CA down-modulate Ache activity.
 | ||
| Fig. 7 Effect of Ci and CA on gross Ache levels; effect of KLO components and donepezil on Ache inhibitory activity; (a) Ci treatment led to significant lowering of Ache activity at 20 and 40 ppm whereas CA exhibited lowering of Ache activity only at 40 ppm. The alteration in Ache activity at 20 ppm was found to be statistically insignificant; (b) dpz treatment exhibited elevated Ache inhibitory activity at 10, 20 and 1000 ppm in a dose-dependent manner; (c) Ci, BCP, CA and Li were devoid of any Ache inhibitory activity at 40 and 100 ppm (*p value < 0.05, **p value < 0.005, ***p value < 0.005). | ||
Donepezil was used as a positive control in this experiment, exhibiting attenuation of Ache activity in a concentration dependent manner at 10 ppm (23.87 ± 1.26, p = 0.147), 20 ppm (52.32 ± 10.45, p = 0.012) and 1000 ppm (79.38 ± 0.43, p = 0.002) in comparison to the control (0.00 ± 2.97) as shown in Fig. 7b.
40 ppm Ci treatment. Upon treatment with 40 ppm Ci, we observed a significant downregulation in the expression of ace-1 (0.82 ± 0.00, p = 0.032) along with upregulation in the expression of unc-17 (2.78 ± 0.03, p = 0.002), unc-38 (1.82 ± 0.15, p = 0.047) and unc-50 (1.34 ± 0.00, p = 0.003). The mRNA expression of the rest of the genes, ace-2, cho-1, cha-1 and unc-29, was close to normal (Fig. 8a).
 | ||
| Fig. 8 Effect of Ci, CA, Li and BCP on expression of genes related to Ach synthesis, transport, degradation and receptors; (a) 40 ppm Ci treatment resulted in upregulation of unc-17 (acetylcholine transporter), unc-38 (alpha subunit of nAchR) and unc-50 (regulator of the nicotinic acetylcholine receptor), while downregulating ace-1 (Ache); 60 ppm dose of Ci showed upregulation of ace-2 (Ache), unc-17 (acetylcholine transporter), unc-29 (non-alpha subunit of nAchR), unc-38 (alpha subunit of nAchR) and unc-50 (regulator of nicotinic acetylcholine receptor), while downregulating ace-1 (Ache); (b) CA at 40 ppm led to significant upregulation of cho-1 (choline transporter), cha-1 (choline acetyltransferase), unc-17 (Ach transporter) and unc-50 (regulator of nAchR trafficking), while downregulating ace-1 and ace-2 (genes coding for Ache); (c) Li at 20 ppm led to significant upregulation in the expression of ace-1 (Ache), unc-29 (non-alpha subunit of nAchR) and unc-50 (regulator of nAchR), while downregulating unc-17 (acetylcholine transporter); (d) BCP at 20 ppm resulted in significant upregulation of ace-1 and ace-2 (genes coding for Ache), cha-1 (choline acetyltransferase), unc-38 (alpha subunit of nAchR) and unc-50 (regulator of nAchR trafficking), while downregulating cho-1 (choline transporter) (*p value < 0.05, **p value < 0.005, ***p value < 0.005). | ||
60 ppm Ci treatment. Upon treatment with 60 ppm Ci, we observed a significant downregulation in the expression of ace-1 (0.61 ± 0.03, p = 0.004) along with upregulation in expression of ace-2 (1.57 ± 0.03, p = 0.001), unc-17 (1.63 ± 0.01, p = 0.032), unc-29 (2.02 ± 0.11, p = 0.008), unc-38 (3.54 ± 0.14, p = 0.002) and unc-50 (1.27 ± 0.03, p = 0.007). The alteration in the expression of rest of the genes, ace-2, cho-1 and cha-1, was statistically insignificant (Fig. 8a).
10 ppm CA treatment. Upon treatment with 10 ppm CA, we found significant downregulation in expression of ace-1 (0.80 ± 0.01, p = 0.012) and ace-2 (0.55 ± 0.10, p = 0.045) along with a significant upregulation in the expression of cho-1 (1.19 ± 0.02, p = 0.018), cha-1 (1.31 ± 0.04, p = 0.041) and unc-50 (1.48 ± 0.03, p = 0.001). The mRNA expression of the rest of the genes, unc-17, unc-29 and unc-38, was close to normal (Fig. 8b).
20 ppm CA treatment. Upon treatment with 20 ppm CA, we observed a significant downregulation in the expression of ace-1 (0.70 ± 0.02, p = 0.003) and ace-2 (0.56 ± 0.04, p = 0.046) along with a significant upregulation in the expression of cho-1 (1.16 ± 0.02, p = 0.030), cha-1 (1.65 ± 0.05, p = 0.005), unc-17 (3.85 ± 0.25, p = 0.002) and unc-50 (1.37 ± 0.014, p = 0.003). The variation in the mRNA expression of unc-29 and unc-38 was found to be statistically insignificant (Fig. 8b).
20 ppm Li treatment. The 20 ppm Li treatment showed significant upregulation in the expression of ace-1 (4.80 ± 0.41, p = 0.012), unc-29 (1.67 ± 0.04, p = 0.011) and unc-50 (1.91 ± 0.09, p = 0.012) along with downregulation in the expression of unc-17 (0.54 ± 0.00, p = 0.043). The alteration in the expression of genes ace-2, cho-1, cha-1 and unc-38 was statistically insignificant (Fig. 8c).
20 ppm BCP treatment. The 20 ppm BCP treatment showed significant upregulation in the expression of ace-1 (1.82 ± 0.08, p = 0.011), ace-2 (1.15 ± 0.00, p = 0.005), cha-1 (1.12 ± 0.02, p = 0.043), unc-38 (1.80 ± 0.11, p = 0.023) and unc-50 (1.44 ± 0.00, p = 0.007) along with downregulation in the expression of cho-1 (0.73 ± 0.00, p = 0.002). The alteration in the expression of unc-17 was statistically insignificant (Fig. 8d).
4. Discussion
Conduction of nerve impulses starts with synthesis of Ach in the presynapse, followed by its transport to the synapse16 where it binds to the specific receptor resulting in the conveying of action potential to the next neuron. The presence of a sufficient amount of Ach in the synapse and its interaction with the receptor is crucial for transmission of nerve impulses across the network of neurons. C. elegans exhibits adequate homology in relation to neurotransmission with mammals and offers a simple and reliable model for studying the aspects of neuronal function and mechanisms. Studies investigating the effect of Ache inhibitors have demonstrated the replication of results observed using rodents in C. elegans.17 We determined the effect of different concentrations of essential oils isolated from leaves and shoots of the C. hystrix plant on cholinergic function. The present study for the first time has identified and elaborated Kaffir lime oil, citronellal and citronellyl acetate as potential therapeutic agents with respect to human pathological conditions associated with cholinergic dysfunction. Initial studies on KLO as a whole employed the use of two assays: the aldicarb and levamisole assays. The former assay provides information regarding the synaptic Ach-mediated conduction of nerve impulses. Aldicarb being an Ache inhibitor12 causes accumulation of Ach by inhibition of Ache resulting in flexion of muscles. On the other hand the levamisole assay provides an index about the health of the nicotinic Ach receptor, a target of Ach. Levamisole, being an agonist to nAchR,18 leads to progressive paralysis. Higher percentage of worms paralyzed at a particular time point in the aldicarb and levamisole assays indicates increased synaptic Ach levels and nAchR responsiveness, respectively. Results from the aldicarb assay exhibit significantly elevated Ach levels at 20 ppm and above, however a slight downward trend (yet significantly higher that control) was observed upon increase of the dose, which could be accredited to either activation of homeostatic feedback mechanisms or some KLO components exhibiting adverse modulation. Next, we tried to address the question regarding the likely role of nAchR towards amassed neurotransmission. Results from the levamisole assay concluded with the absence of any nAchR involvement. In order to compare the efficacy of KLO with that of conventional drugs, we also studied the effect of dpz using the aldicarb assay at 10, 20 and 1000 ppm concentrations. Interestingly only 1000 ppm dpz exhibited significantly elevated levels of synaptic Ach. With our next objective being to deduce the mechanism involved, we questioned if KLO possesses any interaction-based Ache inhibitor activity similar to conventional drugs such as donepezil. Hence, we performed the Ache inhibition assay for 20 and 100 ppm concentrations of KLO. Interestingly, we observed that KLO was devoid of any inherent Ache inhibitor activity. The findings so far seemed quite lucrative since it established the efficacy of a low dose of KLO (20 ppm) in comparison to a relatively higher dose of dpz (1000 ppm) in promoting cholinergic neurotransmission. The above results indeed compelled further exploration for experimental clues so as to justify the observed activity of KLO.Hence, we performed qPCR studies to investigate the effect of 20 ppm KLO on the expression of genes pertaining to Ach synthesis, transport, degradation and nAchR. We observed a significant downregulation of genes ace-1 and ace-2, which code for Ache,19 along with upregulation in the expression of cho-1, which codes for high affinity choline transporters and is responsible for the transport of choline from synapse back to presynapse, acting as a rate limiting step in Ach synthesis.20 Paradoxically, we observed downregulation of cha-1 which codes for choline acetyltransferase (Alfonso et al., 1994). The downregulation of cha-1 could be attributed as a feedback response towards elevated Ach levels in the synapse. In addition, we also observed upregulation in the expression of unc-29 (non-alpha subunit of nAchR)21 and unc-50 (regulator of nAchR trafficking)22 with the expression of unc-17 and unc-50 close to normal. qPCR studies were sufficient to claim the mechanism to be down-modulation of acetylcholinesterase and the possible role of elevated cho-1. The observed upregulation in unc-29 and unc-50 could be ruled out in light of the results obtained from the levamisole assay which were devoid of any significant variation.
In order to completely justify our hypothesis regarding downregulation of ace-1 and ace-2 as the mechanism behind the observed phenotype we further directed our study from a genomic to a biochemical approach by quantifying total Ache activity in vivo, where we observed that 20 ppm KLO treatment resulted in significant lowering of Ache activity, corroborating the results obtained from the overall study. Our findings provided sufficient evidence in favor of KLO as a potential therapeutic agent for improvement of cholinergic neurotransmission.
4.1. Studies on Ci, CA, BCP and Li
The results obtained from the studies on KLO compelled further work towards identification of individual components and mechanisms thereof. Citronellal (Ci), citronellyl acetate (CA), beta caryophyllene (BCP) and linalool (Li) were identified as individual components with Ci comprising the highest fraction.We conducted the aldicarb and levamisole assays for Ci, CA, BCP and Li, leading to identification of Ci and CA as the components responsible. Ci, being the major component, exhibited the highest efficacy at 40 ppm. On the other hand, as expected, CA, being the minor component, exhibited an optimum efficacy at only 20 ppm. Surprisingly, neither Ci nor CA (like their parent mixture KLO) exhibited any significant effect on nAchR responsiveness, implying that the observed effects were due to enhanced synaptic Ach. Paradoxically, on the other hand, Li and BCP exhibited significant mitigation of aldicarb sensitivity at lower doses, indicating negative modulation of Ach levels. BCP reduced Ach levels at 10 and 20 ppm however, exhibited the reverse trend at 60, 80 and 100 ppm. On the other hand, Li curtailed synaptic Ach levels at 20, 40, 60 and 80 ppm (10 and 100 ppm Li being close to the control). We next questioned the probable role of nAchR in the case of BCP and Li. Interestingly, BCP exhibited lowered nAchR responsiveness at 10 and 20 ppm, coinciding with that of the aldicarb assay, implying the significant role of lowered nAchR responsiveness in reduced sensitivity towards aldicarb. Comparatively, Li lacked any significant alteration up to 80 ppm, rather showing elevated nAchR responsiveness at 100 ppm. The aldicarb and levamisole assays conducted for BCP and Li identified them as negative modulators of neurotransmission (at lower doses) and can also be linked with the slight downward trend observed for aldicarb sensitivity upon dose exaltation in the case of KLO treatment.
Since the results observed in the case of Ci and CA were independent of nAchR modulation, we speculated mechanisms similar to the parent mixture, KLO. Hence, we checked the effect of Ci and CA on gross Ache activity at 20 and 40 ppm. As expected, we observed significant lowering of Ache activity in case of Ci at both 20 and 40 ppm. However, a decrease in Ache activity in case of CA was observed only at 40 ppm. We expanded our studies by exploring for any interaction-based Ache mitigation compared to dpz and observed that all of the components, viz. Ci, BCP, CA and Li, are devoid of any Ache inhibitor activity at 40 and 100 ppm. Comparatively, dpz exhibited considerable dose dependent Ache inhibition at 10, 20 and 1000 ppm.
The studies were further extended in order to identify the genomic modulators effecting the observed activity in case of Ci, CA, BCP and Li, simultaneously drawing comparison with the observed synaptic Ach levels.
In case of Ci, where we observed the highest synaptic Ach at 40 ppm with a slight decrease at 60 ppm, we observed a decrease in mRNA expression of ace-1, which further decreased at 60 ppm. The observed downregulation of ace-1 corroborated the findings pertaining to reduced Ache activity. Paradoxically, we observed upregulation of ace-2 as the dose was increased from 40 to 60 ppm. The anomaly regarding ace-2 upregulation, which indeed is a contributing factor in Ache activity, could be justified on the basis of fact that ace-1 is the principle enzyme being expressed in body wall neurons,23 and is known to act as a major synaptic enzyme, reported in Pardosa pseudoannulata.24 We also observed an increase in the expression of unc-29, unc-38 and unc-50 upon change in concentration from 40 to 60 ppm. However, the upregulation in these genes, which contribute as structural and regulatory components of nAchR, can be ruled out in the light of insignificant nAchR responsiveness deduced through the levamisole assay. The results so far were lacking justification for the observed marginal decrease in Ach levels upon increase from 40 to 60 ppm Ci. We hypothesized that this could possibly be due to feedback inhibition which apparently could also be linked with the decrease in the upregulation of unc-17, which codes for acetylcholine transporters,25 (from 178% upregulation at 40 ppm to 63% upregulation at 60 ppm) along with upregulation of ace-2. The qPCR studies in the case of Ci pointed towards the plausible role of downregulated ace-1 and upregulation of unc-17 towards the execution of the observed phenomenon.
In case of CA we observed significant downregulation of both ace-1 and ace-2. The downregulation in ace-1 increased with an increase in dose (from 20% decrease at 10 ppm to 30% decrease at 20 ppm). Although a trivial upregulation was observed at both the concentrations, a significant dose-dependent upregulation was observed in the case of cha-1 and unc-17, which might very well be accredited as a possible mechanism for the efficacy of CA at lower doses compared to its counterpart Ci. These studies established upregulation of cha-1 and unc-17 along with downregulation of ace-1 and ace-2 as an apparent mechanism for the observed activity in the case of CA.
We also attempted to look for clues for lowered cholinergic function in the case of BCP and Li through qPCR studies. We observed a significant upregulation of ace-1 in the case of BCP. 20 ppm BCP exhibited significant downregulation in unc-17 along with upregulation of unc-29 and unc-50. The observed effect of BCP on aldicarb sensitivity could be attributed to the upregulation of ace-1 and downregulation of unc-17. BCP treatment also exhibited increased expression of unc-29 and unc-50. The qPCR studies on BCP explained the observed decrease in aldicarb sensitivity. The observed upregulation of unc-29 and unc-50 can be ruled out in light of the observed reduction in nAchR activity, indicating the possible role of some transcript-independent mechanisms.
In comparison to the BCP, Li also exhibited upregulation of ace-1 and downregulation of cho-1 along with upregulation in unc-29, unc-38 and unc-50. The effects observed upon Li treatment could be explained on the basis of upregulated ace-1 and downregulated cho-1, which codes for high affinity choline transporters and is a rate-limiting step in Ach synthesis.20 The summary of the events underlying effect of KLO, Ci, CA, BCP and Li has been depicted in Fig. 9.
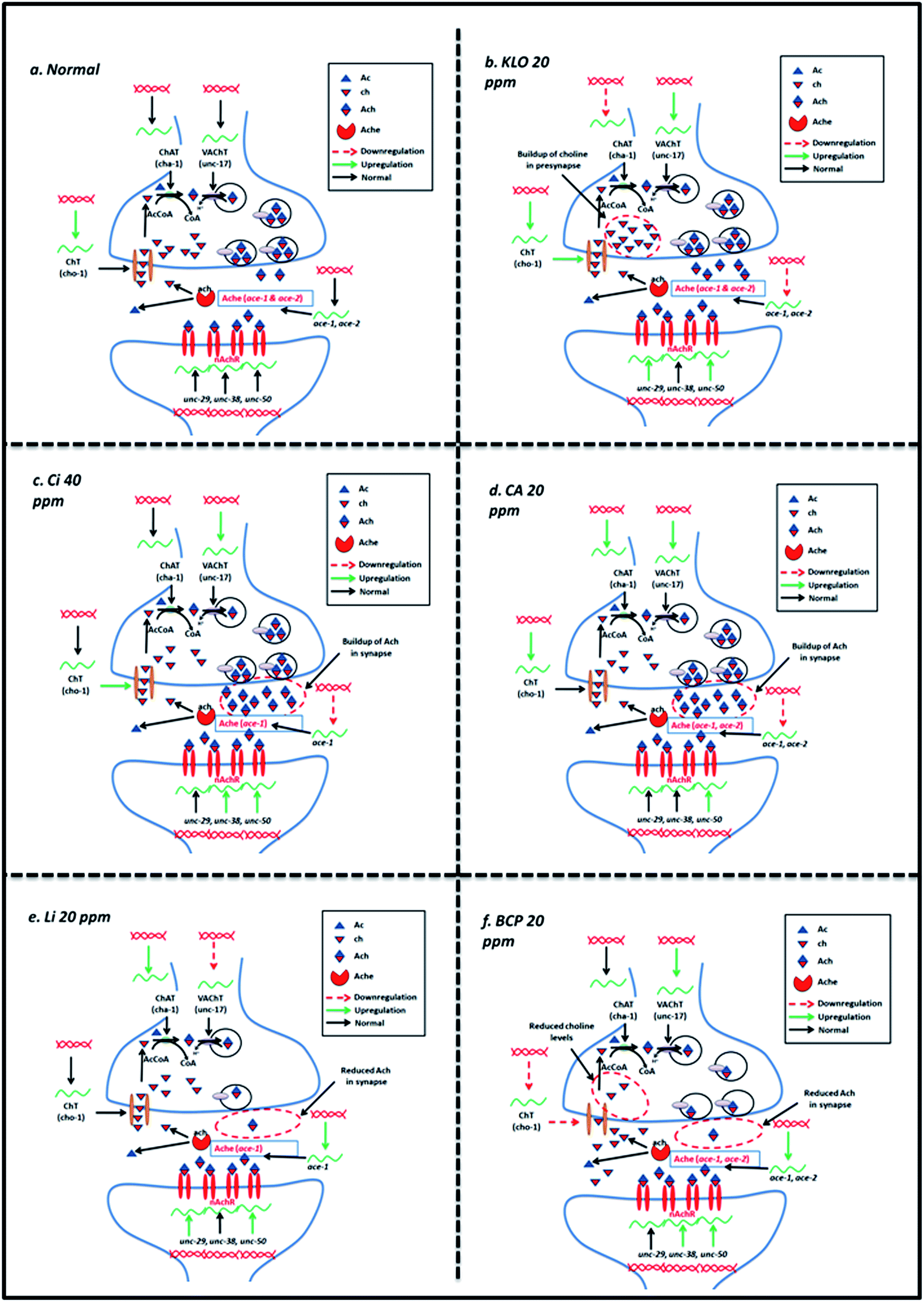 | ||
| Fig. 9 A schematic representation of the effects of KLO, Ci, CA, Li and BCP treatment on cholinergic transmission: (a) in the normal condition the choline transporter (encoded by cho-1) facilitates choline availaibility in the presynapse where it is converted to Ach by the action of choline acetyl transferase, ChAT (encoded by the gene cha-1). Ach is packaged and transported to the synapse by the vesicular acetylcholine transporter, VAChT (encoded by the gene unc-17). Acetylcholine binds to the acetylcholine receptor (ach receptor), allowing transmission of the nerve impulse. Acetylcholine present in the synapse is degraded by acetylcholinesterases (encoded by the genes ace-1 and ace-2). The choline thus generated is transported back to the presynapse; (b) 40 ppm KLO treatment stimulates the upregulation of cho-1, unc-17, unc-29 and unc-50 (indicated by green arrows) resulting in elevated uptake of choline to the presynapse for the synthesis of Ach. However, downregulation of cha-1 is observed (which can be attributed as a feedback inhibitory response towards enhanced Ach in the synapse). Upregulated unc-17 can facilitate enhanced transport of Ach to the synapse. The enhanced cholinergic function is also corroborated by downregulation of ace-1 and ace-2 (indicated by dotted red arrows); (c) 40 ppm Ci treatment results in the upregulation of unc-17, unc-38 and unc-50 (indicated by green arrows). The upregulation in unc-17 is expected to enhance transport of synthesized Ach from the presynapse to the synapse. Similarly, downregulation in ace-1 (indicated by the dotted red arrow) also corroborates the accumulation of Ach augmenting the neurotransmission; (d) 20 ppm CA treatment results in the upregulation of cho-1, cha-1, unc-17 and unc-50 (indicated by green arrows), possibly resulting in an elevated uptake of choline to the presynapse, synthesis of Ach and enhanced transport of synthesized Ach to the synapse, although the possibility of increased nAchR activity could be evidently ruled out from the observations recorded through the levamisole assay. The enhanced cholinergic function is further strengthened by the downregulation of ace-1 and ace-2 (indicated by dotted red arrows) leading to sustenance of transported Ach in the synapse; (e) Li at 20 ppm negatively modulates neurotransmission, possibly resulting from the upregulation of ace-1 contributing to the enhanced degradation of Ach in the synapse. Besides, it was also observed to downregulate unc-17, hence impairing the transport of synthesized Ach to the synapse. Upregulation in the expression of unc-29 and unc-50 does not seem to have an effect at the studied concentration but is expected to enhance nAchR activity at higher doses (100 ppm) as evident from the levamisole assay; (f) BCP at 20 ppm, which resulted in diminished neurotransmission, probably acted through the upregulation of ace-1 and ace-2 while downregulating cho-1, resulting in a decreased uptake of choline to the presynapse, hence impairing Ach synthesis. Upregulation in the expression of unc-38 and unc-50 does not seem to have an effect as evident from the mitigated nAchR activity in the levamisole assay. | ||
5. Conclusion
The present study has for the first time established the potential of Kaffir lime oil and identified its active components for ameliorative potential pertaining to cholinergic dysfunction, a critical and pharmacologically relevant manifestation of patients suffering from neurodegenerative disorders. The present study is speculated to promote research associated with the discovery of natural entities as novel therapeutic candidates, while also unleashing two crucial molecules, viz. 3,7-dimethyloct-6-enal and 3,7-dimethyloct-6-enyl acetate. Notably, Ci and CA share a similar pharmacophore, however CA supercedes Ci in terms of efficacy at relatively lower concentrations. These findings are interesting from the point of view of chemical modification of the identified leads for enhanced pharmacological properties such as augmented efficacy and bioavailability as well as towards research in light of a novel mode of action other than the conventional Ache inhibitory regimens. Since a majority of the previous studies exploring Ache inhibitors employ in vitro assays for discovery/identification of novel lead molecules, our study also highlights the importance of employing in vivo models as a better approach for the reason that a huge number of potential molecules are liable to remain hidden due to a lack of interaction-based Ache inhibitory potential. We speculate that our studies will certainly pave way for research using alternative in vivo models, thus reducing the burden on mammalian models in a cost and time effective manner.Conflict of interest
The authors declare no conflict of interest.Abbreviations
| Ach | Acetylcholine |
| Ache | Acetylcholinesterase |
| nAchR | Nicotionic acetylcholine receptor |
| KLO | Kaffir lime oil |
| Ci | Citronellal (3,7-dimethyloct-6-enal) |
| CA | Citronellyl acetate (3,7-dimethyloct-6-enyl acetate) |
| BCP | Beta caryophyllene (−4,11,11-trimethyl-8-methylidenebicyclo[7.2.0]undec-4-ene) |
| Li | Linalool (2-[(2S,5S)-5-ethenyl-5-methyloxolan-2-yl]propan-2-ol) |
| qPCR | Qualitative polymerase chain reaction |
Acknowledgements
Authors are highly grateful to Director CSIR-CIMAP, Lucknow for his encouragement. Nematode strains employed in this study were provided by the C. elegans Genetics Center (CGC) University of Minnesota, MN, USA, funded by the NIH National Center for Research Resources (NCRR). We are also thankful to CSIR New Delhi, for providing vital monetary support under the project – BSC-0117.References
- B. Li, N. P. Peet, M. M. Butler, J. C. Burnett, D. T. Moir and T. L. Bowlin, Small molecule inhibitors as countermeasures for botulinum neurotoxin intoxication, Molecules, 2010, 16, 202–220 CrossRef PubMed.
- J. C. Parker, D. Sarkar, M. W. Quick and R. A. Lester, Interactions of atropine with heterologously expressed and native alpha 3 subunit-containing nicotinic acetylcholine receptors, Br. J. Pharmacol., 2003, 138, 801–810 CrossRef CAS PubMed.
- P. T. Francis, A. M. Palmer, M. Snape and G. K. Wilcock, The cholinergic hypothesis of Alzheimer’s disease: a review of progress, J. Neurol., Neurosurg. Psychiatry, 1999, 66, 137–147 CrossRef CAS.
- R. Schliebs, Basal forebrain cholinergic dysfunction in Alzheimer’s disease – interrelationship with beta-amyloid, inflammation and neurotrophin signaling, Neurochem. Res., 2005, 30, 895–908 CrossRef CAS PubMed.
- M. L. Müller and N. I. Bohnen, Cholinergic dysfunction in Parkinson’s disease, Curr. Neurol. Neurosci. Rep., 2013, 13, 377 CrossRef PubMed.
- H. Hall, S. Reyes, N. Landeck, C. Bye, G. Leanza, K. Double, L. Thompson, G. Halliday and D. Kirik, Hippocampal Lewy pathology and cholinergic dysfunction are associated with dementia in Parkinson’s disease, Brain, 2014, 137, 2493–2508 CrossRef PubMed.
- R. Smith, H. Chung, S. Rundquist, M. L. Maat-Schieman, L. Colgan, E. Englund, Y. J. Liu, R. A. Roos, R. L. Faull, P. Brundin and Y. J. Li, Cholinergic neuronal defect without cell loss in Huntington’s disease, Hum. Mol. Genet., 2006, 15, 3119–3131 CrossRef CAS PubMed.
- A. Gibbons and B. Dean, The cholinergic system: an emerging drug target for schizophrenia, Curr. Pharm. Des., 2016, 22, 1–10 CrossRef.
- H. Wang, H. Wang, H. Cheng and Z. Che, Ameliorating effect of luteolin on memory impairment in an Alzheimer’s disease model, Mol. Med. Rep., 2016, 13, 4215–4220 Search PubMed.
- M. Pohanka, Cholinesterases, a target of pharmacology and toxicology, Biomed. Pap., 2011, 155, 219–229 CrossRef CAS PubMed.
- J. F. Clevenger, Apparatus for determination of volatile oils, J. Am. Pharm. Assoc., 1928, 17, 345–349 Search PubMed.
- T. R. Mahoney, S. Luo and M. L. Nonet, Analysis of synaptic transmission in Caenorhabditis elegans using an aldicarb-sensitivity assay, Nat. Protoc., 2006, 1, 1772–1777 CrossRef CAS PubMed.
- H. Qian, A. P. Robertson, J. A. Powell-Coffman and R. J. Martin, Levamisole resistance resolved at the single-channel level in Caenorhabditis elegans, FASEB J., 2008, 22, 3247–3254 CrossRef CAS PubMed.
- K. J. Livak and T. D. Schmittgen, Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) method, Methods, 2001, 25, 402–408 CrossRef CAS PubMed.
- T. Kihara and S. Shimohama, Alzheimer’s disease and acetylcholine receptors, Acta Neurobiol. Exp., 2004, 64, 99–105 Search PubMed.
- S. Tuchek, V. Dolezhal and I. Richny, Regulation of acetylcholine synthesis in presynaptic endings of cholinergic neurons of the central nervous system, Neirofiziologiia, 1984, 16, 603–611 CAS.
- S. S. Chaudhaery, K. K. Roy, N. Shakya, G. Saxena, S. R. Sammi, A. Nazir, C. Nath and A. K. Saxena, Novel carbamates as orally active acetylcholinesterase inhibitors found to improve scopolamine-induced cognition impairment: pharmacophore-based virtual screening, synthesis, and pharmacology, J. Med. Chem., 2010, 9, 6490–6505 CrossRef PubMed.
- S. J. Robertson and R. J. Martin, Levamisole-activated single-channel currents from muscle of the nematode parasite Ascaris suum, Br. J. Pharmacol., 1993, 108, 170–178 CrossRef CAS PubMed.
- D. Combes, Y. Fedon, J. P. Toutant and M. Arpagaus, Acetylcholinesterase genes in the nematode Caenorhabditis elegans, Int. Rev. Cytol., 2001, 209, 207–239 CAS.
- T. Okuda, T. Haga, Y. Kanai, H. Endou and T. Ishihara, et al., Identification and characterization of the high-affinity choline transporter, Nat. Neurosci., 2000, 3, 120–125 CrossRef CAS PubMed.
- J. T. Fleming, M. D. Squire, T. M. Barnes, C. Tornoe and K. Matsuda, et al., Caenorhabditis elegans levamisole resistance genes lev-1, unc-29, and unc-38 encode functional nicotinic acetylcholine receptor subunits, J. Neurosci., 1997, 17, 5843–5857 CAS.
- S. Eimer, A. Gottschalk, M. Hengartner, H. R. Horvitz and J. Richmond, et al., Regulation of nicotinic receptor trafficking by the transmembrane Golgi protein UNC-50, EMBO J., 2007, 26, 4313–4323 CrossRef CAS PubMed.
- E. Culetto, D. Combes, Y. Fedon, A. Roig, J. P. Toutant and M. Arpagaus, Structure and promoter activity of the 5’ flanking region of ace-1, the gene encoding acetylcholinesterase of class A in Caenorhabditis elegans, J. Mol. Biol., 1999, 30(290), 951–966 CrossRef PubMed.
- Y. Zhang, Y. Shao, F. Jiang, J. Li and Z. Liu, Identification of pseudoannulata and the sensitivity to insecticides, Insect Biochem. Mol. Biol., 2014, 46, 25–30 CrossRef PubMed.
- A. Alfonso, K. Grundahl, J. S. Duerr, H. P. Han and J. B. Rand, The Caenorhabditis elegans unc-17 gene: a putative vesicular acetylcholine transporter, Science, 1993, 261, 617–619 CAS.
| This journal is © The Royal Society of Chemistry 2016 |