
Unravelling the mechanisms of nanomedicines: analytical tools to characterise the interaction between synthetic macromolecules and lipid membranes
Ayomi Vidana Pathiranage
a,
Mark-Jefferson Buer Boyetey
 bc,
Oluwatoosin B. A. Agbaje
bc and
Nathan R. B. Boase
*bc
bc,
Oluwatoosin B. A. Agbaje
bc and
Nathan R. B. Boase
*bc
aSchool of Bio-Chemical Engineering and Technology, Sirindhorn International Institute of Technology, Thammasat University, Pathum Thani 12120, Thailand
bCentre for Materials Science, Queensland University of Technology, Brisbane, QLD, Australia. E-mail: nathan.boase@qut.edu.au
cSchool of Chemistry and Physics, Queensland University of Technology, Brisbane, QLD, Australia
First published on 13th November 2025
Abstract
Polymer nanomedicines are a transformative class of therapeutics, offering tunable architectures for targeted delivery, controlled release, and improved pharmacokinetics. However, limited understanding of how these systems interact with cellular membranes, undergo internalization, and release their cargo continues to hinder their clinical translation. Membrane interactions are a key determinant of these processes yet remain underexplored in nanomedicine research. This review provides an overview of analytical techniques used to study the interaction of polymers with lipid membranes, drawing on methods from biophysics, physical chemistry, and colloid science. We introduce common model membrane systems and explain how they can complement in vitro studies. A wide range of characterization approaches are discussed, from microscopy and spectroscopy to more advanced scattering and nanomechanical techniques. By illustrating how these methods can be integrated to build a complete mechanistic understanding of dynamic interfacial behaviour, this review aims to bridge disciplinary gaps and support the development of more effective nanomedicines.
 Ayomi Vidana Pathiranage | Ayomi Vidana Pathiranage is a researcher and tutor based in United Arab Emirates. She obtained her M. Sc. in Engineering and Technology from the Sirindhorn International Institute of Technology, Thammasat University, Thailand. Her current research focuses on the molecular mechanisms of nanomedicines, particularly the interactions between synthetic macromolecules and lipid membranes. By combining molecular modeling, medicinal and polymer chemistry, her work aims to investigate how nanoscale materials interact with biological systems to inform the rational design of advanced drug delivery systems and therapeutic agents. |
 Mark-Jefferson Buer Boyetey | Mark-Jefferson Buer Boyetey is a Ph.D. student at the Queensland University of Technology (QUT), Australia, and a member of the Medicinal Molecules and Materials Group. His doctoral research focuses on the design, synthesis and characterization of antiviral polymers, with an emphasis on elucidating their interactions with biological membranes. By integrating principles of polymer chemistry and biophysics, his work seeks to uncover structure–activity relationships governing antiviral efficacy. His research aims to advance the rational development of functional polymeric materials for biomedical and antiviral applications. |
 Oluwatoosin B. A. Agbaje | Oluwatoosin B. A. Agbaje is a research fellow at the Queensland University of Technology and a member of the Centre for Materials. He earned his Ph.D. at Macquarie University, Sydney, where he studied biomineralization processes and designed biomimetic materials. In 2019, he transferred a Swedish VR grant/Uppsala University project to Australia. He was awarded a Marie Curie individual Fellowship in 2021 and a GEOCENTER Denmark startup grant in 2024 at the GLOBE Institute, University of Copenhagen, Denmark. His current research focuses on the sustainable synthesis of biopolymer-derived materials and nanoscale characterization of synthetic ceramics, supporting research and development in radiopharmaceutical applications. |
 Nathan R. B. Boase | Nathan R. B. Boase is a senior lecturer at the Queensland University of Technology and a member of the Centre for Materials Science, and a co-leader of the Medicinal Molecules and Materials Group. He completed his Ph.D. at the University of Queensland in 2015. In 2019 he was recognized as a CAS Future Leader in chemistry and in 2023 with a AIPS Tall Poppy Science Award. His current research explores high-throughput synthesis and machine learning to design materials that respond to their environment. These materials are applied to solve significant challenges in healthcare, such as antibiotic coatings, nanomedicines and antiviral therapies. |
Introduction
Polymeric nanomedicines are important technologies being developed in biomedical science for targeted therapeutic and diagnostic applications, due to their ease of chemical modification, biocompatibility, and ability to interact selectively with biological systems.1,2 Polymers can act as carriers to protect the active pharmaceutical ingredient (API, e.g. genes, proteins or drugs) and improve their targeted biodistribution to particular cells or tissues. Polymers can also be the active pharmaceutical ingredient themselves, harnessing their unique physicochemical properties to alter the native physiology of a cell or tissue, to elicit a therapeutic response. However, for polymers employed in nanomedicine, it is critical to understand their physicochemical interactions with cellular components and tissues, in order to identify the major obstacles that must be addressed for effective clinical translation.3 The specific physicochemical interaction of any given polymer nanomedicine is going to be defined by its intended therapeutic application.Polymers are commonly developed as drug delivery vectors, enabling encapsulation, and controlled release for site-specific targeting to enhance efficacy or decrease side effects.4 Polyethylene glycol is widely established as a biocompatible polymer, able to reduce fouling and enhance circulation of therapeutics, with polyoxazoline5,6 and polysarcosine7,8 materials emerging as attractive alternatives. Biodegradable and bioabsorbable polymers such as poly(lactic acid), poly(glycolic acid), and their copolymers are biocompatible and have the added advantage of being able to safely degrade within the body to small metabolites.4 Polymeric nanocarriers can be designed to take advantage of biological triggers including pH, enzymes or temperature to induce drug release specifically at sites of disease.4 It is important to consider how the changing chemistry of these responsive systems is going to dynamically impact on cell and tissue physiology.9 Often overlooked is how subtle changes in the architecture or chemical structure of the polymer may alter its propensity to interact with cell membranes and the effect this may have on drug delivery efficacy.10
Polymer nanomedicines are being investigated as next generation gene delivery agents, building on the success of lipid nanoparticles. Nanocarriers are critical for gene delivery, to prevent degradation of the genetic material before delivery to the desired site of action. Cationic polymers have been investigated widely for gene delivery, commonly polyethyleneimine (PEI) and polyamidoamine (PAMAM) dendrimers, due to their ability to complex with negatively charged small interfering ribonucleic acid (siRNA).11 These complexes protect the siRNA from degradation and facilitate the internalization of siRNA into cells.11 Critically, their interaction with cellular lipid membranes plays a crucial role in defining delivery efficiency. It has been shown that PEI and PAMAM cross cell membranes through different internalization pathways. However, successful cellular uptake alone is not sufficient for therapeutic action.11 Upon internalization, nanocarriers are often entrapped within endosomal vesicles, where the cargo risks degradation as they mature into lysosomes. Endosomal escape is therefore a crucial step that directly impacts therapeutic efficacy, to allow the siRNA to reach the cytoplasm to exert its biological effect.12 Understanding how endosomal escape occurs for cationic polymers is an area of debate.3,13,14 Therefore, detailed understanding of the full sequence of interactions from ligand binding, internalization, polymer–membrane interactions, and endosomal escape is essential for understanding this mechanism, and in turn designing better gene delivery systems.15
Polymer nanomedicines can be more than just carriers; they can function directly as the active pharmaceutical ingredient. Antimicrobial polymers are an excellent example of this class. Antimicrobial polymers have been engineered to selectively disrupt the membranes of bacteria, fungi, or viruses, providing a mechanism of action to minimize the risk of resistance developing.16 Cationic polymers such as poly(ethyleneimine) (PEI), poly[2-(N,N-dimethylamino)ethyl methacrylate] (PDMAEMA), and polydimethylsiloxane (PDMS)-based derivatives are among the most widely investigated antimicrobial agents.17 Recently, polysiloxane-methacrylate copolymers have attracted attention due to their unique physicochemical properties and high antimicrobial activity.18 Their efficacy depends on multiple factors, including the type and density of antimicrobial functional groups, alkyl chain length, and the structure of the counterion.19–22 These copolymers are effective against both Gram-positive and Gram-negative bacteria, with higher potency observed against Gram-positive strains. This is likely due to differences in cell wall structures, as Gram-negative bacteria possess an outer membrane that can act as a barrier. However, polymers can exhibit varying affinities toward Gram negative bacterial membranes and can cause significant disruption to the outer membrane of Gram-negative bacteria, contributing to their antimicrobial effect.17 Antimicrobials polymers have been designed to be potent against microbiota, while exhibiting low toxicity toward mammalian cells due to differences in lipid membrane composition.
In the context of antimicrobial polymers, membrane disruption is relatively well-characterized for bacterial cells due to their structurally distinct lipid compositions, however, interactions with viral and mammalian membranes are far less predictable, raising questions about mechanisms and concerns about cytotoxicity. There is a growing body of work demonstrating that polymers and nanoparticles can be potent virucidals.23–28 It has been proposed that multivalent binding of viral proteins by anionic sulfonate ligands on these nanomaterials, leads to deformation of the viruses and irreversible viral deactivation, rather than reversible competitive inhibition seen with small molecule or softer ligands.23 While evidence about the length of spacers between nanomaterial and ligand has been used to support this hypothesis, there have been conflicting results presented in these reports that are not entirely consistent with this hypothesis.27 Recent work has shown that purely amphipathic copolymers with long alkyl sidechains can be antiviral, and that addition of sulfonate ligands does not further potentiate their activity.29 This may suggest that increasing linker length in previous reports may increase hydrophobicity of the materials tested, contributing to lipid membrane interactions and measured antiviral activity. While these reports have focused on in vitro analysis to measure antiviral activity, further detailed characterisation of the biophysical interactions of these antiviral materials with viral particles and models of their lipid envelopes are needed to fully elucidate their antiviral mechanisms.
Despite the growing promise of polymer nanomedicines, this short survey of the literature has highlighted a major gap in understanding how these materials interact with lipid membranes at the molecular level. Given these challenges, it's essential to deepen the mechanistic insight into biophysical polymer–membrane interactions using robust, reproducible analytical tools. While in vitro cellular assays offer valuable biological information, they often lack the precision to capture specific biophysical processes. Hence, model membrane systems offer powerful complementary platforms to investigate polymer activity at the molecular level. Such systems enable controlled studies of membrane disruption, fusion, binding affinity, and permeability changes, helping to link structure–function relationships. Analytical techniques can be designed to precisely determine the energy and structure of the polymer–membrane complex at thermodynamic equilibrium, or to follow dynamic processes in real time to understand kinetic processes. Using models allows for precise control over the composition of the lipid membrane, to understand the role individual lipids play in driving interactions in a complex membrane. This review focuses on the biophysical characterization techniques for polymer–membrane interactions using model systems, with the aim of designing better polymeric nanomedicines across gene therapy, drug delivery, antimicrobials, and diagnostics.
Models of cellular membranes
In vitro studies of the membranes of living cells
Studying the membranes of living cells presents significant challenges due to their complex and dynamic nature.30 Probing the interaction of synthetic macromolecules with cellular membranes in vitro aims to provide insights into their function within complex biological systems. Various imaging and fluorescence-based techniques, including confocal microscopy, fluorescence microscopy, and membrane-permeabilization assays are commonly used to investigate polymer localization and morphological changes in cellular membranes.31–33 Fluorescence confocal microscopy can be applied to both fixed and live cells and is particularly advantageous for directly visualising interactions between polymers and cell membranes, or for detecting subtle changes in environmental conditions (for example pH, viscosity, and molecular binding). Fluorescence confocal microscopy achieves these measurements by detecting fluorophore decay times independently of concentration and excitation intensity.34 While confocal microscopy is useful for identifying location and spatial distribution of the polymers, fluorescent labelling can alter polymer behaviour and introduce artefacts.35,36 In live-cell imaging, photobleaching and phototoxicity can affect cell viability and signal quality, limiting its ability to follow dynamic events. Furthermore, fluorescence intensity is only semi-quantitative and can be influenced by dye concentration, photobleaching, cellular environment, and instrument settings. Other fluorescent based assays are designed to overcome some of these limitations by significantly enhancing signal stability, reproducibility, and the accuracy of quantitative measurements.Fluorescence assays have been developed to measure the dynamic processes of cell membrane disruption by polymers. For example, SYTOX Green is a cationic DNA binding dye that only enters cells with damaged membranes, making it a useful tool for detecting membrane disruption. SYTOX Green staining has been used to show that polyhexamethylene biguanide (PHMB), a cationic polymer disinfectant can disrupt nuclear membranes and binds to intracellular DNA. This suggested successful endosomal escape, a critical step for therapeutic polymer efficiency.31 However, this method does not provide any insight into early events of membrane disruption or specific polymer-membrane interactions. Another commonly used method is the lactate dehydrogenase (LDH) leakage assay, which detects membrane damage upon polymer binding. LDH assays are useful for confirming significant permeability and amphiphilic translocation, able to distinguish between large pores (exceeding the hydrodynamic diameter of LDH ∼8.4 nm) which allows complete enzyme leakage, and smaller transient pores that may permit ions or small molecule passage, and amphiphilic translocation in which polymers traverse the membrane without the formation of discrete pores.32 However, they cannot identify the precise site of membrane disruption or reveal the mechanism by which the disruption occurs. Haemolysis assays are also widely used to evaluate the membrane-disruptive activity of polymers under pH conditions that mimic the endosomal environment.37,38 Erythrocytes are incubated with the material under investigation, and membrane disruption is quantified by measuring haemoglobin release spectrophotometrically.39 While haemolysis assays offer a cost effective, sensitive and simple platform for screening such activity, their physiological relevance is limited, as RBC membranes differ significantly in lipid composition and curvature from other cell types and endosomes. Haemolysis assays measure only the end-point release of haemoglobin, therefore offers limited mechanistic insight into the molecular nature of the polymer–membrane interaction. Factors such as incubation time, erythrocyte concentration, and nanoparticle aggregation can significantly affect the results.39 These examples highlight the limitations of relying solely on complex in vitro experimentation. It spotlights the opportunity for complementary approaches when interpreting polymer-membrane interactions in biologically relevant environments.
Possible high-resolution approaches to characterize the morphological and structural features of in vitro cell membranes are electron microscopy (EM) based techniques. Scanning electron microscopy (SEM) and transmission electron microscopy (TEM) enable detailed visualization of surface topography, pore architecture, and internal layering in polymeric membranes at the nanometre scale, in contrast to the micrometre-scale resolution typically used for whole-cell imaging. Despite these advantages, EM techniques are not without limitations. Biological specimens, including membranes, are predominantly composed of water, necessitating careful sample preparation in a way that prevents structural collapse during dehydration under the vacuum conditions required for EM imaging. Moreover, biological materials primarily consist of light atoms, and the electron density of proteins is close to that of vitrified ice, and so these samples often exhibit inherently low contrast.40,41 To enhance imaging contrast and improve visibility, thin biological membranes are often subjected to staining, gold labelling of proteins and lipids, dehydration, or coating with conductive materials.42,43 While these steps are essential for effective imaging, these processes can significantly alter the native architecture of hydrated or soft membranes. The vacuum environment necessary for EM operation may also induce shrinkage or collapse of delicate structures. TEM, particularly cryo-electron microscopy, offers superior spatial resolution for biological samples, however it presents certain challenges when applied to macromolecular systems. These include radiation sensitivity of materials, conformational flexibility, and difficulties in orientation determination, especially in particles exhibiting preferred orientation or low contrast. Although molecules of 500 kDa and above are within the resolution range of modern cryo-EM techniques, accurate orientation assignment can still be problematic depending on sample preparation and intrinsic particle properties.44,45 Additionally, EM provides static images and lacks the capability to monitor dynamic processes in real-time, highlighting the need for complementary techniques to achieve a comprehensive understanding of membrane function.
To further investigate the polymer–membrane interactions beyond spatial localization, and to overcome complexity and variability of living cell membranes, model lipid membranes are complementary alternatives. While they may not accurately recapitulate every aspect of a living cell membrane, model lipid platforms allow for controlled manipulation of membrane composition, membrane structure, and the local environment. With simpler structures and ability to produce them at scale on a variety of interfaces, they can enable detailed analysis of the binding, insertion, and membrane disruption mechanisms of synthetic macromolecules under well-defined conditions. What they lack in realistic complexity is offset by the ease of measurements, analysis and interpretation of physicochemical processes. There are different models that can be used, that increase in the complexity of their composition and architecture, each with their own unique pros and cons (Fig. 1). It is critical to understand the complementary role of each of these models, as well as their suitability to different analytical techniques.
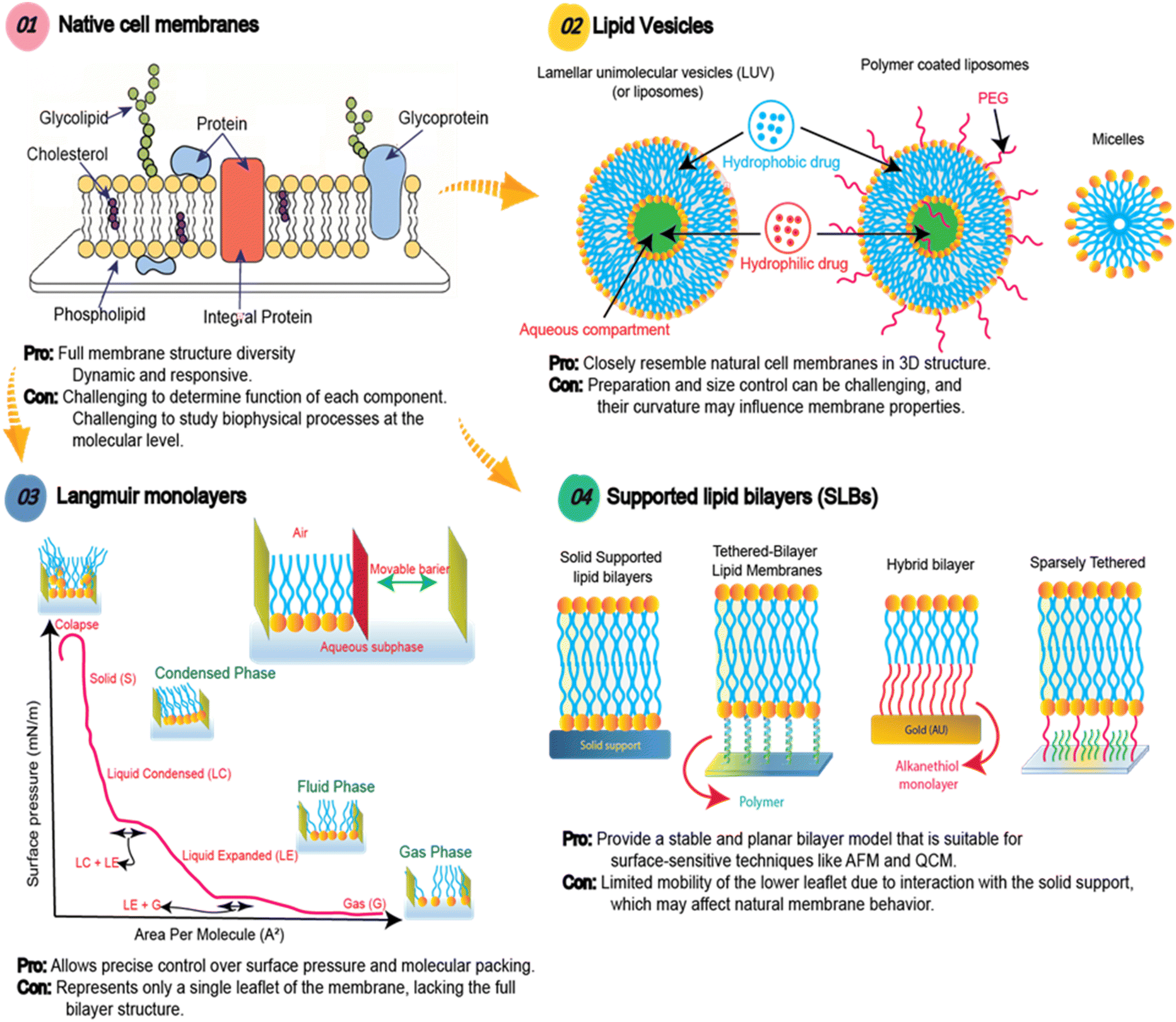 | ||
| Fig. 1 Cellular membranes are complex environments, which makes studying interactions of synthetic macromolecules difficult. Using various lipid models of these membranes, including Langmuir monolayers, lipid vesicles and supported lipid bilayers, aims to reduce the complexity and enable thorough analytical characterization to provide in depth mechanistic insights. | ||
Vesicle models
Lipid vesicles are soluble and spherical assemblies commonly used as in vitro membrane models due to their ability to mimic the structural and functional properties of biological membranes, encapsulating an aqueous compartment.46 The two primary categories are liposomes and micelles, both of which form through the self-assembly of amphiphilic molecules in aqueous environments.46Liposomes consist of one or more phospholipid bilayers enclosing an aqueous core and are classified based on size and lamellarity.46 Unilamellar vesicles (UVs), which contain a single lipid bilayer, are further subdivided into small (SUV < diameter 100 nm), large (LUV diameter of 100 nm–1 µm), and giant unilamellar vesicles (GUV diameter > 1 µm).47 Multilamellar vesicles (MVs), which consist of multiple concentric bilayers, offer enhanced complexity and structural resemblance to some organelles, but are hindered by heterogeneity of size and limited reproducibility. UVs can be formed using a range of molecular components, including phospholipids, amphiphilic polymers, proteins and carbohydrates.47 The physicochemical properties of these components affect membrane structure and determine the characteristics such as stability, permeability, and fluidity, allowing for tailored functions and applications.47 Producing UVs typically begins with dissolving lamellae-forming amphiphiles in an organic solvent, and distributing them onto a solid substrate as a thin film.47 Upon the addition of an aqueous solution, these amphiphiles undergo rehydration, leading to the formation of UVs.47 Downsizing of large UVs to smaller and more consistent populations can be achieved by cycles of snap freezing and sonicating,48 or extrusion through polycarbonate filters of a desired size. Extrusion must be performed at a temperature above the lipid-phase transition point to ensure vesicle stability and prevent formation of rigid or unstable structures.49
Lipid micelles consists of one lipid layer featuring a hydrophobic core surrounded by hydrophilic head groups.46 The size (typically in the range 10–100 nm) and shape of micelles depends on lipid and buffer properties.50 Micelles are formed at the critical micelle concentration (CMC). The small size and high curvature of lipid micelles provides different membrane architecture and properties to larger liposomes and 2D membrane models.46 Lipid micelles and liposomes can be characterized using different techniques, such as atomic force microscopy (AFM), small-angle X-ray scattering (SAXS), transmission electron microscopy (TEM), dynamic light scattering (DLS), and electron paramagnetic resonance (EPR) spectroscopy.50
Langmuir monolayers
Langmuir monolayers are single lipid monolayers at the air–liquid interface used to study membrane structure and interactions.51,52 The amphiphilic nature of some lipids leads to their spontaneous self-assembly at the interface, with polar head groups interacting with the aqueous sub-phase and hydrophobic tails aligning towards the air, thereby stabilizing the monolayer.51 Their physicochemical properties are primarily characterized using surface pressure-area (π-A) isotherms, which can reveal distinct phase transitions, such as the gas phase, liquid-expanded (LE) phase, and liquid-condensed (LC) phase, until a collapse pressure is reached.52 Langmuir–Blodgett troughs allow for precise control over lipid composition and packing, which is harder to achieve in bilayer systems.52 These monolayers enable detailed investigation of physicochemical parameters, such as molecular charge and hydrophobicity, influence of membrane insertion, lipid packing, and structural stability.51–54Langmuir monolayers can be used to probe changes in membrane rigidity, mechanical properties, and thermodynamic stability induced by the intercalation of nanoparticles or polymers.52 As an example, Langmuir monolayers were employed to study the interactions and effects of amphipathic methacrylate copolymers on membrane biomechanics.51 The findings revealed that hydrophobic side chains of the copolymer rapidly intercalated into the lipid monolayer and reduced its stability.51 Another study reported that deuterated styrene maleic acid copolymers synthesised by RAFT polymerisation (d-RAFT-SMA) embedded in both head and tail regions of lipid monolayers, with stronger effects seen with zwitterionic 1,2-dimyristoyl-sn-glycero-3 phosphocholine (DMPC) than with anionic lipids, pointing to the critical role of lipid charge.54 These examples highlight how these simple models can be used to directly quantify interactions between synthetic macromolecules and lipid membranes. They can be further enhanced by combining with surface-sensitive analytical techniques, including Brewster angle microscopy (BAM) and neutron reflectometry (NR), allowing for high-resolution structural and compositional analysis at the air–water interface.51 However, as they represent only a single lipid leaflet, Langmuir monolayers are inherently limited in their ability to model transmembrane processes such as translocation.52 Therefore, while they provide valuable insights into interfacial phenomena, their application should be complemented with bilayer systems to achieve a more comprehensive understanding of polymer–membrane interactions.
Lipid bilayers at solid–liquid interface
Supported lipid bilayers (SLBs) aim to serve as more realistic model membrane systems than lipid monolayers, as they possess both lipid leaflets. These systems are composed of a single lipid bilayer, often deposited onto solid surfaces, typically glass, mica, silicon, sapphire, titanium di oxide or gold.52 SLBs are primarily formed through two key methodologies: vesicle fusion and Langmuir–Blodgett/Langmuir–Schaefer (LB/LS) deposition. The vesicle fusion method involves the adsorption and subsequent rupture of small unilamellar vesicles (SUVs) on a hydrophilic surface, followed by the coalescence of the resulting lipid patches into a continuous bilayer.46–48 The success of this approach depends on various parameters, including vesicle size, lipid composition, temperature (usually above the lipid's chain melting temperature, Tm), hydrophilicity/hydrophobicity of the solid surface, and ionic strength of solution. Divalent cations like Ca2+ and Mg2+ can promote vesicle adhesion and fusion through electrostatic interactions, although excessive vesicle aggregation may occur if not properly controlled.46,52 Alternatively, the LB/LS technique enables the sequential deposition of lipid monolayers at the air–water interface onto a solid support.52 This method provides control over leaflet composition and is especially suited for the formation of asymmetric bilayers, which more closely resemble native biological membranes.52Supported lipid bilayers have been used to model complex systems like the outer membrane of Gram-negative bacteria and fungal membranes.52 Despite their advantages, SLBs have some limitations. The proximity of the bilayer to the solid support restricts lipid and protein diffusion, especially in the proximal leaflet, which interacts directly with the substrate. This can differ from the more native-like dynamics observed in freestanding bilayers. The degree of this effect depends on substrate roughness and the interplay between surface charge and lipid composition. Nevertheless, the ease of preparation, mechanical stability, and compatibility with surface-sensitive analytical techniques, such as AFM, quartz crystal microbalance (QCM), and surface plasmon resonance (SPR) make SLBs a powerful tool for probing the biophysical properties of membranes and biomolecule–membrane interactions.
To overcome the limitations of conventional supported lipid bilayers (SLBs), several advanced membrane models have been developed that offer improved biomimicry, mechanical stability, and compatibility with membrane proteins. These include tethered lipid bilayer membranes, polymer-cushioned membranes, and hybrid bilayer membranes. Tethered bilayers have emerged as advanced biomimetic platforms that overcome the limitations of traditional SLBs by introducing a molecular spacer between the bilayer and the solid support.46,55 The spacers in tethered bilayers are often composed of short oligomers, polyethylene glycol (PEG), or peptides. These spacers can control surface interactions and facilitate the incorporation of functional membrane proteins. For instance, sparsely tethered bilayers, which are attached to a substrate via a few flexible linkers, allow greater lateral mobility and better mimic natural membrane fluidity.55 In contrast, protein-tethered systems using His-tagged proteins to anchor the bilayer more rigidly through direct protein–substrate interactions, reduces membrane fluidity but enables the incorporation of specific membrane-associated proteins in a stable configuration.46 Polymer-cushioned membranes address the issue of restricted membrane-substrate spacing in SLBs by introducing a soft, hydrophilic polymer layer, such as PEG, cellulose, dextran, or polyelectrolytes between the bilayer and the substrate.46 This configuration reduces frictional coupling and supports the stable reconstitution of transmembrane proteins while preserving lateral mobility.46 In contrast, hybrid monolayers combine a self-assembled monolayer of alkanethiols on conductive substrates, such as gold, with a phospholipid monolayer adsorbed above.46 The bilayer assembly is driven by hydrophobic interactions between the acyl chains of the lipids and the self-assembled monolayer. While hybrid bilayer membranes are generally more robust than traditional SLBs, they offer less membrane fluidity due to the crystalline nature of the self-assembled monolayer. However, their structural stability and tuneable electrochemical properties make them particularly valuable for applications in biosensing, redox studies, and electrocatalysis.46 As with all of the models discussed in this review, it is critical to understand the complementary strengths of each individual model, and select to address specific research questions. The use of multiple different models, complementary analytical techniques, and in vitro testing is also encouraged, to demonstrate generalizability of conclusions drawn.
Characterisation techniques for studying the interactions of polymers and lipid membranes
Understanding how polymers interact with lipid membranes is essential for the rational design of polymer nanomedicines. To capture the complexity of these interactions, a variety of microscopic and spectroscopic techniques are employed, each providing unique insights into different facets of the polymer–membrane interface. For example, some methods are ideally suited for studying equilibrium structures of polymers and lipid membranes, while others are better suited to follow dynamic processes. This review will give an overview of the breadth of these methods and compare their complementary roles in understanding polymer interactions with lipid membranes. This review aims to give new researchers insight into how to select appropriate analytical techniques to study the interaction of their chosen polymer nanomedicine with lipid membranes. Critically it will highlight the need for multiple complementary techniques to fully elucidate any mechanism of action. It also aims to highlight new and emerging methods, that have not yet been applied to polymer nanomedicines, but could provide unique perspective on current challenges in the field.Optical microscopy and spectroscopy to study polymer interactions with lipid membranes
Ultraviolet (UV), visible (Vis) and near-infrared (NIR) light are commonly used light sources to study polymer–lipid membrane interactions, as they often require low cost and simple instrumentation, allow for rapid measurements to follow dynamic processes, and can provide spectroscopic information of different molecules involved in these processes.UV-Vis and fluorescence spectroscopic assays are among the simplest approaches available. Fluorescence leakage assays are designed to monitor the leakage of dyes from vesicles to determine simple structure–activity relationship. Calcein, a water-soluble fluorescent dye, is widely used in dye leakage assays due to its self-quenching behaviour at high concentrations inside vesicles.56 When the membrane is disrupted, calcein is released into the surrounding buffer, where dilution reduces quenching and results in a marked increase in fluorescence intensity. Most studies have used calcein dye leakage assays to investigate membrane permeabilization. As an example, calcein has been used to study the ability of new antibacterial ionenes to disrupt lipid vesicles to understand their membrane lytic behaviour on mammalian and bacterial cells.57,58 Two lead candidates, C0-T-p and C8-T-p (Fig. 2a) were tested against vesicles composed of different ratios of cardiolipin (TOCL) and phosphatidylglycerol (POPG). C0-T-p had higher activity for cardiolipin containing vesicles (Fig. 2c, V1 and V5), while C8-T-p which had an additional alkyl sidechain showed greater leakage for more negatively charged vesicles (Fig. 2d, V5 and V6). This suggested that C0-T-p's activity is lipid-specific, whereas C8-T-p's effect is charge-driven. The amphiphilic C8-T-p induced membrane leakage up to a 1.4–2.8 polymer-to-lipid charge ratio, however, at higher ratios the polymer stabilized the membrane.58 The results highlight that lipid composition as well as polymer architecture are both critical for membrane interactions, with cardiolipin playing a significant role in modulating the lytic activity of these polycations.58 The fluorescence-based dye leakage assay has been widely used because it is simple, sensitive, and can be applied in solution-phase systems and, thus, avoids artefacts caused by supported membranes.56 These methods are especially useful to determine significant membrane damage, and give qualitative or semi-quantitative information about the membrane perturbation. However, they are limited in understanding the structural changes at the polymer–membrane interface.
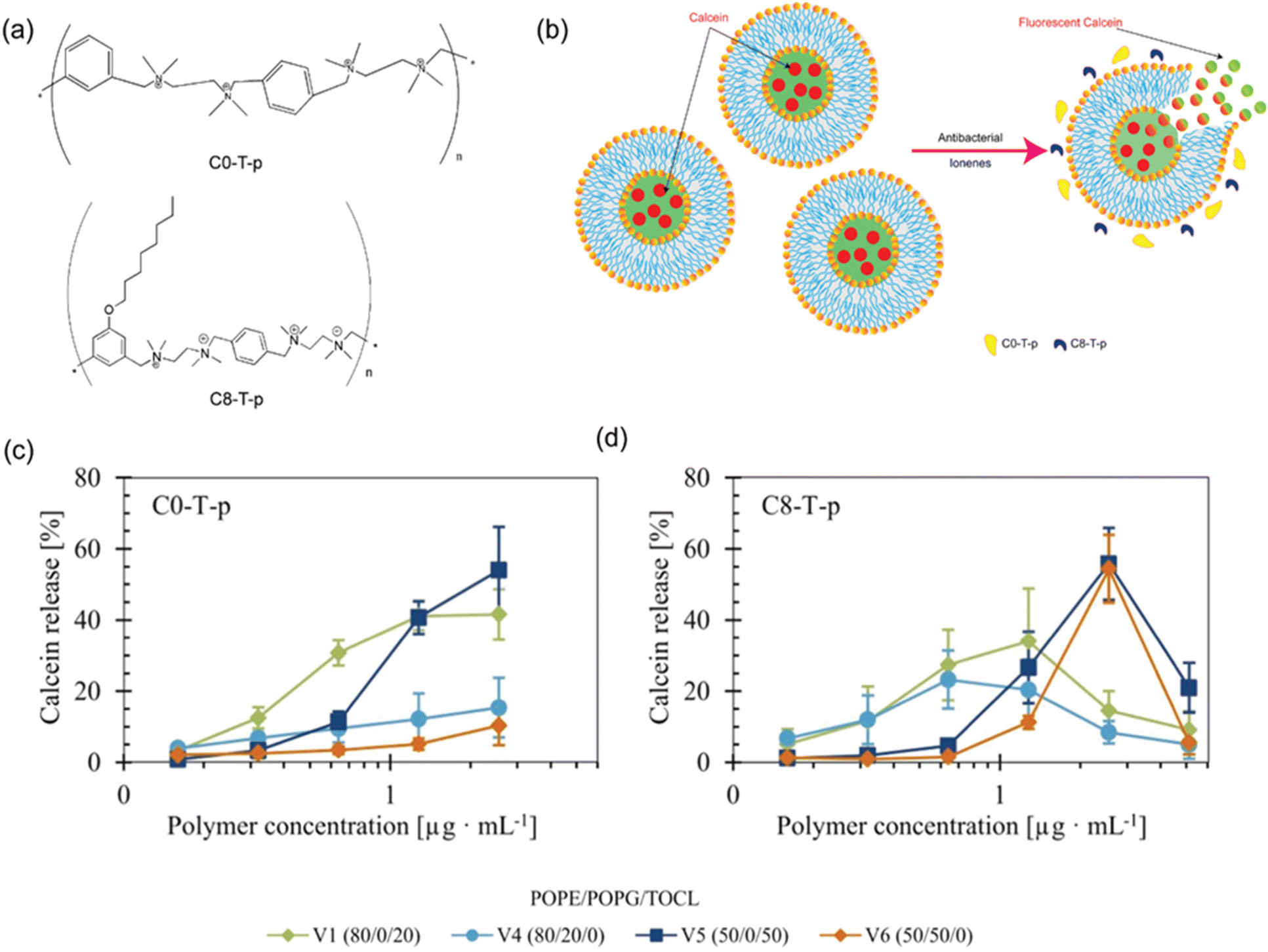 | ||
| Fig. 2 (a) Chemical structures of antibacterial ionenes (b) schematic illustration of lipid vesicles encapsulating calcein at a self-quenched concentration. Upon polymer addition, the vesicles destabilize, releasing calcein into the surrounding aqueous environment, where it becomes fluorescent. Influence of vesicle charge and anionic lipid composition on calcein release induced by (c) C0-T-p and (d) C8-T-p. C0-T-p is more active against vesicles composed of TOCL, while C8-T-p is more active against vesicles composed of higher fraction of anionic lipids. Reproduced with permission. Adapted with permission from ref. 58. Licensed under a Creative Commons Attribution-Non-Commercial (CC BY-NC) License. | ||
Confocal fluorescence microscopy has been used to visualise the interaction of polymers with model lipid membranes.59,60 As a brief example, confocal fluorescence microscopy was used to investigate the adsorption and wrapping of fluorescent microgel particles with spherical (MG1: aspect ratio = 1) and ellipsoidal (MG2: aspect ratio = 2, and MG3: aspect ratio = 6) shapes on model membranes with different lipid compositions (Fig. 3). Confocal microscopy revealed that spherical microgels (MG1) were found to adsorb to all membrane types, exhibiting shallow membrane wrapping, with less deformation seen in the most rigid (DMPC/chol) membranes (Fig. 3b, A–C). In contrast, ellipsoidal microgels, MG2 and MG3 showed deeper wrapping, especially in more fluid membranes like 1,2-dioleoyl-sn-glycero 3-phosphocholine (DOPC), with the extent of wrapping increasing with microgel aspect ratio (Fig. 3b, F, H, and I). The deep wrapping of ellipsoidal particles occurs when they align their long axis perpendicular to the membrane, maximizing interfacial contact, whereas shallow-wrapped ellipsoids align parallel. These behaviours suggest that hydrophobic interactions, rather than electrostatic forces, primarily drive adsorption, as the membranes consist of neutral zwitterionic lipids and microgels are only slightly positively charged. Additionally, 3D confocal reconstructions showed that adsorbed microgels displayed organized arrangements, for instance MG1 formed hexagonal patterns (Fig. 3c, A–C), while MG2 showed local smectic-like ordering on fluid membranes (Fig. 3c, D and E). MG3 particles with the highest aspect ratio displayed random distribution (Fig. 3c, F), highlighting the combined influence of particle shape, hydrophobicity and membrane mechanics on microgel–membrane interactions.61 While fluorescence confocal microscopy can clearly identify where polymers are interacting with membranes, they do not capture subtle changes in membrane properties, nor do they accurately quantify the amount of polymer at the membrane interface or the extent of interfacial changes.
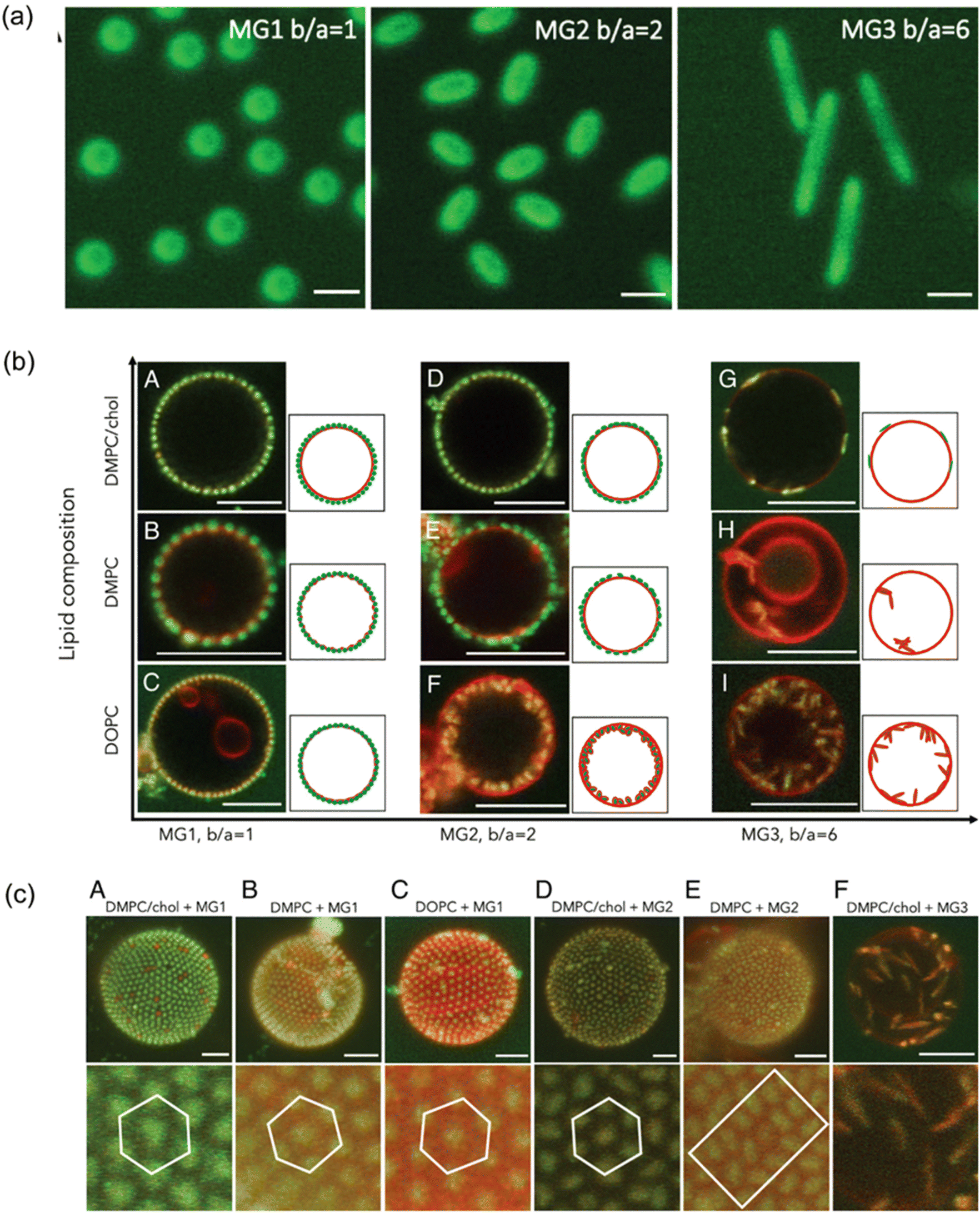 | ||
| Fig. 3 Lipid composition affects membrane bending rigidity and headgroup area. (a) 2D confocal laser scanning microscopy (CLSM) images of three microgels: spherical MG1 and ellipsoidal MG2 and MG3. Scale bar: 1 µm. (b) 2D CLSM images showing adsorption and wrapping of green-labeled microgels on red-labeled lipid membranes with varying compositions: DMPC/chol, DMPC, and DOPC. (c) Top: 3D reconstructed CLSM images of microgel-covered GUVs, illustrating particle–membrane interactions for each microgel type and lipid composition. Bottom: zoom-in views highlighting microgel assembly on membranes. Scale bars: 5 µm. Adapted with permission from ref. 61. Licensed under a Creative Commons Attribution (CC BY) License. | ||
Moving beyond simple dye leakage assays and correlative microscopy, a range of spectroscopic fluorescence methods have been developed to probe membrane properties and polymer dynamics with high sensitivity. These include fluorescence anisotropy to monitor membrane fluidity; fluorescence recovery after photobleaching (FRAP) for assessing lateral membrane mobility; fluorescence lifetime correlation spectroscopy (FLCS) and two-focus fluorescence correlation spectroscopy (2fFCS) to measure diffusion and binding kinetics; and pulsed interleaved excitation fluorescence cross-correlation spectroscopy (PIE-FCCS) for detecting co-diffusion and molecular interactions with high temporal resolution. Together, these techniques offer a valuable toolkit for investigating the mechanisms by which synthetic polymers interact with, stabilize, or disrupt lipid bilayers.
Fluorescence anisotropy is widely used due to its sensitivity to changes in lipid packing and membrane fluidity by measuring the rotational mobility of embedded probes.62 The fluorescence anisotropy study of amphiphilic carbosilane dendrons demonstrated how variations in dendron structure influence the dynamic behaviour of lipid membranes.63 One probe, 1,6-diphenyl-1,3,5-hexatriene (DPH) was embedded within the hydrophobic core of the liposome bilayer, providing information on the fluidity of the inner membrane region. In contrast, trimethylammonium-DPH (TMA-DPH), a derivative of DPH with a positively charged amino group, was anchored at the bilayer surface and remained exposed to the hydrophilic aqueous environment.63 The results showed no significant changes in anisotropy across different liposome types until the highest concentrations (30![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 and above) of the amphiphilic dendrimers were reached, which led to decreased membrane fluidity.63 Dendrons with biotin had minimal effect at lower concentrations, whereas dendrons with azadibenzocyclooctyne caused more pronounced changes on lipid membranes even at low concentrations. The effect also varied with liposome composition. Dendrons with biotin affected neutral DMPC liposomes most, while those with azadibenzocyclooctyne induced the strongest changes in negatively charged liposomes consisting of 30% 1,2-dimyristoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (DMPG) and DMPC. The variations in anisotropy based on liposome charge indicate that electrostatic interactions play a crucial role in modulating membrane–nanoparticle interactions. These findings once again highlight the importance of both molecular design and membrane composition in developing lipid-interacting nanomedicines. While fluorescence anisotropy provides detailed information on membrane fluidity, it does not directly investigate the lateral movement of membrane components.
:1 and above) of the amphiphilic dendrimers were reached, which led to decreased membrane fluidity.63 Dendrons with biotin had minimal effect at lower concentrations, whereas dendrons with azadibenzocyclooctyne caused more pronounced changes on lipid membranes even at low concentrations. The effect also varied with liposome composition. Dendrons with biotin affected neutral DMPC liposomes most, while those with azadibenzocyclooctyne induced the strongest changes in negatively charged liposomes consisting of 30% 1,2-dimyristoyl-sn-glycero-3-phospho-rac-(1-glycerol) sodium salt (DMPG) and DMPC. The variations in anisotropy based on liposome charge indicate that electrostatic interactions play a crucial role in modulating membrane–nanoparticle interactions. These findings once again highlight the importance of both molecular design and membrane composition in developing lipid-interacting nanomedicines. While fluorescence anisotropy provides detailed information on membrane fluidity, it does not directly investigate the lateral movement of membrane components.
Fluorescence recovery after photobleaching (FRAP) can measure molecular mobility and diffusion dynamics in biological membranes. In a FRAP experiment, a specific region of a fluorescently labelled sample is irreversibly photobleached using a high-intensity laser, thereby eliminating fluorescence in that area. Over time, unbleached fluorescent molecules from surrounding regions diffuse into the bleached area, leading to fluorescence recovery. By analysing the kinetics of this recovery, the diffusion coefficient, mobile fraction, and immobile fraction of the fluorescent species can be extracted.64 FRAP has been used to investigate the formation of lipid bilayers on both poly(3,4-ethyl enedioxythiophene) polystyrene sulfonate (PEDOT:PSS) and glass using solvent-assisted lipid bilayer (SALB) and vesicle fusion methods.65 FRAP confirmed the successful formation of planar and fluid bilayers on glass by vesicle fusion, but this method failed on PEDOT:PSS due to its negative charge and greater surface roughness. In contrast, SALB consistently produced fluid SLBs on both surfaces, confirmed by high mobile fractions and recovery of fluorescence. However, the diffusion coefficients were significantly lower on PEDOT:PSS than on glass, despite similar mobile fractions. This reduced diffusivity may result from either the swollen polymer surface increasing the photobleached area, potentially leading to lower diffusion coefficient, or changes in the hydrophilicity of the polymer layer leading to lipid pinning caused by interactions between the lipids and the polymer surface. These findings highlight the advantages of FRAP in investigating bilayer dynamics but also reveal its sensitivity to substrate properties, especially on complex or non-rigid surfaces like conductive polymers.65 FRAP is useful both for qualitative and quantitative measurements including diffusion coefficients and mobile fractions, but its temporal and spatial resolution are limited.
Fluorescence lifetime correlation spectroscopy (FLCS) can provide a more sensitive and detailed view of molecular dynamics in heterogeneous systems, with higher sensitivity and resolution at the membrane interface, by resolving the overlapping fluorescence signals by differences in fluorescence lifetimes. This technique is especially valuable when using spectrally indistinguishable fluorophores, as it eliminates spectral crosstalk, background noise, and detector artefacts.66 By analysing the unique fluorescence decay patterns of individual species, FLCS calculates separate autocorrelation functions for each component. It requires a confocal microscope and Time-Correlated Single Photon Counting instrumentation, making it a powerful tool for resolving complex fluorescence signals at the single-photon level. Demonstrating FLCS capabilities for measuring polymer-membrane interactions, the pH dependent interactions of an amphiphilic co-polymer poly(L-lysine iso-phthalamide) (PLP) grafted with L-phenylalanine (PP50) with DOPC bilayers were studied.67 In solution, the copolymer exhibited fast diffusion but upon binding to the bilayer, its diffusion slowed significantly, suggesting surface association. This interaction also led to a decrease in the anomalous diffusion exponent, indicating sub-diffusive behaviour likely due to membrane obstacles or adsorbed polymers.67 Lipid diffusion within the bilayer was also affected after polymer association, in a pH-dependent and time-dependent manner. The greatest reduction in lipid mobility occurred at pH 6.5, where the copolymer is fully protonated and more hydrophobic, indicating stronger membrane binding. At pH 7.05, moderate and partially reversible effects were seen, while at pH 7.5, the effects were weak and temporary.67 Despite these changes, the lipid probe maintained normal diffusion behaviour (α ≈ 1), and its fluorescence lifetime remained unchanged, suggesting that the copolymer affects membrane fluidity at the surface without deeply penetrating the bilayer core. Although FLCS offers important information about diffusion dynamics at membrane interfaces, it depends on calibrated observation volumes which can leads to uncertainty.
Two-focus fluorescence correlation spectroscopy (2fFCS) can be used to determine absolute diffusion coefficients, by using two laterally shifted, spatially distinct excitation foci generated through orthogonally polarized pulsed lasers and a Nomarski prism.68 The method relies on the known distance between the two foci, making it robust against artefacts typically associated with confocal volume variations due to optical aberrations or sample conditions. A 2fFCS study of PEO-b-PPO star-shaped polymer interactions with lipid liposomes confirmed the multiliposomal nature of the complexes.68 Liposomes labelled with a fluorescent dye were prepared to track their aggregation state upon addition of the amphiphilic polymer (PEO12-b-PPO45)4.68 The 2fFCS cross-correlation curves showed a clear increase in lag time after polymer addition, indicating a significant increase in aggregate size.68 This confirms that the polymer caused the liposome aggregation, forming multicompartmental complexes. The hydrodynamic radius of the liposome–polymer complex was calculated using the Stokes–Einstein equation and found to be approximately 160 nm, compared to ∼30 nm for individual liposomes.
Pulsed interleaved excitation fluorescence cross-correlation spectroscopy (PIE-FCCS) is a dual-colour fluorescence spectroscopy that translates fluctuations in fluorescence signal into a measurement of diffusion and colocalization. PIE-FCCS offers superior specificity and accuracy by temporally resolving excitation events and eliminating spectral crosstalk. This allows researchers to separate real molecular interactions from artefacts, making it particularly powerful for studying weak, transient, or dynamic interactions like those between lipids and polymers. PIE-FCCS was employed to investigate the effects of electrostatic interactions between cationic quaternized polyvinylpyridine and supported lipid bilayers.69 The results demonstrated that the polymer adsorption significantly reduced the lateral diffusion of negatively charged lipids, with the strongest effect observed for fluorescently labelled phosphatidylinositol phosphate lipids under low ionic strength conditions, indicating a charge-dependent interaction. Labelled phosphatidylserine lipids, with fewer negative charges, exhibited only a modest reduction in mobility. Despite these changes in lipid dynamics, PIE-FCCS detected no stable cross-correlation between the polymer and the lipids, suggesting that the interactions are transient rather than involving stable complex formation. Furthermore, lipid molecules near immobile polymer clusters displayed slower diffusion compared to those in regions with mobile polymer, and that lipid diffusion became anomalous upon polymer adsorption. These findings highlight the use of PIE-FCCS in resolving subtle, transient polymer–lipid interactions and spatial heterogeneities in membrane systems.69 While fluorescence spectroscopy is a powerful tool for studying membrane dynamics, it does require labelling each component with fluorophores, changing their molecular structure and properties. This can potentially affect the nature of the physicochemical interactions that are being studied.
Brewster angle microscopy (BAM) is a label free microscopic technique used to investigate the structure and interactions of Langmuir monolayers at the air–liquid interface in real time.70 When a laser beam is polarized parallel to the plane of incidence and is directed at the air–liquid interface at the Brewster angle, no light is reflected from a pure interface. However, the presence of a surface film changes the refractive index, resulting in reflected light that can be captured to produce real-time images of the interfacial structure.70 BAM can provide quantitative structural analysis in addition to qualitative optical information. This is because of its sensitivity to anisotropy caused by variations in the reflective properties of lipid polar head groups. BAM studies have proven effective in revealing how polymer interactions with lipid monolayers are influenced by factors such as monolayer composition, charge, phase state, as well as polymer charge and hydrophobicity. As an example, 1,2-myristoyl-sn-glycero-3-phosphoethanolamine (DMPE) monolayers were used to mimic biological membranes and examine the interaction of a binary mixture of polyhedral oligomeric silsesquioxanes (POSS) and poly(ethylene glycol) (POSS-PEG).71 BAM was conducted with both pure DMPE monolayers and DMPE/POSS-PEG mixtures. In the pure system, BAM revealed the formation of distinct condensed domains during the liquid-expanded to liquid-condensed (LE–LC) phase transition. However, upon incorporation of POSS-PEG, the BAM images showed a reduction in domain size, which eventually disappeared, indicating that the polymer disrupts the native domain structure of the DMPE monolayer.71 BAM effectively confirmed the presence of ordered domains in pure DMPE monolayers and showed the gradual loss of these structures upon addition of POSS-PEG, supporting the interpretation of phase behaviour and polymer–lipid interactions observed in π–A isotherms. Thus, BAM serves as an essential tool for correlating molecular organization.
Vibrational spectroscopy can characterise molecular structures based on the absorption (IR spectroscopy) or scattering (Raman spectroscopy) of infrared light by specific vibrational modes within functional groups. When infrared radiation matches the frequency of a molecular vibration that causes a change in dipole moment, absorption occurs, producing wavelength-specific bands in the IR spectrum. Among IR spectroscopic methods, transmission Fourier transform infrared (FTIR) spectroscopy is a powerful, label-free and nonperturbing technique that has been used for the detection and characterization of lipid composition and phase transitions in model and natural membranes. However, the strong IR absorption of water presents a challenge, often requiring short pathlengths and concentrated lipid samples, which may be impractical for certain systems like micelle-forming lipids.72 Attenuated Total Reflectance (ATR) FTIR is an alternative, which minimizes solvent interference by probing the sample through an evanescent wave at the surface of an ATR crystal.72 While ATR-FTIR minimizes solvent absorption issues, it also requires high lipid concentrations and may introduce artefacts due to interactions between the sample and the polarized crystal surface, especially with charged lipids.72
Raman spectroscopy detects the inelastic scattering of monochromatic light, that matches particular molecular vibrational frequencies. Raman spectroscopy is complementary to IR spectroscopy, as it is particularly sensitive to non-polar functional groups, and less sensitive to polar molecules. This makes Raman spectroscopy compatible with aqueous environments, because water possesses weak Raman scattering, making it ideal for analysing biological membranes in their native state. Raman spectroscopy peak characteristics including spectral position, width, and intensity are highly sensitive to changes in lipid packing, membrane phase, and chain order, particularly within the hydrocarbon tails. Thus, Raman spectroscopy is a valuable label-free tool for probing polymer–lipid membrane interactions to probe structural perturbations at the molecular level. Raman spectroscopy has been used to examine interactions of positively charged amine terminated polyamidoamine (PAMAM) dendrimers (G = 2.0 and G = 4.0) on 1,2-dipalmitoylphosphatidylcholine (DPPC) vesicles.73 Analysis of the Raman spectrum detected structural changes produced by inclusion of PAMAM dendrimers in DPPC lipid membranes structures. Raman band intensity ratios are useful indicators of structural changes in lipid membranes including I(2935/2880 cm−1) and I(1090/1130 cm−1). An increase in the I(2935/2880 cm−1) ratio reflects vibrations of terminal –CH3 groups and lipid alkyl chains which suggests greater lateral steric hindrance and disorder upon dendrimer insertion. Similarly, a rise in the I(1090/1130 cm−1) ratio indicates an increase in disordered (gauche) conformations compared to ordered (trans) ones, further supporting the disruption of lipid chain packing.73 These spectral changes indicate a transition from ordered to disordered states in the bilayer structure, driven by the insertion and interaction of PAMAM dendrimers with the hydrophobic core of DPPC membranes. The extent of membrane disruption correlates with dendrimer generation, with G4.0 dendrimers causing more pronounced disorder than G2.0. Furthermore, the Raman spectra of anionic (G2.5) and cationic (G3.0) PAMAM dendrimers showed that both dendrimers disrupted DPPC membrane order, but the anionic G2.5 dendrimers had greater impact on the bilayer by perturbing DPPC alkyl chain packing. This difference is responsible for distinct interactions and hydration environments associated with their surface functional groups (–COO− vs. –NH3+), highlighting the role of dendrimer charge and chemistry in modulating membrane structure.74 This study highlights the utility of Raman spectroscopy in monitoring polymer-induced conformational transitions and membrane destabilization at the molecular level.
Dynamic Light Scattering (DLS) can measure the hydrodynamic diameter and surface charge distribution of particles in solution by detecting the random fluctuations in the intensity of scattered light that arise from Brownian motion.75 In polymer–lipid membrane studies, DLS has been used to investigate how polymer influences vesicle properties, including vesicle aggregation, fusion, or disruption, detected by changes in hydrodynamic diameter of the samples. As an example, liposomes were used as model cell membranes to study the mechanism of action of guanidinium copolymers loaded with silver nanoparticles, as a function of nanocomposite concentration.76 A significant rise in liposome size indicated aggregation with a peak size at charge neutralization, followed by size reduction at higher concentrations.76 As has been discussed earlier, surface charge of lipid membranes plays a critical role in determining the nature of polymer–membrane interactions, which can be measured by DLS. For example, it was discovered that highly negatively charged liposomes (those comprised of 30% DMPG and DMPC) could destabilize carbosilane dendron aggregates. This indicates that electrostatic factors may mediate not only binding, but also the structural integrity of polymer aggregates, raising the possibility of their disassembly upon membrane contact.63 DLS is a quick, non-invasive, label-free, and sensitive method to measure particle size changes which can give real time results.67 However, the technique is limited by its sensitivity to sample concentration, requiring high dilution to prevent multiple scattering effects. Additionally, DLS is less effective when analysing complex systems, such as those containing a mix of nanoparticles, high concentrations, or non-spherical particles. In polydisperse samples or those with large aggregates, the scattering signal from larger particles can dominate, potentially obscuring the presence of smaller populations and leading to skewed results.75
Analytical tools to study dynamic interfacial interactions and structures
The dynamic interactions between polymers and biological membranes are governed by a delicate balance of forces. These include electrostatic attraction or repulsion, hydrophobic effects, hydrogen bonding, and steric hindrance, all of which can vary depending on the polymer's architecture, charge density, and functional groups, as well as the molecular composition and phase state of the membrane.77 To unravel the complexity of these interactions, analytical techniques that can accurately quantify interfacial phenomena with high sensitivity, spatial resolution in three dimensions, and temporal resolution are needed. Techniques such as Quartz Crystal Microbalance (QCM), Isothermal Titration Calorimetry (ITC) and Surface Plasmon Resonance (SPR) provide real-time, label-free insights into adsorption kinetics, adsorption thermodynamics, binding affinities, and conformational changes at the interface. Meanwhile, scattering techniques like Small-Angle X-ray Scattering (SAXS), Small-Angle Neutron Scattering (SANS), X-ray Reflectivity (XRR) and Neutron Reflectivity (NR) offer structural and dynamic information at the nanoscale, capturing changes in membrane morphology, polymer aggregation, and vesicle stability. Progressively, these techniques have become essential for dissecting the nuanced behaviours of polymers at membrane interfaces, offering critical insights into the mechanisms that drive their functional performance in biological environments.Surface Plasmon Resonance (SPR) is an optical detection technique that enables label-free, real-time monitoring of molecular interactions with high sensitivity.78 The technique relies on the excitation of surface plasmons (collective oscillations of free electrons) at the interface between a metal and a dielectric medium.79 When polarized light hits the metal surface at a specific angle, it triggers a resonant energy transfer to these plasmons, causing a measurable dip in reflected light intensity. Since this effect is highly sensitive to refractive index changes near the metal surface, SPR can precisely detect molecular binding events as they alter the local refractive index.80,81 A key strength of SPR is its capacity to quantify the kinetics of biomolecular interactions, including association and dissociation rates, and affinity constants without requiring fluorescent or radioactive labels.
In lipid membrane studies, SPR is frequently combined with sensor chips coated with lipid bilayers or supported membranes, which closely replicate the natural environment of cell membranes. The polymers are injected into the microfluidic system, making it possible to characterize their binding to the membrane as a function of time (Fig. 4a). SPR has been employed to investigate the binding dynamics of polymers such as poloxamer 188 (P188) and polyethylene oxide (PEO) to supported lipid bilayers (SLBs). While earlier research indicated PEO requires hydrophobic modifications to bind lipid membranes, their SPR measurements challenged this established view. They demonstrated that unmodified 8.6 kDa PEO adheres to SLBs with similar affinity as P188. Upon introduction to SLBs, both P188 and PEO induced substantial SPR response shifts. Continuous monitoring during a 45-minute binding phase and subsequent 30-minute buffer rinse revealed persistent signal, demonstrating essentially irreversible membrane association. The enhanced sensitivity of SPR proved instrumental in these findings, overcoming the detection limitations of other analytical methods employed in prior studies.82
 | ||
Fig. 4 Characterisation of the interactions of polymers with lipid membranes with surface-sensitive analytical techniques. (a) Schematic of the SPR principle illustrating light interaction with a metal surface via a prism to monitor polymer–lipid bilayer interactions in a flow channel. (b) SPR sensorgrams showing ΔRU of HE-PEG adsorption onto gold-supported lipid vesicles (c) QCM-D monitoring of supported lipid bilayer formation, showing frequency and dissipation changes through vesicle adsorption, coverage, and rupture. (d) QCM-D analysis of amphipathic copolymer interactions with POPC bilayers: frequency and mass measurement ( : injection of polymer, : injection of polymer,  : rinsing with buffer). (b) adapted with permission from ref. 83. Copyright 2010, American Chemical Society. (c) and (d) Adapted with permission from ref. 51. Licensed under a Creative Commons Attribution-Non-Commercial (CC BY-NC) License. : rinsing with buffer). (b) adapted with permission from ref. 83. Copyright 2010, American Chemical Society. (c) and (d) Adapted with permission from ref. 51. Licensed under a Creative Commons Attribution-Non-Commercial (CC BY-NC) License. | ||
Insights of polymer–membrane interaction gained through SPR analysis has been leveraged to establish how incremental changes in alkyl chain length influence hydrophobically end-capped polyethylene glycol's (HE-PEG) capacity to stabilize liposomes and whether its binding behaviour mimics membrane-binding molecules.83 The SPR data revealed distinct interaction patterns based on hydrocarbon chain length. Polymers with shorter end-cap chains (carbon number <12) showed negligible adsorption, as evidenced by minimal Response Unit (RU) changes. PEG derivatives with intermediate chain lengths (12–14 carbons) exhibited a reversible binding profile. RU increased during polymer injection but returned to baseline after rinsing, indicating weak, transient interactions with the membrane surface. Unlike the shorter analogues, the PEG derivative with 16 carbon hydrophobic-end cap chains produced significant, irreversible RU shifts that remained stable below the baseline after buffer washing. This persistent signal indicated deep penetration of the hydrocarbon tails into the bilayer's hydrophobic core, transforming the intact vesicles into planar bilayer structure (Fig. 4b). SPR analysis further uncovered concentration-dependent effects. At elevated concentrations, all HE-PEG variants with chains ≥12 carbons caused RU values to fall below baseline after rinsing, except for the shortest (11-carbon) chain (Fig. 4b). This phenomenon was attributed to the combined action of hydrophobic insertion and osmotic pressure, which synergistically disrupted vesicle integrity. In this study, SPR's unique capability enabled real time tracking of binding kinetics, material loss and concentration dependent transitions, providing an understanding that revealed the cooperative effects between hydrophobic insertion and osmotic pressure in membrane remodeling.83
While SPR offers numerous advantages, it also has limitations. While it can monitor interfacial interactions and real-time changes without the need for markers, it has difficulty detecting very low concentrations, distinguishing nonspecific binding events, and analysing electron transfer, redox ion replacement, or functional group modifications.84 Additionally, other experimental factors that alter the surface refractive index can interfere with data interpretation, leading to inaccurate kinetic or equilibrium measurements. A common issue arises when the analyte is contained in a buffer that differs in refractive index from the running buffer. During injection, this new buffer displaces the running buffer near the chip surface, creating a refractive index shift unrelated to actual binding or interaction events.85 While SPR excels in analysing binding kinetics and quantifying association, it provides limited information about changes in the physicochemical properties of the lipid membranes.
Quartz Crystal Microbalance (QCM) measures mass per unit area by detecting variations in the oscillation frequency of a piezoelectric quartz crystal resonator. Since the frequency is directly related to the thickness of the quartz crystal, the deposition of films on its surface causes a measurable decrease in frequency.86 The exceptional sensitivity of QCM to minute mass changes at the nanoscale make it uniquely suited to quantifying mass changes in lipid membrane interfaces. Its applications have expanded to include simultaneous measurements of both the sensor's resonance frequency and the dissipation factor (QCM-D), enabling independent determination of the adlayer's mass and viscoelastic properties.51,87 QCM-D technology has revolutionized the study of soft, hydrated biological materials in solutions, enabling detailed analysis of surface interactions between nanomaterials and membranes, quantifying membrane formation and binding dynamics in real time. QCM-D can allow for real-time measurement of the formation of a lipid bilayer from vesicle fusion at the solid–liquid interface (Fig. 4c).48,51 These bilayers formed on QCM-D sensors have been used to study the binding of a series of amphipathic methacrylic copolymers to 1-palmitoyl-2-oleoyl-sn-glycero-3-phosphocholine (POPC) bilayers, and their effect on the viscoelastic properties (Fig. 4d).51 When copolymer solutions (100 µg mL−1) were introduced to the lipid bilayers, frequency decreased as copolymers adsorbed to the bilayer surface. During association the dissipation increased, signalling an increase in the viscoelastic properties of the bilayer upon formation of a weakly adsorbed polymer layer. Upon washing the bilayers with pure water, loosely bound copolymer was removed, and the frequency showed a slight increase, but remained lower than the original bilayer, corresponding to the amount of copolymer integrated into the membrane. Meanwhile, dissipation returned to the original bilayer values, confirming the copolymers tight integration into the bilayer structure and adopting its elastic characteristic.51 Similar associations have been found with polymer coated nanoparticles, where three parameters were investigated: (1) polymer grafting density, (2) nanoparticle surface charge (controlled by pH), and (3) bilayer charge composition.88 Nanoparticles with low-to-moderate grafting densities displayed rapid frequency decreases indicating fast membrane adsorption enabled by flexible polymer chains forming multiple attachment points. In contrast, high-density grafts showed negligible frequency shifts, demonstrating complete steric inhibition due to brush rigidity. Post-rinse frequency change confirmed irreversible binding, while the dissipation shifts again returned to baseline demonstrating full integration of nanoparticles into the bilayer.88 These studies highlight the versatility and precision of QCM-D as a powerful tool for investigating dynamic surface interactions, including polymer–membrane kinetics and the viscoelastic properties of membrane interfaces. Despite this, both QCM-D and SPR do not provide direct information about the energetics of association and the relative thermodynamic contributions of each of the species involved.
Isothermal Titration Calorimetry (ITC) measures the heat released (exotherm) or absorbed (endotherm) during binding events, providing direct thermodynamic data on the interaction process. While QCM and SPR analyses binding kinetics, ITC provides a more complete thermodynamic perspective by quantifying both enthalpic (heat-related) and entropic (disorder-related) contributions to binding interactions. This enables researchers to identify the fundamental driving forces behind nanoparticle–membrane interactions.89 The technique measures heat changes as a ligand is titrated into an analyte solution under isothermal conditions. The ITC instrument features a dual-cell design: a sample cell containing the analyte of interest and a reference cell filled with solvent for baseline measurements. The ligand solution is drawn into a syringe and titrated into the sample cell. When the ligand binds to the analyte, the interaction generates either exothermic or endothermic effects, creating temperature fluctuations in the sample cell, providing robust measurement of binding thermodynamics.8,90 Isothermal titration calorimetry is another label-free approach, which preserves the native state of the molecules under investigation. The interactions of a series of ionenes with varying spacer lengths (3,3-, 4,4-, 6,6- and 10,10-) with lipid vesicles was studied by ITC.91 The analysis revealed that charge density and hydrophobicity of the ionenes dictated their interactions. While all ionenes neutralized membrane charge suggesting equivalent electrostatic binding affinity regardless of the spacer length, the 10,10-ionene exhibited a larger exotherm in ITC analysis. This distinctive thermodynamic signature implied additional non-electrostatic contributions, as its long spacer can embed into the bilayer, suggesting hydrophobic interactions. The study stands as a good demonstration of how ITC can resolve subtle but biologically critical differences in molecular interactions between materials.91 As another example, ITC can distinctly identify hydrogen bonding versus hydrophobic contributions of peptide amphiphiles interacting with lipid membranes.92 This capability positions ITC as a powerful tool for developing optimized membrane-active biomaterials. Despite this potential, there are few examples of using ITC in polymer–membrane interaction research, highlighting an important opportunity for deeper understanding, complementing techniques to measure association and structure at the interface.
X-ray and neutron scattering techniques have become indispensable tools in the investigation of nanoscale interfacial structures, offering unique capabilities based on their distinct contrasts. X-ray and neutron sources provide complementary information about macromolecular structure, due to the difference in energy of their photons and contrasting interactions with matter. X-ray scattering is particularly effective for interactions with atomic clouds due to its sensitivity to electron distribution. This property makes X-ray techniques suitable for studying materials with significant contrast in electron density.93,94 Neutron scattering on the other hand interacts with atomic nuclei rather than electrons, enabling differentiation between light elements and isotopes like hydrogen and deuterium. Small-angle X-ray/neutron scattering (SAXS/SANS) and neutron/X-ray reflectometry (NR/XRR) are commonly used to investigate structural properties at molecular and atomic levels. While small angle scattering and reflectometry can use the same radiation sources, they differ fundamentally in their measurement principles and sample requirements (Fig. 5a and b).95
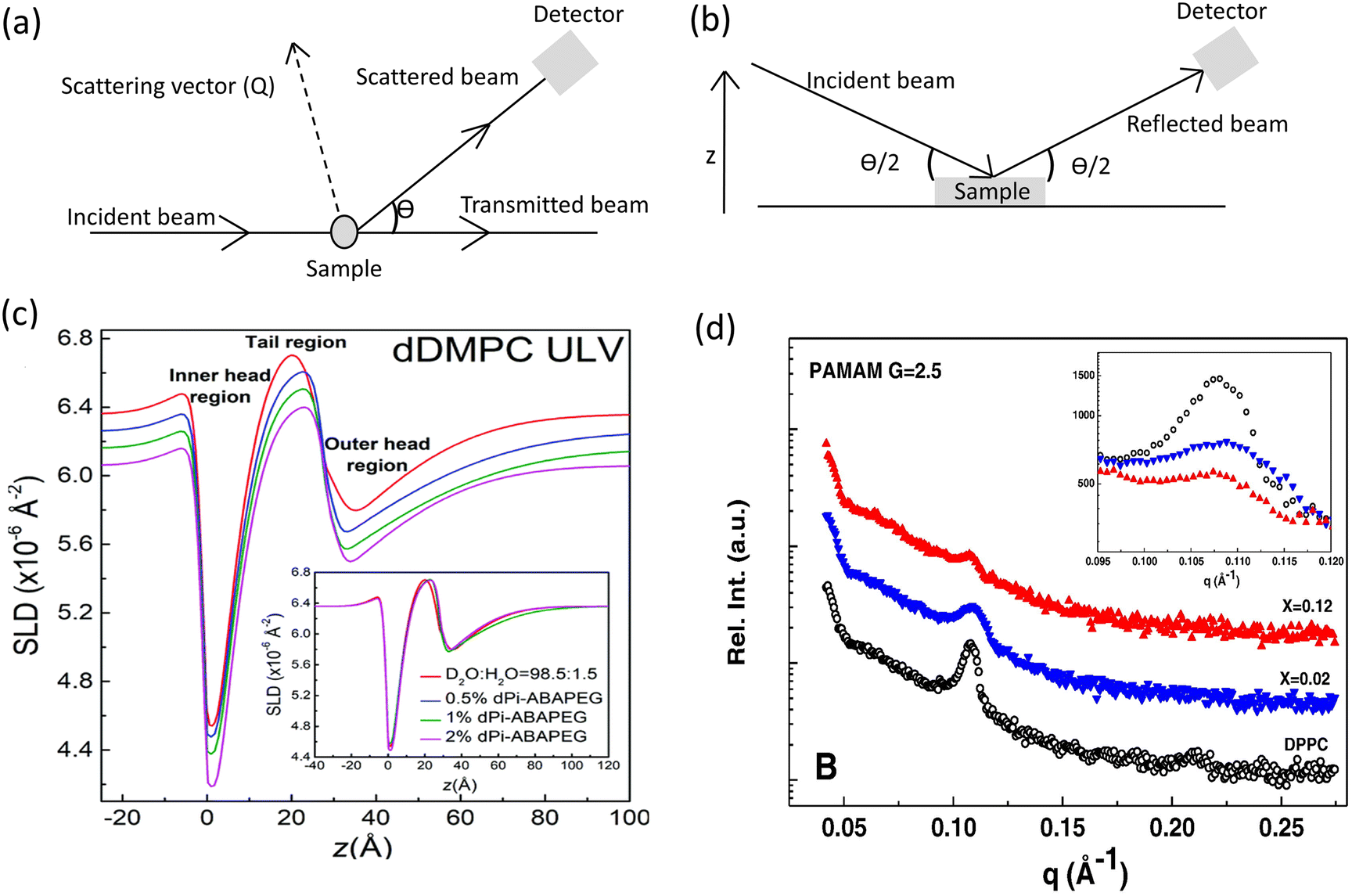 | ||
| Fig. 5 Comparison of the experimental principles behind (a) small-angle scattering and (b) reflectometry techniques. (c) SLD profiles measured by SANS of lipid vesicles interacting with varying mass fractions of phosphorylated end-capped PEG triblock copolymer measured by SANS. (d) SAXS spectra of dendrimers/DPPC interaction during vesicle formation at different dendrimer molar fractions. Magnified view of the Bragg peaks is reported in the insets. (c) Adapted with permission from ref. 98. Copyright 2005, Royal Society of Chemistry. (d) Adapted with permission from ref. 74. Copyright 2016, Elsevier. | ||
Small-angle neutron scattering (SANS) and small-angle X-ray scattering (SAXS) measures elastic scattering at low angles from bulk samples or solutions. These techniques provide statistical averages of nanostructures, such as vesicles, micelles, or membrane pores, by analysing the intensity and distribution of scattered radiation. The resulting data reveals information about particle size, shape, and spatial arrangement in three-dimensional space.96,97 A recent example of polymer–membrane interactions studied using SANS characterized how the chemical modification of phosphorylated polyethylene glycol (PEG) triblock copolymers affect their binding behaviour with lipid vesicles.98 Their SANS analysis specifically addressed whether phosphorylation induced structural perturbations in membranes. Analysis of the low *q* region showed identical scattering patterns across all polymer concentrations, confirming the overall membrane integrity remained intact. However, nanoscale changes emerged in the scattering length density (SLD) profiles, revealing slight outer-leaflet thickening, approximately 1–2 Å (Fig. 5c). These differences in SLD profile were restricted to the membrane surface with no evidence of deep penetration, demonstrating the polymer merely adsorbed to the bilayer exterior without deep insertion.99 Similarly, SANS studies of the interactions of styrene oligomers with DPPC bilayers revealed significant nanoscale membrane perturbations with important biological implications.100 Using contrast-matched samples of deuterated (d-DPPC) and hydrogenated (DPPC) lipids in D2O/H2O solvent systems, researchers observed increased scattering intensity in oligomer-containing membranes, confirming their integration into both gel and fluid phases. In the fluid phase, SANS data showed that styrene oligomers intercalate uniformly between lipid tails, disrupting packing without forming large aggregates. This behaviour was evidenced by the absence of deviations in bilayer-model fitting of SANS curves, alongside a measurable reduction in membrane rigidity. Such fluid-phase remodelling is physiologically relevant, as most cell membranes exist in a fluid state at body temperature.100 The findings suggest that styrene oligomers may destabilize lipid rafts or increase membrane permeability, providing a plausible mechanism for their cellular toxicity.
Similar to SANS, SAXS has provided critical structural insights into how polymer properties influence membrane organization, a key consideration for optimizing nanomedicines For example, SAXS was used to investigate the impact of both anionic and cationic polyamidoamine (PAMAM) dendrimers (generations 2.5 and 3.0) on model cell membranes.74 The study examined two scenarios: dendrimers added during liposome preparation and dendrimers introduced to preformed membranes. While incorporating dendrimers during liposome formation does not fully mimic in vivo conditions, it was employed to enable comprehensive analysis of dendrimer–lipid interactions, with particular focus on structural modifications and electrostatic effects. When added during preparation, both dendrimer generations at different molar fractions altered bilayer organization. The anionic dendrimers (generation 2.5) exhibited a stronger disruptive effect shown in the Bragg reflections on the SAXS pattern located in the low q region (Fig. 5d). This was attributed to electrostatic repulsion and partial embedding of the dendrimers into the bilayer, which perturbed lipid packing in the hydrophobic core. In contrast, introducing dendrimers to preformed liposomes caused no significant structural changes. Through its ability to detect nanoscale structural changes, SAXS proved essential in linking dendrimer-induced membrane disruption to drug delivery design strategies.74 In a follow-up study, the same team investigated two generations (G = 2.0 and G = 4.0) of amine-terminated PAMAM dendrimers varying molar fractions of the two generations of dendrimers, which confirmed and extended prior SAXS finding by providing quantitative insights into dendrimer penetration depth, and membrane structural reorganization. This offered a complete understanding of dendrimer–membrane interactions relevant to drug delivery and cytotoxicity.73
Neutron Reflectometry (NR) and X-ray Reflectometry (XRR) focus on specular reflection from flat surfaces or interfaces. By measuring the intensity of reflected beams as a function of incident angle, these techniques generate depth profiles perpendicular to the surface and accurately measure layer thickness, density, and roughness.101 NR/XRR is particularly adept at resolving interfacial phenomena, such as lipid bilayer formation or polymer adsorption, with sub-nanometre precision in depth profiles. NR has been used to provide molecular-scale insights into the interface between amphipathic copolymers with lipid monolayers at the air–liquid interface.51 The study illustrated how NR bridges the gap between nanoscale structural details such as intercalation depth revealed by SLD profiles and changes in membrane properties like monolayer stability and polymer binding affinity measured by tensiometry and QCM-D. Most significantly, the NR data provided direct evidence of intercalation of hydrophobic sidechains of the copolymers, explaining the strong copolymer binding rather than mere surface adsorption found in the QCM-D experiments (Fig. 6a and b).51 This study highlights the advantage of using multiple complementary techniques to fully understand the interactions between synthetic macromolecules and lipid membranes. Together, these techniques can provide complementary structural insights for the rational design of membrane-active polymers as nanomedicines.
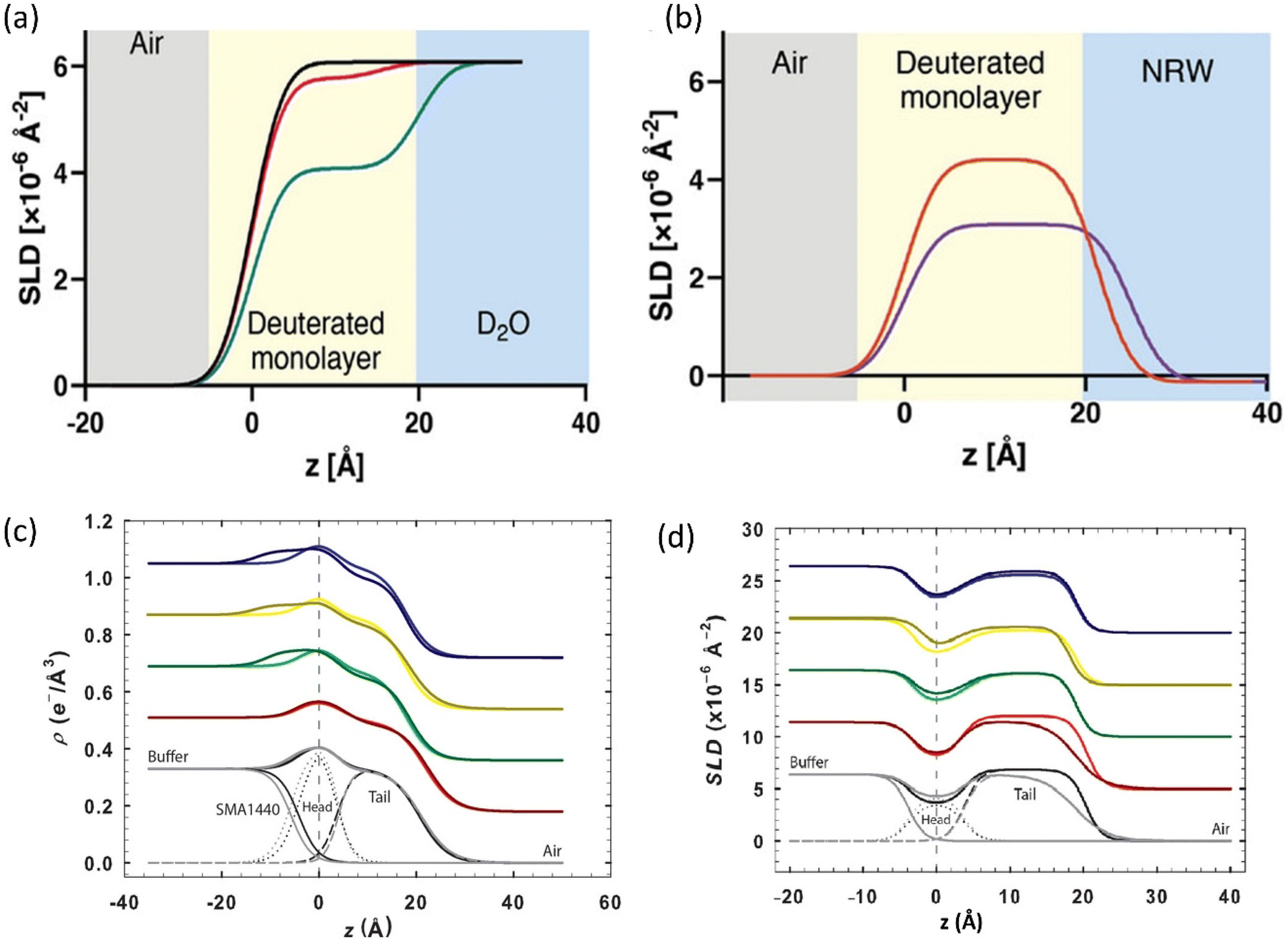 | ||
| Fig. 6 Neutron reflectometry is a powerful tool for studying polymer–membrane interactions. SLD profiles using (a) D2O subphase reveal structural reorganization of the lipid interface upon association of amphipathic copolymers, and no evidence of a polymer rich underlayer. Profiles show: D2O (black), [D77]-POPC monolayer (pink), and with copolymer (green). (b) Null reflecting water subphase isolates nanoscale structural changes in the deuterated [D77]-POPC monolayer upon copolymer association. Profiles show: [D77]-POPC monolayer (pink), and with copolymer (violet). (c) Electron density profiles from X-ray reflectometry showing SMA copolymer interactions with various binary lipid mixtures and (d) complementary NR SLD profile highlighting the spatial distribution of polymer components across the lipid interface. Curve colours indicate lipid compositions: black (DPPC), blue (DPPC/DGDG), yellow (DPPC/SQDG), green (DPPC/MGDG), and red (DPPC/DPPG). (a) and (b) Adapted with permission from ref. 51. Licensed under a Creative Commons Attribution-Non-Commercial (CC BY-NC). (c) and (d) adapted with permission from ref. 102. Copyright 2020, American Chemical Society. | ||
Neutron reflectometry and X-ray reflectometry have also been utilized as complementary techniques to probe the molecular interactions between a styrene–maleic acid (SMA 1440) copolymer derivative, and lipid monolayers containing galactolipids representative of thylakoid membranes.102 SMA copolymers are widely recognized for their ability to isolate membrane proteins by forming nanoscale SMA-lipid particles (SMALPs) that preserve native protein–lipid interactions without the use of detergents. In this study, NR and XRR were used to characterize structural and compositional changes in monolayers upon SMA 1440 adsorption, offering insight into its membrane disruptive behaviour. The study utilized binary lipid mixtures composed of 80 mol% DPPC and 20 mol% of either monogalactosyldiacylglycerol (MGDG), digalactosyldiacylglycerol (DGDG), or sulfoquinovosyldiacylglycerol (SQDG) to mimic natural membrane environments. Key findings from the XRR analysis showed that incorporation of galactolipids reduced membrane thickness and electron density relative to pure DPPC (Fig. 6c), reflecting increased fluidity due to the flexible galactosyl headgroups. Upon SMA 1440 addition, further reductions in electron density were observed in both headgroup and tail regions, indicating disrupted packing from polymer insertion. Additionally, it was shown that SMA formed distinct interfacial layers beneath the monolayers, with more pronounced effects in DGDG and SQDG systems than in MGDG. These changes were less pronounced in phospholipid-rich membranes, highlighting SMA's preference for galactolipids. XRR provided high-resolution nano-structural mapping that revealed the layer thickness changes and copolymer distribution. NR measurements also provided critical molecular-level insights into how SMA copolymers interact with and remodel lipid membranes by leveraging the unique contrast capabilities of neutron scattering. The NR data demonstrated that maleic acid groups of SMA, consistently localized near the lipid headgroups, as evidenced by increased scattering length density (SLD) in this region across all membrane compositions (Fig. 6d). The tail region of galactolipid-rich membranes also exhibited an increase in SLD, in surprising contrast to phospholipids membranes. This rise was attributed to the penetration of butoxyethanol, a hydrophobic yet partially hydrated side chain of SMA 1440. The elevated SLD suggested that butoxyethanol, unlike pure styrene, carries associated water molecules into the galactolipid tails, further enhancing membrane fluidity.102
The study of polymer–membrane interactions has advanced significantly through the complementary use of scattering and mass sensitive techniques. While SPR and QCM capture real-time interaction kinetics and scattering techniques offer ensemble-averaged structural insights, nanoscale microscopy is a technique that uniquely bridges these perspectives by providing direct spatiotemporal visualization of interfacial dynamics. Integrating these advanced microscopy techniques with conventional interfacial methods promises to transform fundamental understanding and accelerate the design of innovative biomaterials.
Nanoscale microscopic imaging to probe interfaces between polymers and lipid
Because seeing is believing, nanoscale microscopic imaging is essential for directly visualizing and mapping the structure, function and dynamics of macromolecular assemblies and membrane organization. These relationships are highly complex and heterogenous at the nanometer scale. High-resolution microscopy techniques, including atomic force microscopy (AFM), transmission electron microscopy (TEM) and scanning electron microscopy (SEM) have emerged as essential tools to probe and provide insights into the complex and dynamic interfacial phenomena between membranes and polymeric materials. TEM offers near-atomic resolution, making it a powerful tool for studying the interactions of lipid membranes with polymers.103,104 The strong electron density contrast between lipid membranes and their surrounding polymer matrix or aqueous media allows clear delineation of vesicle boundaries. This allows determination of membrane thicknesses and subtle structural variations induced by polymer incorporation, facilitating precise nanoscale characterization.105,106 Accordingly, TEM has been used to study the lyotropic behavior of polyhydroxy alkyl amide and its interaction with the bilayer forming phospholipid DPPC.107 Furthermore, TEM was employed to examine the miscibility of bolalipid with bilayer-forming phosphatidylcholines, specifically 1,2-distearoyl-sn-glycero-3-phosphocholine (DSPC), DMPC and DPPC.108 Varying degrees of phase separation and mixing were demonstrated, suggesting that the compatibility between bolalipids and these phospholipids depends strongly on their acyl chain characteristics and membrane packing properties. These findings provide valuable insights into the structural organization and potential functionality of mixed lipid systems in biomimetic membranes.Cryogenic-transmission electron microscopy (cryo-TEM) further enhances capabilities by preserving the native hydration and nanoscale organization of lipid–polymer interactions.109–112 Cryo-TEM allows high-resolution imaging of vesicle morphology, bilayer thickness, and the spatial distribution of lipid and polymer components without dehydration artefacts, making it ideal for capturing unilamellar and multilamellar membranes, degree of polymer incorporation, and the presence of inhomogeneities or phase-separated domains.113–115 Cryo-TEM also supports tomographic reconstruction, allowing visualization of fine molecular details and capturing population-level structural variations.111 Compared to conventional super-resolution optical techniques that lack the spatial resolution required to resolve sub-nanometer structural components, and techniques like expansion microscopy that depend on crosslinked proteins and cannot resolve polymer-based architectures, cryo-TEM provides a uniquely powerful tool for studying supported lipid bilayers (SLBs) and related nanostructures.
Studies using cryo-TEM have demonstrated the formation of well-defined bilayers from liposomes composed of various lipid types, including positively charged DOTAP (1,2-dioleoyl-3-trimethylammonium-propane), neutral zwitterionic lipids like DOPC, POPC, and DMPC, as well as negatively charged lipids such as DOPS (1,2-dioleoyl-sn-glycero-3-phospho-L-serine).103,116 These SLBs were formed on silica nanoparticles (Fig. 7a) through vesicle adsorption, rupture, and spreading on the nanoparticle surface.116 Furthermore, cryo-electron tomography imaging of POPC vesicles containing increasing mole fractions (0–100 mol%) of the block copolymer PBd22-PEO14 showed increasing membrane thickness with polymer content (Fig. 7b). At intermediate compositions (50 mol% and 75 mol%), the quantitative membrane thickness displayed bimodal distribution, corresponding to distinct populations of thin (lipid-like) and thick (polymer-like) membrane regions (Fig. 7b, far right image). The data suggested that vesicles adopt either thin or thick membrane configurations, with no intermediate structures observed, indicating a bistable membrane architecture.111 Overall, cryo-TEM enables direct, artifact-free visualization of membrane morphology, bilayer coverage, and nanoscale structural variation, offering critical insights into how lipid composition and polymer incorporation influence membrane organization and interactions.
 | ||
| Fig. 7 (a) Cryo-transmission electron microscopy (cryo-TEM) images of supported lipid bilayers (SLBs) on silica nanoparticles formed with small unilamellar vesicles (SUVs) composed of neutral lipids: (i) DOPC and (ii) POPC. (iii) Schematic image of silica nanoparticles coated with a SLB. Surrounding the nanoparticle is ring of electron-dense material (green circle) representing the outer leaflet of the SLB. Scale bar: 20 nm. (b) Cryo-electron tomography (cryo-ET) images of POPC vesicles containing increasing mole fractions of 0%, 25%, 50%, 75%, and 100 mol% PBd22-PEO14. The image at the far right represents membrane thickness measurements, determined by identifying the outer edges of contrast in cryo-ET images of individual membranes across the full range (0–100%) of PBd22-PEO14 vesicle samples. The analysis of individual membrane images from vesicle samples containing 0–100% PBd22-PEO14 reveals a clear increase in membrane thickness with increasing PBd22-PEO14 mole fraction. (a) Adapted with permission from ref. 116 Copyright 2005, American Chemical Society. (b) Adapted with permission from ref. 111. Licensed under a Creative Commons Attribution (CC BY) License. | ||
Scanning electron microscopy (SEM) can provide complementary information to TEM, on the surface morphology of lipid bilayers, particularly in dry states. SEM is often less suited for soft matter due to the risk of dehydration and structural alteration during sample preparation. Despite this fact, it is useful for visualizing lipid membrane-substrate interfaces and surface topography, offering insights into membrane uniformity and defect formation.117,118 In contrast, environmental SEM (ESEM) allows imaging of hydrated lipid bilayer systems, avoiding the need for extensive sample preparation. For instance, ESEM has been successfully used to visualize phosphatidylcholine-containing cholesterol liposomes, clearly revealing the formation of spherical vesicular structures.119,120 ESEM is effective for observing POPC liposomes in their hydrated state, preserving their morphology without drying or coating.119 To minimize damage and imaging artefacts under ESEM, gradual reduction of pressure in the sample chamber is essential when imaging delicate structures like supported lipid bilayers.119 These precautions help maintain bilayer integrity and enable clearer visualization of surface features. Moreover, achieving meaningful insights from SEM analysis relies heavily on meticulous sample preparation, including techniques like etching, fracturing, and staining, which are critical for revealing these otherwise hidden structural features.121
Electron microscopic techniques present notable limitations, including complex sample preparation procedures. For example, TEM requires the preparation of ultrathin sections of the sample films, a process that often introduces artefacts that can distort structural integrity. This ultrathin sectioning also limits compositional sensitivity, as only surface accessible molecules can be effectively analyzed. A commonly used method of visualizing submicron liquid domains in TEM is the gold labeling of either lipids or proteins,42,43,122 but this labeling approach can lead to overcounting or other interpretation challenges.123 For example, multiple labels conjugated to a single antibody, or the combined use of primary and secondary antibodies may produce an apparent overcounting of molecular species, which can be mistakenly interpreted as self-clustering protein rather than an artifact of the labeling protocol. While cryo-TEM tomography delivers unparalleled three-dimensional nanometer-scale imaging of hydrated membranes, its effectiveness is limited by dependence on thickness contrast, representativeness of samples, probe accessibility, and low throughput. Typically, TEM benefits from being complemented by other techniques (e.g., SAXS or fluorescence microscopy) to overcome interpretation or sampling limitations.40,111 Again, SEM suffers from limited compositional sensitivity, and the inherently low electrical conductivity of polymer and lipid often results in significant charging issues during imaging.124–126 The multiscale challenge persists in correlating structural hierarchies with functional mechanisms. While computational modeling bridges atomic-scale interactions (via molecular dynamics simulations) to bulk polymer behavior,127,128 experimental limitations remain in simultaneously capturing: (1) nanoscale dynamics (e.g., lipid–polymer membrane reorganization or polymer-sized pores in lipid bilayers); (2) macroscopic performance (e.g., mechanical properties of polymer-lipid interfaces). Emerging super-resolution optical techniques like lipid expansion microscopy (LExM) can complement electron microscopy and are helping to close these gaps. LExM now achieves ∼70 nm resolution for membrane phospholipids using hydrogel-anchoring strategies.129 Structured illumination techniques have been developed to enable live-cell lipid droplet tracking via carbonized polymer. However, integrating these advances with traditional microscopy and multiscale simulations remains critical for deciphering structure–function relationships in hybrid systems.130 Nanomechanical microscopic techniques are able to directly address some of the challenges of electron microscopy.
Atomic force microscopy (AFM) is a widely adopted technique in both physical and biological sciences, particularly valued for its capacity to image soft material interfaces between macromolecules and supported lipid bilayers under ambient or physiological conditions.131,132 An advanced AFM operates as a nanomechanical probe-based technique, wherein interaction forces between a sharp nanoscale tip and the sample surface are transduced into cantilever deflections (Fig. 8a), enabling high-resolution mapping of surface topography and physical properties of materials without the need for chemical fixation, fluorescence staining or sample labeling.133–136 AFM enables dynamic measurements with temporal resolution from microsecond (μs) to minutes, making it particularly well-suited for studying the biophysical behavior of polymers and lipid membranes. For instance, AFM has revealed nanoscale phase separation within DPPC–DOPC lipid bilayers supported on PDMS substrates and has been used to characterize phase domain separation in polymer-lipid membranes, such as DPPC with poly(dimethyl siloxane)-poly(2-methyl-2-oxazoline) (PDMS-PMOXA) copolymers.137,138 Additionally, AFM was used to determine the average diameters and domain structures of lipid membranes incorporating polybutadiene-poly(ethylene oxide) (PBd-b-PEO) block copolymers in vesicle formulations.106 AFM combines high-resolution 3D imaging with measurement in native conditions, large statistical sampling, and minimal sample preparation. This capability enables detailed analysis of nanoscale mechanics, viscoelasticity, and topographical features that are otherwise inaccessible through conventional microscopy techniques.
 | ||
| Fig. 8 (a) Schematic description of atomic force microscopy (AFM) showing the fundamental measurement principle. A laser is focused on a soft and flexible cantilever (AFM tip), and its reflected beam is captured by a photodiode. When the AFM tip operates in contact mode with the sample, any deflection of the tip causes the laser spot to shift on the photodiode, allowing precise movement of tip. (b) An exemplary AFM image of an integral membrane protein complex in a supported lipid bilayer. Images (i–iii) shown in magenta depict individual membrane protein, distinguished by their height as either periplasmic-facing or cytoplasmic-facing components as illustrated in images (iv–vi) in green. Scale bars measure 200 nm (c) and 10 nm (i–iv). (c) Smoothed histogram representing the maximum height of membrane protein protrusions above the lipid bilayer surface, based on measurements from N = 15957 individual protrusions. The peak positions for each population are as follows: peak 1 = 14 Å (green); peak 2 = 27 Å (magenta); and peak 3 = 60 Å (blue). The inset in (c) displays a comparison of histogram profiles between wild type integral membrane protein complex and integral membrane protein complex without the periplasmic domain P1 (ΔP1). (b) and (c) Adapted with permission from ref. 141. Licensed under a Creative Commons Attribution (CC BY) License. | ||
The advancement of high speed-AFM (HS-AFM) tools can enable real-time visualization of a single molecule at nanometer and microsecond scales.139–141 HS-AFM offers lateral resolutions down to ∼10 Å (1 nm) and vertical resolutions around ∼1 Å (0.1 nm). In force-based HS-AFM, a single molecule may be tethered to both the tip and the substrate through mechanisms such as physical adsorption, ligand–receptor binding, or covalent bonding.142 As the cantilever moves vertically and stretches the molecule, the force exerted is recorded until rupture occurs.143 These techniques have been instrumental in studying dynamic processes like membrane remodeling, molecular motor activity, and polymer assembly allowing high-resolution imaging of membrane complexes within supported lipid bilayers and revealing a multimodal height distribution (Fig. 8b and c). Its capacity to characterize across a broad range of environments, from vacuum to physiological liquids, and across diverse temperature conditions, has made it invaluable for capturing lipid phase transitions and polymer-induced membrane remodeling.144–146
Emerging multimodal AFM approaches now integrate mechanical (e.g., adhesion, elasticity, dissipation), electrical, thermal properties and spectroscopic mapping including tip-enhanced Raman spectroscopy (TERS) and infrared-based techniques.147–150 They have significantly expanded the functional range of conventional AFM. These innovations offer unprecedented opportunities to probe structure and function of membranes and polymers at high spatiotemporal resolution, bridging the gap between static imaging techniques (e.g., TEM, SEM) and dynamic, real-time observation. Force spectroscopy (i.e., force–distance curve-based AFM), for instance, has been used to quantify polymer binding kinetics while simultaneously mapping the nanomechanical landscape of membranes.139 This dual capability provides detailed insights into both the chemical and physical nature of molecular interactions at sub-nanoscale resolution. Although the application of HS-AFM to study lipid bilayer-polymer systems remain in its early stages, its potential to uncover complex biological and synthetic membrane systems, such as drug delivery platforms and mechanobiological interfaces is increasingly evident.11,151,152
Despite its capabilities, AFM remains underutilized in areas of membrane biophysics and polymer interface research, largely due to challenges associated with nanoscale imaging in liquid environments and the need for careful sample preparation. The interaction between an AFM tip and sample can induce considerable damage or deformation to the lipid membranes, vesicles or polymer surface during scanning process.49,106,153,154 While supported lipid bilayers and polymer films, and their interactions can be imaged on flat substrates like mica or silicon wafer, achieving uniformity and artifact-free imaging requires precise handling.131,135,138,155 Surface charge density of commonly used substrates in AFM analysis, such as mica, silicon wafers and gold, can impact the structure and interactions of polymer, peptides and lipid bilayers in aqueous buffer environments.134,156,157 These electrostatic properties can significantly influence not only the adsorption and organization of biomolecules at the interface but also the overall stability and morphology of the resulting films or membranes. To the best of our knowledge, there is currently little to no information available in literature regarding the interaction of either lipid or protein membranes with calcite surfaces, largely due to challenges posed by the substrate's atomic-scale irregularities (e.g. steps or defect sites) and heterogenous charge distributions. While foundational work highlights these complexities, the field lacks systematic investigations into how membranes adapt to such surfaces, particularly in synthetic contexts.158,159
Buffer selection further amplifies or stabilizes these effects, as the ionic composition and pH of the buffer can modulate both substrate charge and the screening of electrostatic interactions. This, in turn, can affect imaging outcomes, including the stability, fluidity, and phase behavior of supported lipid bilayers. A particularly striking example is observed with gel-phase domains in supported lipid bilayers on mica. The atomically flat and negatively charged mica surface forms strong interactions with the lipid domains, effectively immobilizing them. This immobilization not only restricts the lateral movement of gel-phase domains but also promotes domain registry across the two bilayer leaflets, stabilizing the overall membrane structure.142,157 In contrast, unsupported (freestanding) membrane systems lack these direct substrate interactions, but instead gel-phase domains in these systems exhibit greater mobility due to the absence of direct interfacial interactions with a solid surface. This difference shows the profound influence of substrate choice and interfacial chemistry on membrane dynamics and organization, factors that must be carefully considered when designing experiments and interpreting results in membrane biophysics and nanotechnology.
Magnetic resonance techniques to probe molecular dynamics and local environments at polymer–lipid membrane interfaces
Magnetic resonance methods are non-destructive, label-free techniques that use the magnetic properties of atomic nuclei or unpaired electrons to probe molecular structure, dynamics, and interactions. When subjected to an external magnetic field, these particles exhibit specific energy transitions that can be detected and analysed using radiofrequency signals. Among these methods nuclear magnetic resonance (NMR) and electron paramagnetic resonance (EPR) have emerged as powerful tools to characterize polymer lipid interactions.Pulsed-field gradient NMR (PFG-NMR) is a solution-state technique that provides quantitative measurements of molecular self-diffusion.160 It is especially valuable in the study of macromolecule–lipid membrane interactions, as it allows monitoring of changes in the translational mobility of macromolecules upon association with lipid assemblies such as micelles and liposomes. By comparing the diffusion coefficients of a macromolecule in the absence and presence of lipid membranes, PFG-NMR can directly infer binding events or membrane insertion. For example, polymers that partition into lipid bilayers exhibit significantly reduced diffusion coefficients, reflective of their association with larger, slower-diffusing membrane structures. This method offers a label-free, non-destructive approach to probe interactions under physiologically relevant conditions. However, its sensitivity to sample concentration and the requirement for relatively homogeneous systems can pose challenges when applied to complex or heterogeneous macromolecular assemblies.
PFG-NMR has been used to probe how temperature and thermal history influence the interactions between amphiphilic block copolymers (Pluronics F68 and F127) and phospholipid liposomes using PFG-NMR.161 The results revealed a strong temperature-dependent enhancement in binding. Samples thermally incubated showed significantly more polymer association (∼29% bound chains) compared to control samples kept at 25 °C (∼4% bound). Binding peaked at 37 °C (∼45%) and decreased at 50 °C due to micelle formation. Notably, a fraction of polymer remained bound even after cooling, indicating kinetic trapping. Further analysis confirmed that this binding state is non-equilibrium but reversible, with desorption occurring slowly over days to weeks. The work modelled desorption kinetics using a first-order process and proposed that elevated temperatures dehydrate PEO/PPO blocks, facilitating transbilayer insertion into liposomes. Upon cooling, rehydration barriers prevent release, leading to trapping. Thermal cycling between 10 °C and 37 °C confirmed this mechanism, where polymers bound at 37 °C remained associated even after cooling, while those bound at 25 °C desorbed completely. Cholesterol incorporation (0–14 mol%) confirmed the role of membrane stiffness, with higher cholesterol levels hindering polymer insertion and slowing desorption kinetics. These results support the idea that membrane mechanical properties regulate transbilayer insertion and kinetic trapping. Finally, polymer architecture also significantly influenced membrane interaction. PEO homopolymers failed to bind, while polymers with small PPO blocks bound only after high-temperature incubation. A bottlebrush polymer with similar composition to F127 bound weakly and did not show thermal trapping, likely due to steric hindrance. This highlights how linear, flexible architectures favour bilayer insertion and trapping, while bulky rigid structures do not.161
PFG-NMR was used to study a complementary series of amphiphilic block copolymers (specifically polaxmers PBO-b-PEO and PPO-b-PEO) with POPC liposomes. The technique enabled differentiation between free polymer chains, micelles, and liposome-bound polymers by analysing their diffusion behaviour. The results showed that PBO-b-PEO copolymers exhibited significant membrane binding, with approximately 30–40% of the polymer population associated with liposomes. This binding increased the liposome hydrodynamic radius, likely due to the formation of a polymer corona.162 In contrast, PPO-based copolymers showed minimal to no detectable binding. The extent of binding was largely independent of the polymer block length. Membrane binding reduced the micellar population without significantly affecting the free chain fraction. Furthermore, PFG-NMR could show how the mechanical properties of cell membranes change upon poloxamer binding. The results demonstrated that the hydrophobic poloxamers with higher molar mass showed greater affinity towards the cell membrane by reducing the stretching modulus.163 Although PFG-NMR effectively captured strong and stable interactions and provided quantitative method for membrane studies, it was less sensitive to weak or transient binding events, which may go undetected due to fast exchange rates.162 Overestimation of membrane-bound polymer and inability to distinguish between outer and inner leaflet binding suggest that PFG-NMR alone may not fully explain mechanical property changes.163
Electron paramagnetic resonance (EPR) is a useful technique for obtaining localized structural and dynamic information on nanoparticle cell interactions. Like nuclear magnetic resonance, EPR offers high detection sensitivity. Two common EPR approaches include using spin probes and spin labels. A spin label is a stable free radical that is covalently attached to a specific site on a molecule, whereas a spin probe is a small, stable paramagnetic molecule that is added to the system to measure structural and dynamic information of their environment. These are effective for characterising the interactions of macromolecules with model membranes, cellular systems and even in living systems.164,165
To investigate polymer–membrane interactions, a dual approach using both a spin probe and a spin label can be used. With this approach, the spin probe can penetrate the lipid membranes, while the spin label is covalently attached to the macromolecule, thereby enabling simultaneous measurements of membrane dynamics and polymer interactions (Fig. 9a). For example, Schiff-based carbosilane dendrimers with Ru(II) complexes were studied with cetyltrimethylammonium bromide micelles (CTAB) and lecithin liposomes (LEC) as model membranes. The study revealed how dendrimer structure, metal coordination, and dendrimer generation modulate the interaction strength and binding mode with membranes (Fig. 9). In this work, 4-(N,N-dimethyl N-dodecyl) ammonium-2,2,6,6-tetramethylpiperidine-1-oxyl bromide (CAT12) was used as the spin probe and 2,2,6,6-tetramethylpiperidine-1-oxyl (TEMPO) was used as the spin label to investigate the polymer dendrimer interactions.164 This approach allowed simultaneous monitoring of interactions from both the dendrimer side and the membrane side. As shown in Fig. 9b (left) the EPR spectra for spin label showed two components including a free component (narrow lines, fast-moving radicals) and an interacting component (broader lines, slower motion), which provided information into local microviscosity and polarity. While Fig. 9b (middle and right) shows the experimental and computed free and interacting components for spin label alone and in the presence of CTAB. The dendrimers without Ru initially presented only the free component, indicating high radical mobility on the dendrimer surface. Upon addition of CTAB micelles, the mobility decreased and microviscosity increased, suggesting partial insertion of the radical into the micellar structure. In contrast, LEC caused minimal change, indicating weak interactions. Both free and interacting components were observed in metallodendrimers even in the absence of lipid membranes. This was due to restricted radical motion caused by interaction with the Ru complex. Addition of membranes, especially CTAB, caused only slight further changes. Furthermore, the study of CAT12 as a spin probe revealed membrane-side effects, such as probe extraction, aggregation, and changes in local environment depending on dendrimer generation and surface chemistry. Notably, the presence of a single –NH2 group significantly altered interaction modes, favouring polar surface interactions over micelle penetration. This dual EPR method provided detailed, real-time insights into nanocarrier–membrane dynamics that are critical for designing efficient drug delivery systems.
 | ||
| Fig. 9 Using EPR to study the interactions of a metallodendrimers with liposomes. (a) Schematic of using the spin label (TEMPO) covalently attached to the dendrimer while the spin probe (4-(N,N-dimethyl N-dodecyl) ammonium-2,2,6,6-tetramethylpiperidine-1-oxyl bromide (CAT12)) was embedded with model membranes. (b) EPR spectra of the labelled dendrimers in the absence and presence of the model membranes. Left: an experimental EPR spectrum of dendrimers containing Ru(II) complexes in phosphate-buffered saline is shown as a representative example. Arrows indicate the presence of both free and interacting components. Middle and right: the experimental and simulated spectra corresponding to the interacting components of the dendrimers with Ru(II) complexes, either alone or in the presence of cetyltrimethylammonium bromide micelles. Adapted with permission from ref. 164. Licensed under a Creative Commons Attribution (CC BY) License. | ||
EPR studies were conducted to investigate the surface interactions of DOTA glycodendrimers with various model lipid membranes.166 Using a spin probe embedded in the liposome bilayers, the study demonstrated that the DOTA glycodendrimers, both in its free form and as a Cu(II) complex, interacts predominantly at the membrane surface without penetrating or disrupting the bilayer structure. EPR revealed noncovalent, surface-driven interactions, including electrostatic, hydrogen bonding, and ion-dipole forces between the positively charged dendrimer and the lipid headgroups. These interactions were especially shown with negatively charged 1,2-dimyristoyl-sn-glycero-3-phospho rac-(1-glycerol) sodium salt (DMPG) and structurally heterogeneous LEC liposomes, where membrane composition and surface heterogeneity modulated the dendrimer's binding affinity and dynamics. Notably, EPR also indicated a weakening of Cu–N2O2 coordination upon liposome addition, attributed to competitive dendrimer–membrane interactions. These findings highlight the strength of EPR in providing real-time, molecular level insight into polymer–membrane interactions, particularly in assessing dynamic surface behaviour and complexation effects.
EPR and NMR offer site-specific and sensitive insights into local environmental properties and molecular interactions. Unlike many optical methods, both techniques can be used with samples that are turbid or opaque, making them ideal for studying complex systems like micelles or liposomes without the need for optical transparency.166 As with fluorescent techniques, EPR is limited by the necessity to label the nanomedicine, unless the polymer has an inherent EPR signal (stable radical or paramagnetic metal centre). Again, this can introduce artificial interactions that may alter or change how the polymer interacts with lipid membranes. Although NMR does not require labelling, this technique heavily relies on components having unique, identifiable resonances. Enhancing selectivity through labelling with unique chemical signatures like 19F or paramagnetic species can also be used, but this method faces similar drawbacks as EPR spin labelling and fluorophore labelling.
Conclusions and future perspectives
We argue that a deeper understanding of the interactions between polymers and lipid membranes is critical for advancing the development and preclinical translation of polymer-based nanomedicines. While extensive research has explored how protein recognition, receptors, and transporters mediate cellular uptake, the complementary role of direct interactions with lipid membranes, where these proteins are embedded, is often overlooked. This gap is partly due to the abundance of established assays focusing on protein-mediated internalization, which often fail to capture the full complexity of uptake pathways, particularly as polymer architecture and chemistry evolve during biological processes. We have highlighted in this review that while some common in vitro and microscopic techniques can provide insight into polymer–lipid membrane interactions, significant limitations arise when their results are interpreted in isolation.In this review, we introduced and contextualized a range of complementary analytical techniques that can be used to probe polymer–lipid membrane interactions (Table 1). Although many of these methods originate from biophysics, physical chemistry and colloid science, they offer valuable insights into interfacial behaviours that are highly relevant to nanomedicine. By highlighting these tools, we aim to encourage researchers in nanomedicine to integrate fundamental biophysical characterisation into their experimental design, fostering a more fundamental understanding of cellular interactions. Conversely, we hope to inspire biophysicists and colloid scientists to engage with the complex challenges posed by applied nanomedicine systems, where their expertise can contribute to a system level understanding of the behaviour of nanomedicines.
| Technique | Model system | Key Insight | Advantages | Limitations |
|---|---|---|---|---|
| FRAP: fluorescence recovery after photobleaching, FLCS: fluorescence lifetime correlation spectroscopy, 2fFCS: two-focus fluorescence correlation spectroscopy, PIE-FCCS: pulsed interleaved excitation fluorescence cross-correlation spectroscopy, SPR: surface plasmon resonance, QCM-D: quartz crystal microbalance, SANS: small angle neutron scattering, SAXS: small angle X-ray scattering, NR: neutron reflectometry, XRR: X-ray reflection, EDX/EDS: energy-dispersive X-ray spectroscopy. | ||||
| Confocal imaging-based techniques (fluorescence microscopy, FRAP) | Living or fixed cells, supported bilayers, fluorescently labelled lipids or polymers | Location and spatial distribution of the polymers and lipids. Measures molecular mobility and lateral diffusion (FRAP) | High spatial resolution, real time imaging, compatible with live cells. FRAP provides quantitative assessment of membrane fluidity and dynamics (FRAP) | Semi quantitative method, artefacts from photobleaching or dye aggregation. FRAP assumes homogeneous diffusion and sensitive to substrate (FRAP) |
| Fluorescence assays (membrane leakage, haemolysis and LDH) | Living cells, model membranes | Cell membrane disruption through dye leakage | Provide qualitative and semi quantitative information about membrane perturbation, distinguishes between full rupture and transient pore formation | Cannot identify the precise site or pathway of disruption |
| Fluorescence anisotropy | Lipid vesicles with embedded fluorescent probes | Measures change in rotational mobility of fluorophores which gives information about lipid packing and membrane fluidity | Sensitive to small changes in membrane order, provides quantitative measurements, can distinguish between the inner or outer leaflet when different probes are used | Does not directly probe lateral diffusion or polymer binding sites, the outcome may be affected by probe location and concentration, requires careful calibration |
| Fluorescence correlation spectroscopy (FLCS, 2fFCS, PIE-FCS) | Live cells, liposomes, supported lipid bilayers, immobilised biological membranes | Molecular diffusion, polymer-lipid interactions, and complex formation, quantification of dynamic heterogeneities | High sensitivity, high resolution, distinguishes interacting and non-interacting species, provides absolute or relative diffusion coefficients | Technically demanding alignment and calibration, multiple fluorescent labels required, data interpretation complex, requires stable fluorescence and low background |
| Brewster angle microscopy | Langmuir lipid monolayers at the air–liquid interface | Visualizes monolayer morphology and domain organization, detects phase transitions and polymer-induced structural rearrangements | Label-free and real-time imaging, sensitive to changes in monolayer order anisotropy | Limited to air–liquid interfaces (monolayers only, not bilayers), does not provide molecular-scale resolution, quantitative analysis requires careful calibration |
| Vibrational spectroscopy | Lipid vesicles, supported bilayers | Lipid composition, phase transitions, polymer-induced changes in lipid packing or hydration | Label-free, sensitive to conformational changes of lipids and hydrogen bonding, ATR-FTIR or Raman minimizes water absorption | Strong IR absorption of water limits transmission FTIR, requires high lipid concentration, ATR-FTIR may introduce artefacts from crystal, limited spatial resolution. Raman is less sensitive than FT-IR and can suffer from background fluorescence |
| Dynamic light scattering | Liposomes, micelles, polymer self-assembly | Measures hydrodynamic diameter and size distribution Detects polymer-induced vesicle aggregation, fusion, disruption, or nanoparticle assembly/disassembly | Rapid, label-free, and non-invasive, sensitive to size changes, can measure polymer effects on vesicle stability, real-time aggregation behaviour | Requires dilute samples to avoid multiple scattering, not accurate on samples with mixed size distributions, provides limited structural or mechanistic detail |
| Mass sensitive techniques (SPR, QCM-D) | Supported lipid bilayer, vesicles, immobilised biological membranes | SPR measures binding affinity and kinetics (associate rate constant, dissociation rate constant and binding capacity) between polymers and membranes. QCM-D provides real-time kinetics of bilayer formation, mechanical properties, association kinetics and structural integrity of bilayers upon interaction with polymers | No labelling of sample is required. Measurements are proportional to adsorbed mass, and structural properties | Limited to surfaces immobilised models. Only measures physical changes, no chemical information. Difficulty detecting very low concentrations and distinguishing nonspecific binding events |
| Isothermal calorimetry | Lipid vesicles, micelles | Measures binding kinetics and provides information about enthalpy and entropy change | Label-free measurement. A complete thermodynamic profile (enthalpy and entropy change, and Gibbs free energy) of binding events | Does not measure binding kinetics. Sensitive to temperature fluctuations, experiments must be done in stable thermal conditions |
| Small angle scattering (SANS, SAXS) | Lipid vesicles, polymer self-assembly | Vesicle size and shape, bilayer thickness, vesicle aggregation, size dispersity. Changes in properties can be measured after polymer association | Label-free measurements. Structural information of vesicles is obtained while in solution. Component specific analysis due to contrast variation is possible. | Do not provide molecular information. May need contrast variation, or isotopic labelling of analyte (SANS). SAXS has low contrast for organic materials. SANS require neutron sources which are limited |
| Reflectometry (NR, XRR) | Langmuir monolayers, supported lipid bilayers, immobilised biological membranes | Structural information about lipid monolayers, bilayers or membranes, while NR can highlight specific components in complex structures using contrast variation | Non-destructive and applicable in situ. NR can be used for component specific analysis due to isotopic labelling. XRR has high resolution and is widely available | Do not provide molecular information. Requires complex fitting of models to interpret. May need contrast variation, or isotopic labelling of analyte (NR). XRR has low contrast for organic materials. NR require neutron sources which are limited. |
| Cryogenic transmission electron microscopy | Fixed cells, micelles, liposomes, supported lipid bilayers, polymer–lipid hybrids, polymer self-assembly | Direct imagining of polymer-liposome interactions, nanoparticle size, shape, and surface morphology. Detects and quantifies bilayer thickness changes, vesicle deformation, fusion, or disruption upon polymer binding | Provides high-spatial resolution of membrane architecture near-native morphology, internal structural detail and direct structural observation | Complex sample preparation (staining, freezing, and sectioning) can introduce artefacts, unable to capture dynamic polymer-bilayer interactions in real time, and electron irradiation can alter soft organic materials |
| Scanning electron microscopy | Fixed cells, supported lipid films, polymer-coated membranes, or dried vesicle aggregates | Reveals surface texture and architecture, polymer coating behaviour and identifies membrane blebbing, deformation, or nanoparticle accumulation on cell membranes | Rapid imaging of large areas, enables chemical composition mapping of polymer-membrane interfaces with EDX/EDS coupling | Non-native conditions imaging with sample preparation that can obscure fine surface details or alter morphology, provides no direct information on internal bilayer organization or polymer encapsulation |
| Atomic force microscopy | Living or fixed cells, lipid vesicles, polymer self-assembly, supported lipid bilayers | Visualizes and quantifies polymer-induced morphological and mechanical alterations at micro- and nano-scale resolution, uncovering mechanisms of adsorption, penetration, or disruption | High-resolution micro- and nano-sized imaging, label-free, liquid-phase compatibility, real-time or high-speed imaging, quantitative force analysis in near-physiological conditions and mechanical mapping | Restricted temporal and spatial scale, possible tip artefacts, less suitable for dynamic or suspended membranes |
| Nuclear magnetic resonance | Lipid vesicles, micelles, polymer self-assembly | Quantifies molecular self-diffusion, detects polymer–membrane association via reduced diffusion coefficients, differentiates free chains and membrane-bound polymers | Label-free, non-destructive, provides absolute diffusion data under physiologically relevant conditions, sensitive to strong/stable binding and trans-bilayer insertion | Less sensitive to weak or fast-exchanging interactions, requires homogeneous samples, diffusion averaging may overestimate bound fractions, cannot distinguish inner vs outer leaflet binding |
| Electron paramagnetic resonance | Lipid vesicles, micelles, supported lipid bilayers | Measures local polarity, microviscosity, mobility, and insertion depth can resolve polymer surface binding vs penetration, detects transient and heterogeneous interactions. | High sensitivity, site-specific information, tolerant of opaque samples, can simultaneously probe polymer and membrane dynamics | Requires spin labelling, labels may alternative interactions, spectral deconvolution can be complex |
By collaborating across disciplines and capturing polymer-lipid membrane interactions into a systems level understanding of the mechanisms of nanomedicines, future research will be able to address some of the most significant challenges in nanomedicine. This includes elucidating cellular uptake mechanisms, and how subtle changes in nanomedicine chemistry or cell populations, can lead to vast differences in uptake. Internalisation of a nanomedicine is only the first step, with endosomal escape for drugs and nucleic acid therapies being the next major challenge. For example, commercial lipid nanoparticle formulations for mRNA vaccine delivery have shown less than 10% endosomal escape. Considering these interactions also offers up new possibilities, outside of the realm of antimicrobials, to consider polymers as APIs rather than only carriers.
This review has aimed to present both routine analysis and cutting-edge developments in the analytical techniques used to study the interactions of polymers with lipid membranes. While technological advancements in each technique will continue, it is the unique insight that can be provided by using multiple complimentary techniques, where new researchers can push the frontier of existing science. Considering how to measure the dynamic processes driving polymer–lipid membrane interactions in real times, with multiple techniques to probe both interfacial dynamics and structural changes in situ.
Finally, while this review presents the relative simplicity of model lipid membranes as advantageous over the complexity of real biological membranes, a true systems level understanding cannot be achieved with siloed approaches. Our review has focused on models of membranes composed only of lipids. While we have presented some attempts to increase the complexity of model systems to better emulate realistic or natural membranes, further progress in this area is still needed. It is critical to consider the role of interfaces between different lipid species, lipid rafts, polysaccharides, or interfaces between lipids and proteins within biological membranes on the internalisation of nanomedicines. Concurrently considering how to translate the analytical techniques reviewed here beyond model systems, to real biological membranes would significantly advance our understanding of nanomedicine function. This includes studying membranes both in isolation and within living systems. Such efforts would further push the envelope of our knowledge, reveal new insights into polymer–membrane interactions and ultimately guide the design of more effective nanomedicines.
Author contributions
The manuscript was prepared through contributions of all authors, with specific contributions recognized below using the CRediT framework. Ayomi V. Pathiranage: data curation, writing – original draft, writing – review and editing. Mark-Jefferson B. Boyetey – writing – original draft, writing – review and editing. Oluwatoosin B. A. Agbaje – writing – original draft, writing – review and editing. Nathan R. B. Boase – data curation, funding acquisition, project administration, supervision, writing – original draft, writing – review and editing.Conflicts of interest
There are no conflicts to declare.Data availability
No primary research results, software or code have been included, and no new data were generated or analysed as part of this review.Acknowledgements
M-J. B. B. would like to acknowledge QUT Postgraduate Research Award (QUTPRA) (International), QUT HDR Tuition Fee Sponsorship and Australian Institute of Nuclear Science and Engineering (AINSE) Postgraduate Research Award (PGRA) Top-Up Scholarship. O. B. A. A. and N. R. B. B. would like to acknowledge Australian Research Council for Funding to support their work (LP220100450). All authors would like to acknowledge the QUT Centre for Materials Science for ongoing funding support for their work.References
- H.-C. Huang, S. Barua, G. Sharma, S. K. Dey and K. Rege, J. Controlled Release, 2011, 155, 344 CrossRef CAS PubMed.
- M. A. Beach, U. Nayanathara, Y. Gao, C. Zhang, Y. Xiong, Y. Wang and G. K. Such, Chem. Rev., 2024, 124, 5505 CrossRef CAS PubMed.
- S. Patel, J. Kim, M. Herrera, A. Mukherjee, A. V. Kabanov and G. Sahay, Adv. Drug Delivery Rev., 2019, 144, 90 CrossRef CAS PubMed.
- Y. K. Sung and S. W. Kim, Biomater. Res., 2020, 24, 12 CrossRef CAS PubMed.
- K. Knop, R. Hoogenboom, D. Fischer and U. S. Schubert, Angew. Chem., Int. Ed., 2010, 49, 6288 CrossRef CAS PubMed.
- J. Humphries, D. Pizzi, S. E. Sonderegger, N. L. Fletcher, Z. H. Houston, C. A. Bell, K. Kempe and K. J. Thurecht, Biomacromolecules, 2020, 21, 3318 CrossRef CAS PubMed.
- A. Birke, J. Ling and M. Barz, Prog. Polym. Sci., 2018, 81, 163 CrossRef CAS.
- M. Menéndez, in eLS, 2020, p. 113, DOI:10.1002/9780470015902.a0028808.
- K. J. Kauffman, C. Do, S. Sharma, M. D. Gallovic, E. M. Bachelder and K. M. Ainslie, ACS Appl. Mater. Interfaces, 2012, 4, 4149 CrossRef CAS PubMed.
- U. Nayanathara, F. Yang, C. Zhang, Y. Wang, B. R. Herling, S. A. Smith, M. A. Beach, A. P. R. Johnston and G. K. Such, Biomater. Sci., 2025, 13, 1335 RSC.
- P. K. C. Chang, C. A. Prestidge and K. E. Bremmell, Pharm. Res., 2022, 39, 1151 CrossRef CAS PubMed.
- Z. Wang, J. Zhang, Y. Wang, J. Zhou, X. Jiao, M. Han, X. Zhang, H. Hu, R. Su, Y. Zhang and W. Qi, ACS Nano, 2024, 18, 10324 CrossRef CAS PubMed.
- L. M. P. Vermeulen, S. C. De Smedt, K. Remaut and K. Braeckmans, Eur. J. Pharm. Biopharm., 2018, 129, 184 CrossRef CAS PubMed.
- Y. Wang, V. Ukwattage, Y. Xiong and G. K. Such, Mater. Horiz., 2025, 12, 3622 RSC.
- S. A. Smith, L. I. Selby, A. P. R. Johnston and G. K. Such, Bioconjugate Chem., 2019, 30, 263 CrossRef CAS PubMed.
- S. Alkarri, H. Bin Saad and M. Soliman, Polymers, 2024, 16, 771 CrossRef CAS PubMed.
- P. Kumari and A. Kumar, in Antiviral and Antimicrobial Smart Coatings, ed. A. Kumar, A. Behera, T. A. Nguyen, M. Bilal and R. K. Gupta, Elsevier, 2023, p. 239, DOI:10.1016/B978-0-323-99291-6.00003-7.
- J. Jonca, C. Tukaj, W. Werel, U. Mizerska, W. Fortuniak and J. Chojnowski, J. Mater. Sci.: Mater. Med., 2016, 27, 55 CrossRef PubMed.
- W. Fortuniak, U. Mizerska, J. Chojnowski, T. Basinska, S. Slomkowski, M. M. Chehimi, A. Konopacka, K. Turecka and W. Werel, J. Inorg. Organomet. Polym. Mater., 2011, 21, 576 CrossRef CAS.
- U. Mizerska, W. Fortuniak, J. Chojnowski, R. Hałasa, A. Konopacka and W. Werel, Eur. Polym. J., 2009, 45, 779 CrossRef CAS.
- U. Mizerska, W. Fortuniak, J. Chojnowski, K. Turecka, A. Konopacka and W. Werel, J. Inorg. Organomet. Polym. Mater., 2010, 20, 554 CrossRef CAS.
- K. Rózga-Wijas, U. Mizerska, W. Fortuniak, J. Chojnowski, R. Hałasa and W. Werel, J. Inorg. Organomet. Polym. Mater., 2007, 17, 605 CrossRef.
- V. Cagno, P. Andreozzi, M. D’Alicarnasso, P. J. Silva, M. Mueller, M. Galloux, R. Le Goffic, S. T. Jones, M. Vallino, J. Hodek, J. Weber, S. Sen, E. R. Janeček, A. Bekdemir, B. Sanavio, C. Martinelli, M. Donalisio, M.-A. R. Welti, J. F. Eleouet, Y. Han, L. Kaiser, L. Vukovic, C. Tapparel, P. Král, S. Krol, D. Lembo and F. Stellacci, Nat. Mater., 2018, 17, 195 CrossRef CAS PubMed.
- R. Groß, L. M. D. Loiola, L. Issmail, N. Uhlig, V. Eberlein, C. Conzelmann, L. R. Olari, L. Rauch, J. Lawrenz, T. Weil, J. A. Müller, M. B. Cardoso, A. Gilg, O. Larsson, U. Höglund, S. A. Pålsson, A. S. Tvilum, K. B. Løvschall, M. M. Kristensen, A. L. Spetz, F. Hontonnou, M. Galloux, T. Grunwald, A. N. Zelikin and J. Münch, Adv. Sci., 2022, 9, e2201378 CrossRef PubMed.
- L. M. Jones, E. H. Super, L. J. Batt, M. Gasbarri, F. Coppola, L. M. Bhebhe, B. T. Cheesman, A. M. Howe, P. Král, R. Coulston and S. T. Jones, ACS Infect. Dis., 2022, 8, 2084 CrossRef CAS PubMed.
- S. T. Jones, V. Cagno, M. Janeček, D. Ortiz, N. Gasilova, J. Piret, M. Gasbarri, D. A. Constant, Y. Han, L. Vuković, P. Král, L. Kaiser, S. Huang, S. Constant, K. Kirkegaard, G. Boivin, F. Stellacci and C. Tapparel, Sci. Adv., 2020, 6, eaax9318 CrossRef CAS PubMed.
- E. Mohammadifar, M. Gasbarri, V. Cagno, K. Achazi, C. Tapparel, R. Haag and F. Stellacci, Biomacromolecules, 2022, 23, 983 CrossRef CAS PubMed.
- E. H. Super, S. M. Lai, U. Cytlak-Chaudhuri, F. Coppola, O. Saouaf, Y. E. Song, K. M. Casey, L. J. Batt, S.-L. Macleod, R. H. T. Bagley, Z. Walsh-Korb, P. Král, E. A. Appel, M. A. Travis and S. T. Jones, bioRxiv, 2024, DOI:10.1101/2024.07.10.602907.
- H. M. Mengist, P. Denman, C. Frost, J. D. J. Sng, S. Logan, T. Yarlagadda, K. M. Spann, L. Barner, K. E. Fairfull-Smith, K. R. Short and N. R. B. Boase, Biomacromolecules, 2024, 25, 7377 CrossRef CAS PubMed.
- A. Luchini and G. Vitiello, Biomimetics, 2021, 6, 3 CrossRef CAS PubMed.
- W. Ntow-Boahene, I. Papandronicou, J. Miculob and L. Good, Sci. Rep., 2023, 13, 2790 CrossRef CAS PubMed.
- E. Boonstra, H. Hatano, Y. Miyahara, S. Uchida, T. Goda and H. Cabral, J. Mater. Chem. B, 2021, 9, 4298 RSC.
- B. Eshaghi, N. Alsharif, X. An, H. Akiyama, K. A. Brown, S. Gummuluru and B. M. Reinhard, Adv. Sci., 2020, 7, 2000649 CrossRef CAS PubMed.
- R. Datta, A. Gillette, M. Stefely and M. C. Skala, Biomed. Opt., 2021, 26, 070603 Search PubMed.
- A. D. Elliott, Curr. Protoc. Cytom., 2020, 92, e68 CrossRef PubMed.
- J. D. Simpson, G. R. Ediriweera, C. B. Howard, N. L. Fletcher, C. A. Bell and K. J. Thurecht, Biomater. Sci., 2019, 7, 4661 RSC.
- S. Chen, S. Wang, M. Kopytynski, M. Bachelet and R. Chen, ACS Appl. Mater. Interfaces, 2017, 9, 8021 CrossRef CAS PubMed.
- S. Chen and R. Chen, ACS Appl. Mater. Interfaces, 2016, 8, 22457 CrossRef CAS PubMed.
- I. P. Sæbø, M. Bjørås, H. Franzyk, E. Helgesen and J. A. Booth, Int. J. Mol. Sci., 2023, 24, 2914 CrossRef PubMed.
- C. E. Cornell, A. Mileant, N. Thakkar, K. K. Lee and S. L. Keller, Proc. Natl. Acad. Sci. U. S. A., 2020, 117, 19713 CrossRef CAS PubMed.
- H. Stahlberg and T. Walz, ACS Chem. Biol., 2008, 3, 268 CrossRef CAS PubMed.
- B. S. Wilson, S. L. Steinberg, K. Liederman, J. R. Pfeiffer, Z. Surviladze, J. Zhang, L. E. Samelson, L.-h. Yang, P. G. Kotula and J. M. Oliver, Mol. Biol. Cell, 2004, 15, 2580 CrossRef CAS PubMed.
- T. J. LaRocca, P. Pathak, S. Chiantia, A. Toledo, J. R. Silvius, J. L. Benach and E. London, PLoS Pathog., 2013, 9, e1003353 CrossRef CAS PubMed.
- R. J. C. Gilbert, in Methods in Enzymology, ed. A. P. Heuck, Academic Press, 2021, vol. 649, p. 71 Search PubMed.
- R. Henderson, S. Chen, J. Z. Chen, N. Grigorieff, L. A. Passmore, L. Ciccarelli, J. L. Rubinstein, R. A. Crowther, P. L. Stewart and P. B. Rosenthal, J. Mol. Biol., 2011, 413, 1028 CrossRef CAS PubMed.
- C. Sebaaly, H. Greige-Gerges and C. Charcosset, in Current Trends and Future Developments on (Bio-) Membranes, ed. A. Basile and C. Charcosset, Elsevier, 2019, p. 311, DOI:10.1016/B978-0-12-813606-5.00011-7.
- Y. Lu, G. Allegri and J. Huskens, Mater. Horiz., 2022, 9, 892 RSC.
- N.-J. Cho, C. W. Frank, B. Kasemo and F. Höök, Nat. Protoc., 2010, 5, 1096 CrossRef CAS PubMed.
- Y. Yan, Y. Sun, Y. Yang, Z. Hu and X. Zhao, Appl. Surf. Sci., 2012, 258, 9656 CrossRef CAS.
- S. Perumal, R. Atchudan and W. Lee, Polymers, 2022, 14, 2510 CrossRef CAS PubMed.
- C. Frost, S. Wiedbrauk, E. Graham, N. R. Yepuri, A. R. J. Nelson and N. R. B. Boase, Macromol. Chem. Phys., 2023, 224, 2300315 CrossRef CAS.
- L. A. Clifton, R. A. Campbell, F. Sebastiani, J. Campos-Terán, J. F. Gonzalez-Martinez, S. Björklund, J. Sotres and M. Cárdenas, Adv. Colloid Interface Sci., 2020, 277, 102118 CrossRef CAS PubMed.
- P. Quan, W. Bu, L. Wang, C. Chen, X. Wu, C. Heffern, K. Y. C. Lee, M. Meron and B. Lin, J. Phys.: Mater., 2021, 4, 034004 CAS.
- S. C. L. Hall, C. Tognoloni, R. A. Campbell, J. Richens, P. O’Shea, A. E. Terry, G. J. Price, T. R. Dafforn, K. J. Edler and T. Arnold, J. Colloid Interface Sci., 2022, 625, 220 CrossRef CAS PubMed.
- A. P. Girard-Egrot and O. Maniti, Appl. Sci., 2021, 11, 4876 CrossRef CAS.
- Y. Liu and J. Liu, Langmuir, 2020, 36, 810 CrossRef CAS PubMed.
- R. J. Kopiasz, A. Rukasz, K. Chreptowicz, R. Podgorski, A. Kuzminska, J. Mierzejewska, W. Tomaszewski, T. Ciach and D. Janczewski, Colloids Surf., B, 2021, 207, 112016 CrossRef CAS PubMed.
- D. Kozon-Markiewicz, R. J. Kopiasz, M. Glusiec, A. Lukasiak, P. Bednarczyk and D. Janczewski, Colloids Surf., B, 2023, 229, 113480 CrossRef CAS PubMed.
- C. M. St. Croix and S. H. Shand, BioTechniques, 2005, 39, S2 CrossRef PubMed.
- M. J. Sanderson, I. Smith, I. Parker and M. D. Bootman, Cold Spring Harb. Protoc., 2014, 2014, pdb–top071795 Search PubMed.
- X. Liu, T. Auth, N. Hazra, M. F. Ebbesen, J. Brewer, G. Gompper, J. J. Crassous and E. Sparr, Proc. Natl. Acad. Sci. U. S. A., 2023, 120, e2217534120 CrossRef CAS PubMed.
- S. Selvaraj, S. Krishnaswamy, V. Devashya, S. Sethuraman and U. M. Krishnan, Prog. Lipid Res., 2015, 58, 1 CrossRef CAS PubMed.
- D. Wrobel, A. Edr, E. Zemanova, T. Strasak, A. Semeradtova and J. Maly, Chem. Phys. Lipids, 2023, 255, 105314 CrossRef CAS PubMed.
- C. A. Day, L. J. Kraft, M. Kang and A. K. Kenworthy, Curr. Protoc. Cytom., 2012, Ch. 2, Unit2.19 Search PubMed.
- H. Su, H. Y. Liu, A. M. Pappa, T. C. Hidalgo, P. Cavassin, S. Inal, R. M. Owens and S. Daniel, ACS Appl. Mater. Interfaces, 2019, 11, 43799 CrossRef CAS PubMed.
- A. Ghosh, N. Karedla, J. C. Thiele, I. Gregor and J. Enderlein, Methods, 2018, 140–141, 32 CrossRef CAS PubMed.
- S. Ramadurai, M. Werner, N. K. H. Slater, A. Martin, V. A. Baulin and T. E. Keyes, Soft Matter, 2017, 13, 3690 RSC.
- C. E. Pinguet, E. Ryll, A. A. Steinschulte, J. M. Hoffmann, M. Brugnoni, A. Sybachin, D. Woll, A. Yaroslavov, W. Richtering and F. A. Plamper, PLoS One, 2019, 14, e0210898 CrossRef CAS PubMed.
- X. Shi, X. Li, M. J. Kaliszewski, X. Zhuang and A. W. Smith, Langmuir, 2015, 31, 1784 CrossRef CAS PubMed.
- W. Daear, M. Mahadeo and E. J. Prenner, Biochim. Biophys. Acta, Biomembr., 2017, 1859, 1749 CrossRef CAS PubMed.
- M. Skrzypiec and K. Prochaska, Colloids Surf., B, 2018, 171, 167 CrossRef CAS PubMed.
- R. N. A. H. Lewis and R. N. McElhaney, in Methods in Membrane Lipids, ed. A. M. Dopico, Humana Press, Totowa, NJ, 2007, pp. 207, DOI:10.1007/978-1-59745-519-0_14.
- D. Lombardo, P. Calandra, S. Magazu, U. Wanderlingh, D. Barreca, L. Pasqua and M. A. Kiselev, Colloids Surf., B, 2018, 170, 609 CrossRef CAS PubMed.
- D. Lombardo, P. Calandra, E. Bellocco, G. Lagana, D. Barreca, S. Magazu, U. Wanderlingh and M. A. Kiselev, Biochim. Biophys. Acta, 2016, 1858, 2769 CrossRef CAS PubMed.
- J. Stetefeld, S. A. McKenna and T. R. Patel, Biophys. Rev., 2016, 8, 409 CrossRef CAS PubMed.
- M. Gorbunova, A. Efimova, T. Grokhovskaya and Y. Beloglazova, Colloid Polym. Sci., 2024, 303, 147 CrossRef.
- M. Schulz, A. Olubummo and W. H. Binder, Soft Matter, 2012, 8, 4849 RSC.
- P. Wang, in Comprehensive Analytical Chemistry, Elsevier, 2021, vol. 95, pp. 55 Search PubMed.
- X. Wang, J. Wang and M. Sun, Appl. Mater. Today, 2019, 15, 212 CrossRef.
- R. B. M. Schasfoort, in Handbook of Surface Plasmon Resonance, ed. R. B. M. Schasfoort, The Royal Society of Chemistry, 2017, 10.1039/9781788010283-00001.
- E. Helmerhorst, D. J. Chandler, M. Nussio and C. D. Mamotte, Clin. Biochem. Rev., 2012, 33, 161 Search PubMed.
- M. Kim, M. Vala, C. T. Ertsgaard, S. H. Oh, T. P. Lodge, F. S. Bates and B. J. Hackel, Langmuir, 2018, 34, 6703 CrossRef CAS PubMed.
- F. Zhao, X. Cheng, G. Liu and G. Zhang, J. Phys. Chem. B, 2010, 114, 1271 CrossRef CAS PubMed.
- T. Chen, J. Xin, S. J. Chang, C. J. Chen and J. T. Liu, Adv. Mater. Interfaces, 2023, 10, 2202202 CrossRef.
- C. S. Schneider, A. G. Bhargav, J. G. Perez, A. S. Wadajkar, J. A. Winkles, G. F. Woodworth and A. J. Kim, J. Controlled Release, 2015, 219, 331 CrossRef CAS PubMed.
- R. J. Mosley, M. V. Talarico and M. E. Byrne, J. Bioact. Compat. Polym., 2021, 36, 261 CrossRef.
- A. Alassi, M. Benammar and D. Brett, Sensors, 2017, 17, 2799 CrossRef PubMed.
- M. Bekir, C. Brückner, S. Zauscher and M. Gradzielski, Colloids Surf., A, 2023, 677, 132354 CrossRef CAS.
- G. K. Krishnamoorthy, P. Alluvada, S. H. M. Sherieff, T. Kwa and J. Krishnamoorthy, Biochem. Biophys. Rep., 2020, 21, 100712 Search PubMed.
- V. K. Srivastava and R. Yadav, in Data Processing Handbook for Complex Biological Data Sources, 2019, p. 125, DOI:10.1016/b978-0-12-816548-5.00009-5.
- P. I. Semenyuk, A. A. Efimova, I. I. Lentin, I. M. Le-Deygen and V. A. Izumrudov, Langmuir, 2020, 36, 14717 CrossRef CAS PubMed.
- N. Zhang, S. Dai, R. Qi and Y. Han, Colloids Surf., A, 2025, 708 CAS.
- R. Santra, J. Phys. B: At., Mol. Opt. Phys., 2009, 42, 169801 CrossRef.
- T. Narayanan and O. Konovalov, Materials, 2020, 13, 752 CrossRef CAS PubMed.
- R. Richardson, in Colloid Science, 2005, p. 228, DOI:10.1002/9781444305395.ch12.
- L. Caselli, L. Conti, I. De Santis and D. Berti, Adv. Colloid Interface Sci., 2024, 327, 103156 CrossRef CAS PubMed.
- E. Metwalli, K. Gotz, S. Lages, C. Bar, T. Zech, D. M. Noll, I. Schuldes, T. Schindler, A. Prihoda, H. Lang, J. Grasser, M. Jacques, L. Didier, A. Cyril, A. Martel, L. Porcar and T. Unruh, J. Appl. Crystallogr., 2020, 53, 722 CrossRef CAS PubMed.
- J. Yu, J. Mao, M. Nagao, W. Bu, B. Lin, K. Hong, Z. Jiang, Y. Liu, S. Qian, M. Tirrell and W. Chen, Soft Matter, 2020, 16, 983 RSC.
- M. Kim, F. Heinrich, G. Haugstad, G. Yu, G. Yuan, S. K. Satija, W. Zhang, H. S. Seo, J. M. Metzger, S. M. Azarin, T. P. Lodge, B. J. Hackel and F. S. Bates, Langmuir, 2020, 36, 3393 CrossRef CAS.
- M. I. Morandi, M. Kluzek, J. Wolff, A. Schroder, F. Thalmann and C. M. Marques, Proc. Natl. Acad. Sci. U. S. A., 2021, 118, e2016037118 CrossRef CAS PubMed.
- S. Basu and S. Singh, Neutron and X-ray Reflectometry: Emerging phenomena at heterostructure interfaces, IOP Publishing, 2022 Search PubMed.
- M. D. Phan, O. I. Korotych, N. G. Brady, M. M. Davis, S. K. Satija, J. F. Ankner and B. D. Bruce, Langmuir, 2020, 36, 3970 CrossRef CAS PubMed.
- A. Meister and A. Blume, Polymers, 2017, 9, 521 CrossRef PubMed.
- K. Mio and C. Sato, Biophys. Rev., 2018, 10, 307–316 CrossRef CAS PubMed.
- Q. Chen, H. Schönherr and G. Vancso, Soft Matter, 2009, 5, 4944–4950 RSC.
- C. Presutti, E. Vreeker, S. Sasidharan, Z. Ferdinando, M. Stuart, J. Juhaniewicz-Dębińska, G. Maglia, W. H. Roos and B. Poolman, Biomacromolecules, 2025, 26, 2868 CrossRef CAS PubMed.
- P. Scholtysek, A. Achilles, C.-V. Hoffmann, B.-D. Lechner, A. Meister, C. Tschierske, K. Saalwächter, K. Edwards and A. Blume, J. Phys. Chem. B, 2012, 116, 4871 CrossRef CAS PubMed.
- S. Drescher, V. M. Garamus, C. J. Garvey, A. Meister and A. Blume, Beilstein J. Org. Chem., 2017, 13, 995 CrossRef CAS PubMed.
- R. Cao, J. Gao, S. Thayumanavan and A. D. Dinsmore, Soft Matter, 2021, 17, 7069 RSC.
- X. Zhang, Z. Ding, G. Ma and W. Wei, Adv. Sci., 2021, 8, 2004506 CrossRef CAS PubMed.
- R. Seneviratne, G. Coates, Z. Xu, C. E. Cornell, R. F. Thompson, A. Sadeghpour, D. P. Maskell, L. J. Jeuken, M. Rappolt and P. A. Beales, Small, 2023, 19, 2206267 CrossRef CAS.
- N. Sen, S. Krüger and W. H. Binder, RSC Chem. Biol., 2024, 5, 1248 RSC.
- M. Chountoulesi, N. Pippa, A. Forys, B. Trzebicka and S. Pispas, Polymers, 2024, 16, 290 CrossRef CAS PubMed.
- E. Brodszkij, I. N. Westensee, S. F. Holleufer, C. Ade, P. D. D. Andres, J. S. Pedersen and B. Städler, Appl. Mater. Today, 2022, 29, 101549 CrossRef.
- O. Le Bihan, P. Bonnafous, L. Marak, T. Bickel, S. Trépout, S. Mornet, F. De Haas, H. Talbot, J. C. Taveau and O. Lambert, J. Struct. Biol., 2009, 168, 419 CrossRef CAS PubMed.
- S. Mornet, O. Lambert, E. Duguet and A. Brisson, Nano Lett., 2005, 5, 281 CrossRef CAS PubMed.
- M. M. Pérez-Madrigal, L. J. del Valle, E. Armelin, C. Michaux, G. Roussel, E. A. Perpète and C. Alemán, ACS Appl. Mater. Interfaces, 2015, 7, 1632 CrossRef PubMed.
- A. Puiggalí-Jou, M. M. Pérez-Madrigal, L. J. del Valle, E. Armelin, M. T. Casas, C. Michaux, E. A. Perpète, F. Estrany and C. Alemán, Nanoscale, 2016, 8, 16922 RSC.
- Y. Perrie, H. Ali, D. J. Kirby, A. U. R. Mohammed, S. E. McNeil and A. Vangala, in Liposomes: Methods and Protocols, ed. G. G. M. D'Souza, Springer New York, New York, NY, 2017, p. 131, DOI:10.1007/978-1-4939-6591-5_11.
- A. R. Mohammed, N. Weston, A. G. Coombes, M. Fitzgerald and Y. Perrie, Int. J. Pharm., 2004, 285, 23 CrossRef CAS PubMed.
- G. Goizueta, T. Chiba and T. Inoue, Polymer, 1992, 33, 886 CrossRef CAS.
- C. P. Mercogliano and D. J. DeRosier, J. Struct. Biol., 2007, 160, 70 CrossRef CAS PubMed.
- S. L. Veatch, B. B. Machta, S. A. Shelby, E. N. Chiang, D. A. Holowka and B. A. Baird, PLoS One, 2012, 7, e31457 CrossRef CAS PubMed.
- D. Lee, U. Jeong and D. Kim, Sci. Adv., 2025, 11, eadt6177 CrossRef CAS.
- B. G. Kopek, G. Shtengel, J. B. Grimm, D. A. Clayton and H. F. Hess, PLoS One, 2013, 8, e77209 CrossRef CAS PubMed.
- P. Wandrol and M. Slouf, Microsc. Microanal., 2017, 23, 1816 CrossRef.
- A. Gooneie, S. Schuschnigg and C. Holzer, Polymer, 2017, 9, 16 Search PubMed.
- K. Lin and Z. Wang, Commun. Mater., 2023, 4, 66 CrossRef CAS.
- B. M. White, P. Kumar, A. N. Conwell, K. Wu and J. M. Baskin, J. Am. Chem. Soc., 2022, 144, 18212 CrossRef CAS PubMed.
- A. Makky, J. Czajor, O. Konovalov, A. Zhakhov, A. Ischenko, A. Behl, S. Singh, W. Abuillan and M. Shevtsov, Sci. Rep., 2023, 13, 19157 CrossRef CAS PubMed.
- P. Guha, B. Roy, P. Nahak, G. Karmakar, A. Karak, A. G. Bykov, A. B. Akentiev, B. A. Noskov, K. Dutta, C. Ghosh and A. K. Panda, J. Oleo Sci., 2024, 73, 547 CrossRef CAS PubMed.
- D. C. Bode, M. Freeley, J. Nield, M. Palma and J. H. Viles, J. Biol. Chem., 2019, 294, 7566 CrossRef CAS PubMed.
- C. L. Freeman, L. Dieudonné, O. B. A. Agbaje, M. Žure, J. Q. Sanz, M. Collins and K. K. Sand, Environ. DNA, 2023, 5, 1691 CrossRef CAS.
- A. E. Pittman, B. P. Marsh and G. M. King, Langmuir, 2018, 34, 8393 CrossRef CAS PubMed.
- K. Schaefer, A. Pittman, F. Barrera and G. King, Methods, 2022, 197, 20 CrossRef CAS PubMed.
- P. K. Johansson, D. Jullesson, A. Elfwing, S. I. Liin, C. Musumeci, E. Zeglio, F. Elinder, N. Solin and O. Inganäs, Sci. Rep., 2015, 5, 11242 CrossRef PubMed.
- S. Di Leone, M. Kyropoulou, J. Köchlin, R. Wehr, W. P. Meier and C. G. Palivan, Langmuir, 2022, 38, 6561 CrossRef CAS PubMed.
- J. A. Goodchild, D. L. Walsh, H. Laurent and S. D. Connell, Langmuir, 2023, 39, 10843 CrossRef CAS.
- M. Pfreundschuh, D. Martinez-Martin, E. Mulvihill, S. Wegmann and D. J. Muller, Nat. Protoc., 2014, 9, 1113 CrossRef CAS PubMed.
- S. Scheuring, J. Seguin, S. Marco, D. Lévy, B. Robert and J.-L. Rigaud, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 1690 CrossRef CAS PubMed.
- D. Weaver, D. Amin and G. King, J. Biol. Chem., 2022, 298, 102412 CrossRef CAS PubMed.
- C. D. Blanchette, T. V. Ratto and M. L. Longo, in Principles of Cellular Engineering, Elsevier, 2006, p. 195 Search PubMed.
- S. Jelavić, A. C. Mitchell and K. K. Sand, Geochem. Perspect. Lett., 2020, 15, 25 CrossRef.
- Y. L. Lyubchenko, J. Phys. D: Appl. Phys., 2018, 51, 403001 CrossRef PubMed.
- H. Takahashi, A. Miyagi, L. Redondo-Morata and S. Scheuring, Small, 2016, 12, 6106 CrossRef CAS.
- L. Redondo-Morata, M. I. Giannotti and F. Sanz, Langmuir, 2012, 28, 12851 CrossRef CAS PubMed.
- Y.-F. Bao, M.-Y. Zhu, X.-J. Zhao, H.-X. Chen, X. Wang and B. Ren, Chem. Soc. Rev., 2024, 53, 10044–10079 RSC.
- Y. Huang, D. Talaga, G. Salinas, P. Garrigue, G. S. Cooney, S. Reculusa, A. Kuhn, S. Bonhommeau and L. Bouffier, Nanoscale, 2025, 17, 1411 RSC.
- L. M. Otter, M. W. Förster, E. Belousova, P. O'Reilly, D. Nowak, S. Park, S. Clark, S. F. Foley and D. E. Jacob, Geostand. Geoanal. Res., 2021, 45, 5 CrossRef CAS.
- R. Zenobi, N. Kumar and P. Verma, Nano Lett., 2025, 25, 3707–3716 CrossRef CAS PubMed.
- S. Karanth, B. Meesaragandla and M. Delcea, Biophys. Chem., 2020, 267, 106465 CrossRef CAS PubMed.
- Y. Navon, B. Jean, L. Coche-Guerente, F. Dahlem, A. Bernheim-Groswasser and L. Heux, Langmuir, 2020, 36, 1474 CrossRef CAS PubMed.
- M. D'Acunto, F. Dinelli and P. Pingue, Beilstein J. Nanotechnol., 2015, 6, 2278 CrossRef PubMed.
- Y. He, Y. Yan and Y. Geng, Ultramicroscopy, 2023, 254, 113832 CrossRef CAS PubMed.
- R. J. Goodband, C. D. Bain and M. Staykova, Langmuir, 2022, 38, 5674 CrossRef CAS PubMed.
- L. Bar, P. Losada-Pérez and J. Troncoso, J. Mol. Liq., 2023, 384, 122196 CrossRef CAS.
- S. Y. Kim, A. E. Pittman, E. Zapata-Mercado, G. M. King, W. C. Wimley and K. Hristova, J. Am. Chem. Soc., 2019, 141, 6706 CrossRef CAS.
- H. Söngen, B. Reischl, K. Miyata, R. Bechstein, P. Raiteri, A. L. Rohl, J. D. Gale, T. Fukuma and A. Kühnle, Phys. Rev. Lett., 2018, 120, 116101 CrossRef PubMed.
- L. Wang and C. V. Putnis, Acc. Chem. Res., 2020, 53, 1196 CrossRef CAS PubMed.
- K. S. Han, J. D. Bazak, Y. Chen, T. R. Graham, N. M. Washton, J. Z. Hu, V. Murugesan and K. T. Mueller, Chem. Mater., 2021, 33, 8562 CrossRef CAS.
- J. F. Hassler, M. Lawson, E. C. Arroyo, F. S. Bates, B. J. Hackel and T. P. Lodge, Langmuir, 2023, 39, 14263 CrossRef CAS PubMed.
- N. J. Van Zee, A. S. Peroutka, A. Crabtree, M. A. Hillmyer and T. P. Lodge, Biomacromolecules, 2022, 23, 1433 CrossRef CAS PubMed.
- N. J. Van Zee, A. S. Peroutka, M. A. Hillmyer and T. P. Lodge, Langmuir, 2023, 39, 7258 CrossRef CAS PubMed.
- R. Carloni, N. Sanz del Olmo, P. Ortega, A. Fattori, R. Gómez, M. F. Ottaviani, S. García-Gallego, M. Cangiotti and F. J. de la Mata, Biomolecules, 2019, 9, 540 CrossRef.
- Z. Qin, Z. Wang, F. Kong, J. Su, Z. Huang, P. Zhao, S. Chen, Q. Zhang, F. Shi and J. Du, Nat. Commun., 2023, 14, 6278 CrossRef CAS PubMed.
- M. Carone, S. Moreno, M. Cangiotti, M. F. Ottaviani, P. Wang, R. Carloni and D. Appelhans, Langmuir, 2020, 36, 12816 CrossRef CAS PubMed.
| This journal is © The Royal Society of Chemistry 2025 |