
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Synthesis and RAFT polymerisation of hydrophobic acrylamide monomers derived from plant oils†
Oliver J.
Harris
 a,
Peter
Tollington
b,
Calum J.
Greenhalgh
c,
Ryan R.
Larder
a,
Helen
Willcock
a and
Fiona L.
Hatton
*a
a,
Peter
Tollington
b,
Calum J.
Greenhalgh
c,
Ryan R.
Larder
a,
Helen
Willcock
a and
Fiona L.
Hatton
*a
aDepartment of Materials, Loughborough University, Loughborough, LE11 3TU, UK. E-mail: f.hatton@lboro.ac.uk
bCargill, Evert van de Beekstraat 378, 1118 CZ Schiphol, The Netherlands
cDepartment of Chemistry, Loughborough University, Loughborough, LE11 3TU, UK
First published on 21st November 2024
Abstract
Polymeric materials based on fatty acids (FAs) have a combination of characteristics (alkene groups, hydrophobicity, tuneable Tg) that give them great potential as renewable, high value materials. Here, we investigate the base catalysed transesterification of four different plant oils (high oleic sunflower, olive, hydrogenated coconut and hydrogenated rapeseed) with N-hydroxyethyl acrylamide. By conducting kinetics experiments, investigating potential side reactions and improving isolation of the target products, we were able to identify reactive impurities (radical inhibitors, unintended co-monomers) that were found to remain in the impure brine washed plant oil-based monomers (POBM). Kinetics experiments were then performed to investigate the RAFT polymerisation of these monomers. It was found that the more sustainable brine washing process was viable for the controlled radical polymerisation of the higher kp app (saturated) monomers, however column purification was necessary for good control of unsaturated monomers. Polymers with values of Mn between 3000 and 12![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 g mol−1 were synthesised and dependent on the FA source exhibited either amorphous or semi-crystalline behaviour (Tg values between −1 and 33 °C, Tm values between 48 and 66 °C). This work demonstrates the first example of RAFT polymerisation of acrylamide monomers derived from plant oils by a one step direct transesterification, opening the door for novel well-defined, functional bio-based polymers.
000 g mol−1 were synthesised and dependent on the FA source exhibited either amorphous or semi-crystalline behaviour (Tg values between −1 and 33 °C, Tm values between 48 and 66 °C). This work demonstrates the first example of RAFT polymerisation of acrylamide monomers derived from plant oils by a one step direct transesterification, opening the door for novel well-defined, functional bio-based polymers.
Introduction
Increasing the use of sustainable chemical feedstocks in place of petrochemicals is a key barrier for reducing global fossil fuel usage; in the chemical feedstocks market crude oil is the basis of 90% of all organic chemicals.1 To that end, new technologies making use of sustainable chemical feedstocks should be explored. However, new technologies will remain a lab-scale novelty unless forethought is paid towards their real-world feasibility as a readily integrated process.2 Fatty acid (FA) based polymers have gained interest due to their attractive characteristics (unsaturations, hydrophobicity, tuneable Tg) and could prove to be exciting materials in technical applications.3–7FAs are readily available, bound as triglycerides (TAG) in fats and oils. They can be found in organisms from multiple branches of life (plants, animals, algae) and the source and variety can produce a range of chemical structures.8,9 The most accessible FA feedstock in terms of availability and existing infrastructure is plant oils. A large oleochemical industry already exists supporting the food and chemical industry with a wide range of reactions employed to generate products from these feedstocks.10,11 Plant oils represent a facile platform for studies at the laboratory scale, however any number of alternative feedstocks could act as drop-in substitutions if desired as their chemical behaviour would be identical (e.g. animal fats, algae derived TAGs, and oils from waste sources12).
Prior studies have highlighted the advantages of modifying the carboxylic acid (COOH) group of the FA, for instance, by functionalising the FA with a polymerisable moiety, while other methods such as modifying the internal unsaturations are less common.4 Most commonly, FA-based monomers have been synthesised by Steglich esterification of FA COOH groups with a (meth)acrylate bearing a primary alcohol, such as hydroxyethyl methacrylate.13–16 Other approaches to functionalise the FA COOH include esterification using carbonyldiimidazole,17 and epoxy ring opening of allyl glycidyl ether.18 Another approach, recently reported by the Voronov group was the synthesis of acrylamide functional plant oil based monomers (POBMs) via the direct transesterification of plant oil TAGs with N-hydroxyethyl acrylamide (HEAA).19–21 A similar approach first conducted amidation of plant oil TAGs to generate N-hydroxyalkyl fatty amides which were subsequently reacted with methacrylic anhydride to give a methacrylate FA monomer.22,23 However, the latter approach requires a two-step synthesis as opposed to the one-step direct transesterification with HEAA and requires the use of more toxic reagents (e.g., 4-dimethylaminopyridine). Additionally, transesterification and related processes are already widely used on TAGs in industry on a large scale (biodiesel, interesterification, wax making, soap making).24–26
Reversible addition–fragmentation chain transfer (RAFT) polymerisation is a well-established, versatile reversible deactivation radical polymerisation technique which allows for the synthesis of well-defined polymers and control of molecular weight and dispersity.27–30 RAFT polymerisation of monomers derived from renewable resources is a growing field,31 and the technique is a useful tool for investigating the possibility of well-defined advanced materials (block copolymers, nanoparticles) based on these monomers. Many studies have performed RAFT polymerisation of similar non-renewable pendant alkyl monomers, most commonly stearyl32–34 and lauryl methacrylates.35,36 However, the use of RAFT polymerisation for FA-based monomers is less well researched.4,31 Maiti et al. investigated the RAFT polymerisation of saturated FA methacrylates (FA with C8–18), achieving homopolymers and block copolymers with narrow dispersities (Đ < 1.22).13 In subsequent work, they investigated the RAFT polymerisation of the unsaturated FA methacrylate 2-(methacryloyloxy)ethyl oleate (MAEO), which resulting in homopolymers with broader dispersities at higher molecular weights (Đ = 1.10–1.57).14 A large proportion of internal unsaturations were reported to remain in the resultant polymer, and post-polymerisation modification of these was demonstrated by epoxidation and then crosslinking.
Here, we report the RAFT solution polymerisation of POBMs (plant oil-based monomers) directly derived from plant oils via base catalysed transesterification with HEAA (Fig. 1). Four plant oil feedstocks were selected: unrefined olive, refined high oleic sunflower (HO-Sun), hydrogenated coconut and hydrogenated rapeseed oil. These were chosen for comparison of their reaction behaviour and material properties dependent on the FA structure. The monomers were subsequently polymerised using free radical and RAFT-mediated polymerisation, including evaluation of polymerisation kinetics. The thermal properties of the resulting POBM polymers were studied by differential scanning calorimetry (DSC) and thermogravimetric analysis (TGA) to investigate their potential use in high-value materials. This work demonstrates the first RAFT polymerisation of acrylamide POBMs derived via direct transesterification of the feedstock, as well as the first synthesis of POBMs from hydrogenated feedstocks.
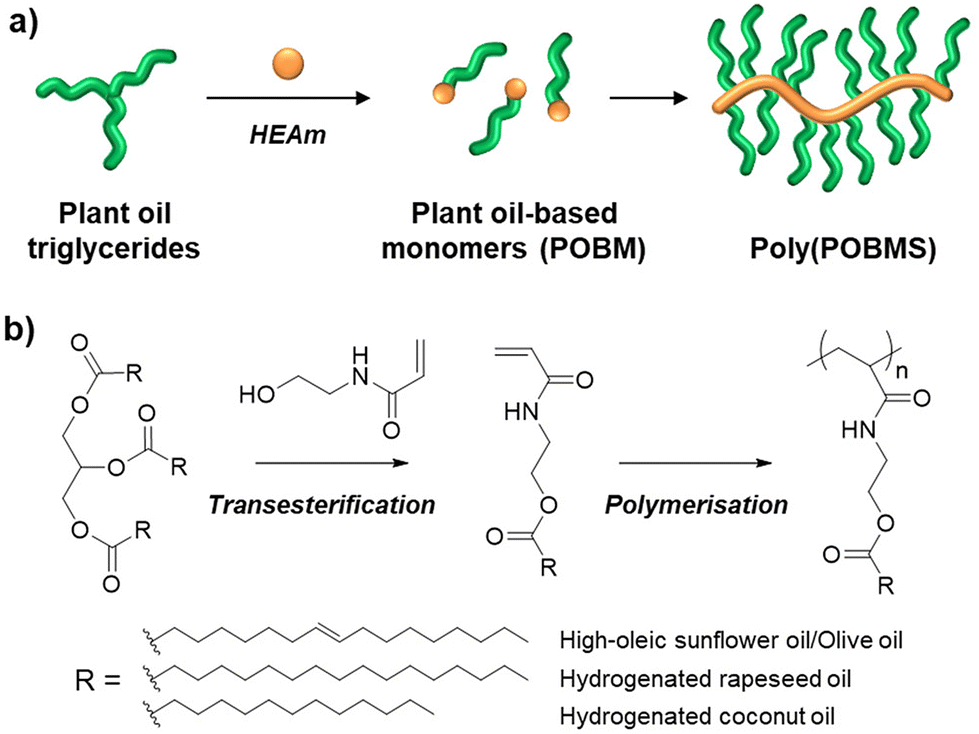 | ||
| Fig. 1 Graphical (a) and schematic (b) representations of the synthesis and polymerisation of plant oil-based monomers in this work. R denotes the most abundant fatty acid hydrocarbon chains present in each feedstock oil used in this study. | ||
Experimental
Materials
All materials in this work were used as received. Olive oil (Filippo Berio, extra virgin, cold extracted) was purchased from a local supermarket. High oleic sunflower oil, hydrogenated coconut oil and hydrogenated rapeseed oil were kindly donated by Cargill. FA distributions of each of the feedstocks can be found in Table S1.†N-Hydroxyethyl acrylamide (HEAA, 97%), butylated hydroxytoluene (BHT, ≥99.0%), dichloromethane (DCM, ≥99.8%), dimethylacrylamide (DMA, 99%), N-isopropylacrylamide (NIPAM, 97%), 2-hydroxyethyl acrylate (HEA, 96%), 2,2′-azobis(2-methylpropionitrile) (AIBN, 98%), dimethyl sulfoxide (DMSO, ≥99.7%), 2-(dodecylthiocarbonothioylthio)-2-methylpropionic acid (DDMAT, 98%), 2-cyano-2-propyl dodecyl trithiocarbonate (CPDT, 97%), 4-cyano-4-[(dodecylsulfanylthiocarbonyl)-sulfanyl]pentanoic acid (CDTPA, 97%), cyanomethyl dodecyl trithiocarbonate (CDT, 98%) and lithium chloride (≥99.0%) were purchased from Sigma Aldrich. 4-((((2-carboxyethyl)thio)-carbonothioyl)thio)-4-cyanopentanoic acid (CECPA, 95%), 2-(dodecylthiocarbonothioylthio)propionic acid (DDTPA, 95%) and cyanomethyl (3,5-dimethyl-1H-pyrazole)-carbodithioate (py-CTA, 95%) were purchased from Boron Molecular. Sodium hydroxide (98.8%), methanol (99.99%), were purchased from Fisher Scientific. Anhydrous tetrahydrofuran (THF, Acros Organics, 99.5%); Sodium chloride (Alfa Aesar, 99+%); Anhydrous magnesium sulphate (Acros Organics, 97%); Silica gel (Apollo Scientific, 40–63 μm); Toluene (Honeywell, >99.9%); Diethyl ether (Honeywell, ≥99.8%); Chloroform-d (Thermo Scientific, 99.8 atom % D).Characterisation
Nuclear magnetic resonance (NMR) experiments (1H and 13C) were conducted using a JEOL ECS 400 MHz spectrometer at 21 °C on sample dissolved in CDCl3 (16 scans). Spectra were analysed using Delta 5.3.1 software.
Gel permeation chromatography (GPC) analyses were performed using an Agilent 1260 Infinity GPC system, equipped with both refractive index and UV detectors. Samples were injected at a flow rate of 1.0 mL min−1 through a guard column, followed by two separation columns (Agilent PL gel 5 μm Mixed-C) at 40 °C. The eluents were chloroform containing 2% triethylamine for non-polymeric samples and THF:MeOH 90:10 (v/v) + 0.5 wt% LiCl for polymeric samples. All samples were prepared using the corresponding eluent solution to an approximate concentration of 5 mg mL−1. The system was calibrated using near-monodisperse poly(styrene) standards (Mp ranging from 162 to 364000 g mol−1). Chromatograms were analysed using Agilent GPC/SEC software.
Mass spectrometry (MS) was obtained using a Thermo Scientific Exactive Orbitrap mass spectrometer. A positive mode ESI mass spectrum of column purified monomer was recorded by diluting a sample to 25 μg mL−1 in MeOH:DCM 90:10 (v/v). A scan range of 100.0 to 1000.0 m/z was performed with a maximum inject time of 500 ms and an AGC target of 5 × 105 ions. Ion source settings were as follows: spray voltage = 4.50 kV, capillary temperature = 300 °C, sheath gas flow = 10, auxiliary gas flow = 5, sweep gas flow = 1.
Fourier transform infrared (FTIR) spectra were collected using an Agilent Cary 630 FTIR Spectrometer with a single reflection attenuated total reflectance (ATR) system using a 45° diamond positioned on the top plate (64 scans, 4000–650 cm−1, resolution 8 cm−1). Spectra were analysed using Agilent MicroLab software.
Differential scanning calorimetry (DSC) was performed using TA Instruments Q200 and Q2000 DSCs in an N2 atmosphere. Oil feedstocks and monomers were analysed using single heat ramps (10 °C min−1) from −60 to 90 °C. For analysis of polymeric materials, a heat-cool-heat program between −70 and 100 or 200 °C (10 °C min−1) was performed in all cases with thermal transition values determined from the second heat cycle. Analysis of results was performed using TA Instruments Universal Analysis software.
Thermogravimetric analysis (TGA) was performed on a TA Instruments TGA 550 using platinum crucibles over a temperature range of 30 to 500 °C, at a heating rate of 10 °C min−1 under an argon atmosphere. Analysis of results was performed using TA Instruments Trios software.
Synthesis of plant oil-based monomers (POBMs) using base-catalysed transesterification
The following method for the synthesis of a high-oleic sunflower oil-based monomer (HOSM) is a representative example for the general synthesis of POBMs via base-catalysed transesterification (Scheme 1). HO-Sun oil (10 g, 11.29 mmol), finely ground NaOH (0.304 g, 7.601 mmol), HEAA (11.6 g, 0.1008 mol), BHT (0.0064 g, 31.02 μmol) and THF (11 mL) were combined in a 100 mL round bottom flask. The reaction mixture was heated to 30 °C for 3 hours under constant agitation via mechanical stirring (4 cm paddle, 500 rpm). The resultant crude reaction mixture was diluted with dichloromethane (50 mL) and washed with 0.1 M brine solution (3 × 200 mL). The organic phase was dried over anhydrous sodium sulphate, filtered and concentrated in vacuo to yield the resulting POBM. The HO-Sun monomer (HOSM, 56% yield) and olive oil monomer (OVM, 66% yield) were obtained as viscous oils that became off-white to yellow butter-like solids after refrigeration. The hydrogenated coconut oil monomer (HCM, 50% yield) formed a white waxy solid, whilst the hydrogenated rapeseed oil monomer (HRM, 49% yield) formed a white powder.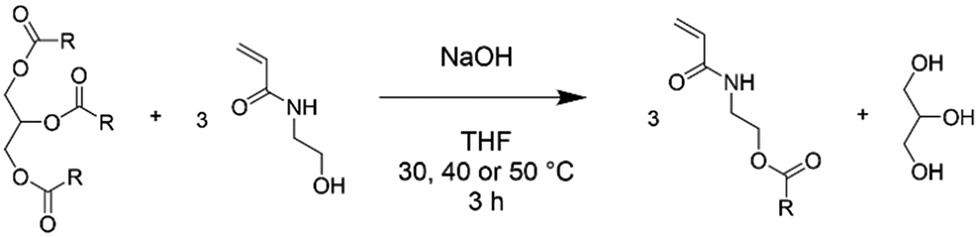 | ||
| Scheme 1 Reaction scheme for the base-catalysed transesterification of triglycerides with N-hydroxyethyl acrylamide. | ||
Another sample of the brine washed HOSM product was further purified via column chromatography using silica gel as a stationary phase and a gradient of hexane and ethyl acetate (90:10 to 50:50 v/v). mp 27.8–32.4 °C. IR (vmax/cm−1): 3260br (amide N–H), 3070, 2920, 2850 (CH stretch), 1730 (ester C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 1660 (conj. CC), 1630 (amide CO), 1550 (amide N–H bend). 1H NMR (400 MHz; CDCl3) δH (ppm): 6.29 (1H, dd, vinyl CH2CH–), 6.09 (1H, dd, vinyl CH2CH–), 5.90 (1H, br s, –NH–), 5.66 (1H, dd, vinyl CH2CH–), 5.35 (2H, m, –CH2CHCHCH2–, mono-unsaturated FA), 4.21 (2H, t, –NH-CH2CH2-O–), 3.61 (2H, q, –NH-CH2CH2-O–), 2.77 (t, HC-CH2-CH, poly-unsaturated FA), 2.32 (2H, t, –OCO-CH2–), 2.03 (4H, m, –CH2-CHCH-CH2–, mono-unsaturated FA), 1.61 (2H, m, –OCO-CH2CH2–), 1.32 (20H, m, –(CH2)n–), 0.88 (3H, t, –CH3). 13C NMR (400 MHz; CDCl3) δC (ppm): 174.2 (–O-CO-CH2–), 165.7 (CH-CO-NH–), 130.7–129.8 (–CH, both conjugated and unconjugated), 126.9 (CH2CH–), 63.1 (–NH-CH2CH2-O–), 39.2 (–NH-CH2CH2-O–), 34.3 (–O-CO-CH2CH2–), 32.1–22.8 (–CH2–, FA chain), 14.3 (–CH3).
O), 1660 (conj. CC), 1630 (amide CO), 1550 (amide N–H bend). 1H NMR (400 MHz; CDCl3) δH (ppm): 6.29 (1H, dd, vinyl CH2CH–), 6.09 (1H, dd, vinyl CH2CH–), 5.90 (1H, br s, –NH–), 5.66 (1H, dd, vinyl CH2CH–), 5.35 (2H, m, –CH2CHCHCH2–, mono-unsaturated FA), 4.21 (2H, t, –NH-CH2CH2-O–), 3.61 (2H, q, –NH-CH2CH2-O–), 2.77 (t, HC-CH2-CH, poly-unsaturated FA), 2.32 (2H, t, –OCO-CH2–), 2.03 (4H, m, –CH2-CHCH-CH2–, mono-unsaturated FA), 1.61 (2H, m, –OCO-CH2CH2–), 1.32 (20H, m, –(CH2)n–), 0.88 (3H, t, –CH3). 13C NMR (400 MHz; CDCl3) δC (ppm): 174.2 (–O-CO-CH2–), 165.7 (CH-CO-NH–), 130.7–129.8 (–CH, both conjugated and unconjugated), 126.9 (CH2CH–), 63.1 (–NH-CH2CH2-O–), 39.2 (–NH-CH2CH2-O–), 34.3 (–O-CO-CH2CH2–), 32.1–22.8 (–CH2–, FA chain), 14.3 (–CH3).
ESI MS: m/z (relative abundance), 781.6064 (9), 418.2715 (3, [M + K]+), 402.2978 (100, [M + Na]+), 380.3157 (2, [M + H]+), 376.2820 (4), 304.2610 (4).
Free radical polymerisation of POBMs
The following method for the free radical polymerisation of HOSM is a representative example of the general polymerisation of each brine washed POBM. Calculations of stoichiometry assumed an 80% w/w of POBM monomer in the brine washed samples (calculated from molar purity determined by 1H NMR, Table S4†). HOSM (0.4738 g, 1.000 mmol) and AIBN (0.0125 g, 76.12 μmol, [M]0:[I]0 ≈ 13:1) were added to a vial with toluene (2 mL), to give an approximate solids content of 25 wt%. The vial was sealed, cooled in an ice bath and purged with N2 for 30 minutes. In the case of HRM, the monomer was purged separately to the initiator in a vial heated to 70 °C, to ensure full dissolution of the HRM. After purging the solution was heated to 70 °C for 7 hours before quenching by exposing the solution to the atmosphere and allowing the reaction mixture to cool to room temperature. The crude reaction mixture was diluted in THF (2 mL) then purified by precipitation into a MeOH:diethyl ether 15:1 (v/v) mixture (40 mL) or a 6:1 (v/v) for P(HRM). Precipitated polymers were collected via centrifugation followed by decanting of the solvent and then dried under vacuum for 24 hours at 50 °C. P(HOSM), P(OVM) and P(HCM) presented as clear viscous liquids whereas P(HRM) presented as a white powder. 1H NMR (400 MHz; CDCl3) δH (ppm): 7.09 (1H, br s, –NH–), 5.33 (2H, m, –CH2CHCHCH2–, mono-unsaturated FA), 4.06 (2H, br, –NH-CH2CH2-O–), 3.70 (br, co-monomer unit), 3.41 (2H, br, –NH-CH2CH2-O–), 2.75 (t, HC-CH2-CH, poly-unsaturated FA), 2.48 (br, co-monomer unit), 2.27 (2H, br, –OCO-CH2–), 1.94 (4H, br, –CH2-CHCH-CH2–, mono-unsaturated FA), 1.58 (2H, br, –OCO-CH2CH2–), 1.19 (20H, m, –(CH2)n–), 0.86 (3H, t, –CH3), 2.75–0.75 (3H, br, p(HOSM) backbone).
RAFT polymerisation of POBMs
The following method for the RAFT polymerisation of HOSM with DDMAT is a representative example of any of the RAFT solution polymerisation of POBMs presented in this work, see Scheme 2. The molar ratio of [M]0:[CTA]0:[I]0 was 50:1:0.2, targeting a DP of 50 (again assuming 80% w/w of POBM monomer in the brine washed samples). HOSM (0.5419 g, 1.144 mmol), DDMAT (8.34 mg, 22.88 μmol) and AIBN (0.75 mg, 4.575 μmol) were added to a vial with toluene (2.5 mL), to give an approximate solids content of 25% w/w. The solutions were purged with N2 for 30 minutes. The solutions were heated at 70 °C using an oil bath for the predetermined reaction time. Aliquots were taken (using a syringe purged with N2) at appropriate intervals to obtain kinetics samples which were quenched by exposing the solution to the atmosphere. Purification of the polymers was performed as described for the free radical polymerisations.
 | ||
| Scheme 2 Reaction scheme for the RAFT solution polymerisation of HOSM in toluene at 70 °C. | ||
The chain transfer agent (CTA), monomer were varied where appropriate, and when targeting different degrees of polymerisation, the relative amounts of monomer, CTA and initiator were varied while maintaining a [CTA]:[I] ratio of 1:0.2, and a total solids content of 25% w/w.
For the end group analysis conducted with py-CTA, the DP by NMR was calculated using the –CH3 for the CTA at 2.67 ppm and the –NH-CH2-CH2-O– peaks corresponding to the repeat monomer unit at 3.45 and 4.14 ppm.
Results and discussion
Previous work reported the direct transesterification of sunflower, linseed, olive and soybean oils, with varying degrees of unsaturations.19–21 Here, we expand this approach, investigating the direct transesterification of olive, high oleic sunflower (HO-Sun), and unexplored hydrogenated oils; coconut and rapeseed (Fig. 1). Initial characterisation of the plant oils used in this work was conducted by 1H NMR Spectroscopy, see Fig. S1–4,† confirming the TAG chemical structures present, bearing in mind the heterogeneous nature of these biobased natural materials.Investigation of the transesterification of plant oils with HEAA
Kinetics experiments of the transesterification of HO-Sun were performed on a 2.5 g scale at several reaction temperatures (30, 40, 50 °C), see Scheme 1. Analyses of the crude reaction mixtures were performed using 1H NMR spectroscopy and gel permeation chromatography (GPC) (Fig. 2). The total conversion of glycerides, including TAGs, diglycerides (DAGs) and monoglycerides (MAGs), to POBMs was assessed by integrating and comparing the triplet at 4.20 ppm, corresponding to the –NH-CH2-CH2-O– environment in the POBM (Fig. 3F) and the triplet at 0.86 ppm corresponding to the pendant –CH3 group of the FA (Fig. 3N) respectively. The integral from the –CH3 of the FA moieties was used as a reference peak as it was observed to remain constant regardless of the molecule the FA moiety was bonded to. From Fig. 2a it is apparent that the maximum conversion of glyceride bound FA to POBM achieved (around 55%) was independent of temperature. Given that the transesterification reaction is reversible and an equilibrium system, it was expected that the temperature of the reaction would impact the equilibrium position.37–39 The rate of reaction was affected by the temperature, with the reaction performed at 50 °C reaching a maximum conversion of ∼55% within 1 h, rather than within 2 h, as observed at 30 and 40 °C. The TAG starting material was fully consumed in all cases, confirmed by 1H NMR analyses, and remaining unreacted glycerides were found to be a mixture of DAGs and MAGs. This was further confirmed by GPC analyses, whereby the higher molecular weight TAGs (17.4 min) were converted to lower molecular weight species, including the target POBM at 18.3 min, see Fig. 2b. However small peaks at higher and lower retention times are also observed (17.8 and 19.2 min) most likely being DAGs/MAGs and free FAs respectively. Kinetics of the direct transesterification of the other feedstock oils to prepare the olive oil monomer (OVM), hydrogenated coconut oil monomer (HCM) and hydrogenated rapeseed oil monomer (HRM) were comparable (Fig. S5–7†). Saponification is a well-understood side reaction in base-catalysed transesterification reactions, however the presence of the reactive acrylamide group in the reaction mixture could lead to side reactions not previously considered (e.g. conjugate additions, autopolymerisation).40–45 To investigate this, HEAA and several other similar monomers (NIPAM, DMA, HEMA) were heated at 50 °C with NaOH in THF (see Table S2†). Conversion of vinyl groups was observed by 1H NMR and an increase in Mn was observed by GPC in each case, indicating oligomerisation. Reaction of HEAA by these means could explain the limitations to the conversion of the transesterification as well as indicate the nature of non-TAG derivative impurities in the final product. | ||
| Fig. 2 Data from the transesterification of HO-Sun oil; (a) conversion of glyceride bound fatty acids to POBMs vs. time at 30, 40 and 50 °C determined by 1H NMR spectroscopy, and (b) GPC chromatograms performed on 0, 3 and 6 h samples from the 30 °C experiment. Peaks have been labelled to highlight the most abundant species. FFA = free fatty acid, DAG = diglyceride, TAG = triglyceride, POBM = plant oil-based monomer. | ||
 | ||
| Fig. 3 1H NMR spectra (400 MHz, CDCl3, 25 °C) of the brine washed and column purified HOSM. Peaks for the column purified sample are assigned to the target structure and integrals (referenced against the CH3 signal at 0.9 ppm) are displayed. Peak H corresponds to allylic protons from linoleic and linolenic FAs (structure not shown). | ||
Plant oil-based monomer synthesis
Based on the results of the kinetics experiments, the direct transesterification of HO-Sun with HEAA was performed at a 10g scale in THF at 30 °C. The HO-Sun monomer (HOSM) was purified by aqueous washing (brine wash) as previously reported.46,47 Characterisation by 1H NMR spectroscopy confirmed the successful synthesis of the desired monomer, see Fig. 3. However, it also confirmed the presence of impurities in the HOSM isolated from brine washings. Therefore, to obtain pure HOSM the crude product was further purified by column chromatography. 1H NMR peak assignments were made using observations from prior studies and peak integrals are in good agreement with expected values (based on the known FA distribution of the feedstock).25,46,48–50 The slightly higher than expected value of L could be due to dissolved water. Two key resonances that support the successful synthesis of HOSM were the peaks of the NH-CH2-CH2-O and NH-CH2-CH2-O environments (seen at 4.20 and 3.60 ppm respectively).The determination of the nature of the impurities in the brine washed HOSM was considered important in order to further understand any limitations in the synthesis, as well as any potential effects in polymerisations. Through comparison with literature sources,51,52 glyceryl protons in MAGs and DAGs (4.15, 4.10, 4.00, 3.90, 3.80, 3.65 ppm) are easily identified in the spectra. Small resonances indicating low concentrations of the radical inhibitors MEHQ (6.76 ppm) and BHT (6.98 ppm), supported by observations in the 1H NMR spectra of some of the fractions separated by column chromatography, were likely introduced from additives in the HEAA and THF reagents. Additional small resonances can be seen near the resonances for vinyl environments (e.g. at 6.15 ppm) that may reflect vinyl groups from unreacted HEAA or may belong to other unintended monomeric products. The peaks at 3.50 and 3.70 ppm appear to correspond to the NH-CH2-CH2-O and NH-CH2-CH2-O environments from remaining HEAA. Additionally, the peak 2.45 ppm could reflect a backbone peak from oligomeric or polymeric acrylamide species.53,54 This all suggests that HEAA and/or unintended acrylamide derivatives (oligomers, monomers) were also present as impurities. These observations from the spectra for the brine washed monomer can also be made in the visually comparable data produced in prior studies.46,47 To further confirm successful isolation of the target HOSM monomer, the sample purified by column chromatography was further characterised by 13C NMR spectroscopy, FTIR and LC-MS (see Fig. S8 and Table S3†). All carbon environments in the target HOSM were identified by 13C NMR, and the validity of the assignments was confirmed by the DEPT 135 phasing. Analysis by LC-MS confirmed that the predominant component was the target HOSM with peaks for the H+, Na+ and K+ adducts visible in the ESI-MS spectrum. Low mass error values (<1 ppm) were calculated for each of the adducts of the POBM ions, showing that the predicted mass of the proposed structure matches the observed m/z values.
The column purified HOSM reported here represents a substantially improved isolation of the target POBM from approximately 70% to >99% purity. However, provided that impurities did not negatively impact their controlled radical polymerisations, conducting purification post-polymerisation would be a more facile and sustainable methodology (as smaller molecule impurities could be more easily separated from larger polymer chains). Subsequently, the synthesis of brine washed monomers OVM, HCM and HRM was conducted, and these isolated monomers were characterised by 1H NMR spectroscopy (Fig. S9–11†), confirming purities between 70–79% (Table S4†).
Free radical polymerisation of POBMs
Initially, POBMs were polymerised by free radical polymerisation in toluene at 70 °C for 7 h, using AIBN as the radical initiator, according to previous reports.14,41 Successful polymerisation was confirmed by 1H NMR and GPC analyses (Table S5†). Near quantitative conversions were determined by 1H NMR spectroscopy, while GPC analyses confirmed high molecular weights (20–48 kg mol−1) and dispersities, Đ, between 1.76–2.52, typical for free radical polymerisation. The resultant POBM free radical polymers were purified by precipitation and their compositions were confirmed by 1H NMR analyses (Fig. S12–15†). Interestingly, the unsaturated P(POBM)s, P(HOSM) and P(OVM), had lower Mn (25.0 and 20.4 kg mol−1) and Đ (1.76 and 1.84) compared with P(HCM) and P(HRM), with Mn of 45.4 and 47.7 kg mol−1, and Đ = 2.33 and 2.52, respectively. This could suggest increased allylic chain transfer events during these polymerisations of unsaturated monomers HOSM and OVM.RAFT polymerisation of POBMs
An initial RAFT agent screening of several trithiocarbonate CTAs was performed using the brine washed OVM (see Table S6†), and DDMAT was selected for further studies. Following this, RAFT polymerisations using DDMAT as the CTA were performed on each of the brine washed monomers targeting DPx of 25, 50, 100 and 200 while maintaining reaction times, Table 1. When conducting the RAFT polymerisations of HOSM and OVM over 7 hours, relatively high conversions were observed when targeting a DP of 25 (84 and 78% respectively). However, at higher target DPs conversion dropped off considerably, to 45–46% for DP50, 30% and 40% for HOSM and OVM respectively at DP100, and negligible conversion was observed at DP300 (3–4%). And dispersities were generally relatively low (Đ = 1.18–1.44) for these P(HOSM)x and P(OVM)x homopolymers. Such comparable behaviour was expected due to their structural similarities. However, significant differences were observed for the polymerisations of the saturated POBMs (HCM, HRM). Generally, higher conversions were observed in shorter reaction times (70 min) for the hydrogenated monomer HCM; targeting DPs between 25–100, higher conversions (>70%) and lower dispersities (Đ = 1.17–1.32) were achieved for P(HCM)x. However, this was not achieved with the HRM monomer where conversion was limited (24–55%) over the same reaction times (70 min). Due to difficulties eliminating effects from practical issues with the use of this high melting point, poorly soluble monomer (e.g. due to higher concentrations of inhibitor impurities after work up, poor degassing) HRM was not used further in this work. Additionally, because of the comparable behaviour of OVM and HOSM, further experiments made use of just HOSM to represent a predominantly mono-unsaturated FA POBM. Consequently, HOSM and HCM were chosen as the focus for the rest of this study.| Target composition | Reaction time | Conversion (%) | M n th (g mol−1) | M n GPC (g mol−1) | Đ |
|---|---|---|---|---|---|
| a Theoretical Mn calculated as follows: Mn th = Mw CTA + (Mw monomer × DPth). b Determined by THF GPC analyses. | |||||
| P(HOSM)25 | 7 h | 84 | 8300 | 6700 | 1.19 |
| P(HOSM)50 | 46 | 9000 | 6600 | 1.26 | |
| P(HOSM)100 | 30 | 11700 |
9400 | 1.44 | |
| P(HOSM)200 | 3 | 2500 | 4200 | 1.36 | |
| P(OVM)25 | 7 h | 78 | 7800 | 6000 | 1.18 |
| P(OVM)50 | 45 | 9000 | 6300 | 1.24 | |
| P(OVM)100 | 20 | 8100 | 6600 | 1.37 | |
| P(OVM)200 | 4 | 3200 | 4200 | 1.38 | |
| P(HCM)25 | 70 min | 91 | 7100 | 6700 | 1.17 |
| P(HCM)50 | 84 | 12900 |
9600 | 1.23 | |
| P(HCM)100 | 70 | 21000 |
13100 |
1.32 | |
| P(HCM)200 | 26 | 15500 |
10300 |
1.58 | |
| P(HRM)25 | 70 min | 55 | 10300 |
9000 | 1.3 |
| P(HRM)50 | 38 | 14400 |
7600 | 1.25 | |
| P(HRM)100 | 29 | 21600 |
6800 | 1.38 | |
| P(HRM)200 | 24 | 36000 |
9000 | 1.49 | |
Kinetics studies on the RAFT polymerisations on the brine washed HOSM and HCM were performed at a target DP of 50 using DDMAT as the CTA (Fig. 4). First order kinetics were observed in the initial stages of each of the reactions as seen in the semilog plots (Fig. 4a and b).30 A deviation from first order kinetics was observed as the polymerisations progressed, most noticeable for the brine washed HCM (Fig. 4a) and brine washed HOSM (Fig. 4b, blue triangles). This indicated a decrease in the number of propagating radicals, likely due to termination events or side reactions with impurities (e.g., radical inhibitors). Similar behaviour has previously been observed for other N-monosubstituted acrylamides,55–58 and in some cases this was attributed to degradation of the trithiocarbonate CTA used.57,58 As RAFT polymerisations of HOSM consistently achieved low conversions, kinetics were also performed on the column purified monomer which increased the maximum conversion obtained of 50% with brine washed HOSM, to 88% with column purified HOSM. Moreover, the rate versus time plot for the column purified HOSM (Fig. 4b, red squares) did not exhibit such a drastic deviation from linearity, suggesting that CTA degradation is not occurring and radical scavenger impurities may be responsible for the deviation from linearity observed for the brine washed monomers. The comparable behaviour of OVM and HOSM in the earlier RAFT polymerisations (targeting DPs between 50-200) suggests that any effects of radical scavengers inherent to unrefined feedstocks (e.g. antioxidants) are negligible compared the effect of any introduced in the course of the monomer's synthesis. It is possible that impurities capable of acting as radical inhibitors could have formed from oxidation of the oil during synthesis, resulting in small concentrations of peroxides.59 However, as the loss of linearity and low conversions were observed for saturated monomers too, it is more likely that inhibitor impurities were accumulated from reagents HEAA and THF during the monomer synthesis.
 | ||
| Fig. 4 Kinetic evaluations of RAFT polymerisations of HOSM and HCM in toluene at 70 °C. (a) Conversion (open circles) and rate (open triangles) versus time plots for brine washed HCM, and (c) corresponding Mn (open diamonds) and Đ (crosses) versus conversion plots. (b) Polymerisation rates for the RAFT polymerisations of column purified HOSM (open red squares) and brine washed HOSM (open blue triangles). (d) Corresponding Mn (closed symbols) and Đ (open symbols) versus conversion plots for column purified HOSM (red circles) and brine washed HOSM (blue diamonds). | ||
The slopes of the ln([M]0/[M]t) plots were used to determine the apparent value of the monomer propagation constant (kp app) for each monomer. The kp app values for the RAFT polymerisation of the saturated HCM (2.32 h−1) were an order of magnitude higher than those of the unsaturated HOSM; 0.12 and 0.24 h−1 for the brine washed and column purified HOSM, respectively. Previous studies have observed that reaction rates in free radical polymerisations decreased with increasing degree of unsaturation of the FA moieties and attributed this to chain transfer mechanisms involving the abstraction of allylic protons from alkenes in the FA moiety (determined via Mayo analysis and 1H NMR).47,60,61 Though similar observations by 1H NMR were not found in this work, to establish the effect of this on the RAFT system further reactions were conducted on the fully saturated HCM (see Fig. S17†). The reaction mixtures were doped with several concentrations of unsaturated HO-Sun oil to provide a source of inactivated alkenes independent of the monomer.62 A 94% reduction in the conversion achieved after 70 min was observed from the addition of the lowest molar ratio of HO-sun oil ([HO-Sun Oil]0/[HCM]0 = 0.083), supporting this hypothesis. Further evidence for reaction of monomers with the RAFT CTA was established by the observation of an induction period in all polymerisations (∼10 min for HCM, ∼40 min for HOSM). Induction periods are a common feature of RAFT polymerisations pertaining to the pre-equilibrium stage of the mechanism and can be indicative of slow re-initiation.63,64 During the induction period peaks were observed at higher retention times in the UV GPC trace that likely correspond to the pre-equilibrium species (most likely not visible in the RI trace due to low concentration, see Fig. S18†).
The proportional relationship between Mn and conversion indicates that chain transfer with the RAFT CTA is more rapid than the polymer propagation. Thus, confirming the polymerisations are proceeding by a controlled RAFT polymerisation mechanism. Retention of the CTA on the end of polymer chains was confirmed by dual RI/UV detection in the GPC chromatograms (see Fig. S19†). Relatively low dispersities (Đ < 1.3) were observed for all resulting plant oil-based polymers and were significantly lower than those obtained by free radical polymerisation, indicating improved control due to the addition of the CTA. Similar observations were also made from kinetic evaluations of the synthesis of P(OVM)50 and P(HRM)50 by RAFT solution polymerisation, see Fig. S16 and Table S7,† and as previously discussed, maximum conversions were limited to 51 and 38% respectively.
These kinetics studies demonstrate that RAFT control of each POBM is viable. We found that where kp app values are sufficiently high thorough purification of the monomer may not be required (as with HCM). However, in systems where the rate is decreased significantly (as with the use of the brine washed HOSM) thorough purification of the monomer is necessary.
Varying the target degree of polymerisation
A second RAFT agent screening was performed using the brine washed HOSM, see Table S8.† This led to the selection of (3,5-dimethyl-1H-pyrazole)-carbodithioate (py-CTA) for the remaining RAFT syntheses. RAFT polymerisations of column purified HOSM and brine washed HCM were performed in toluene at 70 °C for 20 h, targeting DPs between 10 and 60 using py-CTA, see Fig. 5 and Table 2. RAFT control was achieved across the range of target DPs for HCM, with increasing Mn with target DP and with low dispersities (Đ < 1.18) obtained. This was also observed for HOSM up to a target DP for 40 (Đ < 1.23), where a higher Đ of 1.29 was obtained for P(HOSM)60. It was considered that chain transfer to the unsaturations of the HOSM monomer may be the reason for lower conversions, lower Mn and broader dispersities when targeting higher DPs of 40 and 60. For these RAFT polymers (Table 2), due to the use of the py-CTA, end group analysis was performed on the purified P(HOSM)x and P(HCM)x homopolymers after purification by precipitation, see Fig. S23 and S24.† There was good correlation between DPth and DP by end group analysis for most of the polymers, suggesting high CTA efficiency. Comparison with the free radical polymerisations conducted under the same conditions without the presence of CTA (Table 2) showed that Mn achieved for P(HOSM)FRP (80.5 kg mol−1) was considerably lower than P(HCM)FRP (124.2 kg mol−1) and the dispersity was broader 2.49 versus 2.26 respectively, supporting the hypothesis that allylic chain transfer is occurring for the unsaturated HOSM monomer. | ||
| Fig. 5 Normalised GPC chromatograms for; (a) P(HOSM)x synthesised using column purified HOSM, and (b) P(HCM)x using brine washed HCM, by RAFT polymerisation in toluene at 70 °C targeting DPs of 10, 20, 40 and 60. | ||
| Monomer batch | Target composition | Conversion (%) | DPtha | DPNMRb | M n th | M n | Đ | T g (°C) | T m (°C) |
|---|---|---|---|---|---|---|---|---|---|
| a Theoretical DP, DPth, calculated as follows: DPth = target DP × (conversion/100). b Determined by 1H NMR end group analyses. c Theoretical Mn calculated as follows: Mn th = Mw CTA + (Mw monomer × DPth). d Determined by THF GPC analyses. e Determined by DSC analyses, from the second heating. f Synthesised by free radical polymerisation. | |||||||||
| Column purified HOSM | P(HOSM)10 | 91 | 9.1 | 10.4 | 3700 | 4800 | 1.16 | −1.2 | — |
| P(HOSM)20 | 83 | 16.6 | 17.2 | 6500 | 6500 | 1.18 | 4.2 | — | |
| P(HOSM)40 | 77 | 30.8 | 28.9 | 11900 |
9400 | 1.23 | 11.2 | — | |
| P(HOSM)60 | 67 | 40.2 | 30.4 | 15400 |
9500 | 1.29 | 11.9 | — | |
| P(HOSM)FRPf | 64 | — | — | — | 80500 |
2.49 | 17.3 | — | |
| Brine washed HCM | P(HCM)10 | 99 | 9.9 | 12.2 | 3200 | 5100 | 1.07 | 22.4 | 48.8 |
| P(HCM)20 | 99 | 19.8 | 19.4 | 6100 | 6800 | 1.10 | 28.0 | 61.4 | |
| P(HCM)40 | 98 | 39.2 | 45.7 | 11900 |
11100 |
1.14 | 33.5 | 71.0 | |
| P(HCM)60 | 99 | 59.4 | 64.7 | 17900 |
14900 |
1.18 | 30.7 | 65.7 | |
| P(HCM)FRPf | 94 | — | — | — | 124200 |
2.26 | 32.1 | 54.2 | |
Thermal characterisation of plant oil-based polymers
Initially, the plant oil-based polymers synthesised by free radical polymerisation were analysed by thermogravimetric analysis (TGA) which revealed similar degradation behaviours, with onset of degradation observed between 253–263 °C, which would be expected due to the common acrylamide backbone (see Table S4 and Fig. S20†).DSC analysis was performed on the P(HOSM)x and P(HCM)x RAFT polymers, where x = 10, 20, 40 and 60, to elucidate their thermal transitions (Table 2 and Fig. S21, S22†). The unsaturated P(HOSM)x polymers displayed a glass transition temperature, Tg, observed between −1.2 to 11.9 °C. Whereas P(HCM)x polymers showed both Tg (22.4–30.7 °C) and melting temperatures, Tm, recorded between 48.8–71.0 °C indicating semi-crystallinity. For both P(HOSM)x and P(HCM)x polymers the Tg transition temperatures were found to be dependent on Mn, whereas the Tm observed for P(HCM) generally increased with increasing Mn, for P(HCM)10, P(HCM)20 and P(HCM)40, this was not the case for the P(HCM)60, where the Tm reduced slightly. The higher Tg observed for P(HCM) polymers compared with P(HOSM) is likely due to the presence of crystalline domains which is known to impact Tg. A broad endothermic feature was observed for all P(HCM)x below the Tg (approximately −10 °C), the peak temperature of which was independent of Mn. Literature studies on similar polymers suggest such a feature could be a phase transition related to side chain crystallisation/alignment.65,66
In summary, the physical properties of these novel well-defined bio-based polymers could make them suitable for investigation in applications such as polyolefin compatibilisers, coatings, or viscosity modifiers, for example. Moreover, the presence of unsaturations allows for subsequent post-polymerisation modifications expanding the capabilities of these plant oil-based polymers.
Conclusions
In this work we have demonstrated isolation and characterisation of plant oil-based acrylamide monomers, synthesised via an industrially relevant base-catalysed transesterification reaction, including two novel monomers derived from fully saturated feedstocks (hydrogenated coconut and rapeseed oils). Limitations of the synthesis were also further elucidated. Evidence for a previously unidentified side reaction, involving the acrylamide functional group, helps to further explain the limited conversion of the transesterification as well as the origin of side products.The RAFT polymerisations of each of the brine washed POBMs were investigated for the first time. Evidence of reaction with the CTA and of RAFT control was demonstrated for each of the monomers by the observation of reduced Đ values as compared to free radical polymerisations, UV-GPC analyses, and growth of Mn proportionally to monomer conversion. Saturated HCM was observed to have a kp app value an order of magnitude higher than that of the unsaturated HOSM. In order to overcome limited conversions/molecular weight in the RAFT polymerisation of brine washed HOSM (caused by a combination of factors including rate reduction due to allylic chain transfer and radical inhibitor impurities in the monomer) it was found that column purification was necessary. However, the more sustainable brine washing method was suitable for the controlled polymerisation of saturated HCM. Using these learnings, samples of P(HOSM) and P(HCM) were synthesised over a range of Mn (3000 to 12000 g mol−1) with low dispersities (Đ <1.3). Thermal analysis of these polymers revealed that polymers with saturated pendant FAs displayed semi-crystalline behaviour whereas polymers bearing unsaturated pendant FAs were fully amorphous. Lower Tg values (−1 to 12 °C) were observed for unsaturated FA polymers than for the saturated P(HCM) (Tg = 22 to 34 °C). These thermal properties were shown to vary with molecular weight. This work advances our understanding of the RAFT polymerisation of fatty acid-based monomers and has elucidated interesting thermal properties of the resulting plant oil-based polymers. Generating such novel materials from readily available biobased feedstocks is important for the move towards more sustainable polymers.
Author contributions
OJH: project administration, methodology, investigation, validation, visualisation, writing. PT: resources, conceptualisation. CJG: investigation. RRL: supervision, investigation, writing. HW: supervision, writing. FLH: project design, writing, funding acquisition, supervision.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
Support for O. H. is gratefully acknowledged from Loughborough University and an EPSRC Doctoral Training Partnership (EP/T518098/1) through UKRI ref 2585422. R. L. also acknowledges support through the EPSRC Doctoral Training Partnership (EP/T518098/1). F. H. acknowledges an EPSRC New Investigator Award (EP/W019175/1). The authors would also like to acknowledge Loughborough University Department of Chemistry for their facilities and expertise in NMR Spectroscopy and LC-MS.References
- S. Griffiths, B. K. Sovacool, J. Kim, M. Bazilian and J. M. Uratani, Energy Res. Soc. Sci., 2022, 89, 102542 CrossRef.
- Petrochemicals Europe interactive flowchart.
- J. Chen, W. Wang, Y. Pan, D. Peng, Y. Li and C. Zou, Polym. Chem., 2023, 14, 1103–1109 RSC.
- J. Lomège, V. Lapinte, C. Negrell, J. J. Robin and S. Caillol, Biomacromolecules, 2019, 20, 4–26 CrossRef PubMed.
- V. Sharma and P. P. Kundu, Prog. Polym. Sci., 2006, 31, 983–1008 CrossRef CAS.
- F. Seniha Güner, Y. Yaǧci and A. Tuncer Erciyes, Prog. Polym. Sci., 2006, 31, 633–670 CrossRef.
- L. Montero De Espinosa and M. A. R. Meier, Eur. Polym. J., 2011, 47, 837–852 CAS.
- U. Biermann, U. T. Bornscheuer, I. Feussner, M. A. R. Meier and J. O. Metzger, Angew. Chem., Int. Ed., 2021, 60, 20144–20165 CrossRef CAS.
- A. S. Carlsson, Biochimie, 2009, 91, 665–670 CrossRef CAS PubMed.
- W. E. Farr and A. Proctor, Green Vegetable Oil Processing: Revised First Edition, Elsevier Inc., 2013 Search PubMed.
- M. K. Gupta, Practical Guide to Vegetable Oil Processing, Elsevier, 2017 Search PubMed.
- S. K. Hess, N. S. Schunck, V. Goldbach, D. Ewe, P. G. Kroth and S. Mecking, J. Am. Chem. Soc., 2017, 139, 13487–13491 CrossRef CAS PubMed.
- B. Maiti and P. De, RSC Adv., 2013, 3, 24983–24990 CAS.
- B. Maiti, S. Kumar and P. De, RSC Adv., 2014, 4, 56415–56423 CAS.
- M. Decostanzi, J. Lomège, Y. Ecochard, A. S. Mora, C. Negrell and S. Caillol, Prog. Org. Coat., 2018, 124, 147–157 CrossRef CAS.
- H. Gan, S. A. Hutchinson, C. Hurren, Q. Liu, X. Wang and R. L. Long, Ind. Crops Prod., 2021, 171, 113882 CrossRef CAS.
- J. T. Windbiel and M. A. R. Meier, Macromol. Chem. Phys., 2021, 223(12), 2100360 CrossRef.
- J. Liu, Z. Cai and Y. Ji, J. Appl. Polym. Sci., 2023, 140(39), 54462 CrossRef.
- I. Tarnavchyk, A. Popadyuk, N. Popadyuk and A. Voronov, ACS Sustainable Chem. Eng., 2015, 3, 1618–1622 CrossRef CAS.
- Z. Demchuk, O. Shevchuk, I. Tarnavchyk, V. Kirianchuk, M. Lorenson, A. Kohut, S. Voronov and A. Voronov, ACS Omega, 2016, 1, 1374–1382 CrossRef CAS.
- Z. Demchuk, O. Shevchuk, I. Tarnavchyk, V. Kirianchuk, A. Kohut, S. Voronov and A. Voronov, ACS Sustainable Chem. Eng., 2016, 4, 6974–6980 CAS.
- W. Liu, M. Wu, C. Ma, C. Liu, X. Zhang, Z. Wang and Z. Wang, ACS Sustainable Chem. Eng., 2022, 10, 13301–13309 CAS.
- L. Yuan, Z. Wang, N. M. Trenor and C. Tang, Macromolecules, 2015, 48, 1320–1328 CAS.
- R. L. Glass, Lipids, 1971, 6, 919–925 CAS.
- M. Morgenstern, J. Cline, S. Meyer and S. Cataldo, Energy Fuels, 2006, 20, 1350–1353 CrossRef CAS.
- Y. Asakuma, K. Maeda, H. Kuramochi and K. Fukui, Fuel, 2009, 88, 786–791 CrossRef CAS.
- G. Moad, E. Rizzardo and S. H. Thang, Polymer, 2008, 49, 1079–1131 CrossRef CAS.
- G. Moad, Y. K. Chong, A. Postma, E. Rizzardo and S. H. Thang, Polymer, 2005, 46, 8458–8468 CrossRef CAS.
- J. Chiefari, Y. K. Chong, F. Ercole, J. Krstina, J. Jeffery, T. P. T. Le, R. T. A. Mayadunne, G. F. Meijs, C. L. Moad, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 1998, 31, 5559–5562 CrossRef CAS.
- S. Perrier, Macromolecules, 2017, 50, 7433–7447 CAS.
- F. L. Hatton, Polym. Chem., 2020, 11, 220–229 Search PubMed.
- J. Zhu, X. Zhu, Z. Cheng, J. Lu and F. Liu, J. Macromol. Sci., Part A:Pure Appl. Chem., 2003, 40, 963–975 Search PubMed.
- J. Gupta, D. J. Keddie, C. Wan, D. M. Haddleton and T. McNally, Polym. Chem., 2016, 7, 3884–3896 RSC.
- M. Demetriou and T. Krasia-Christoforou, J. Polym. Sci., Part A:Polym. Chem., 2008, 46, 5442–5451 CrossRef CAS.
- M. Semsarilar, N. J. W. Penfold, E. R. Jones and S. P. Armes, Polym. Chem., 2015, 6, 1751–1757 RSC.
- Y. Pei, L. Thurairajah, O. R. Sugita and A. B. Lowe, Macromolecules, 2015, 48, 236–244 CrossRef CAS.
- F. Ma, L. D. Clements and M. A. Hanna, Trans. Am. Soc. Agric. Eng., 1998, 41, 1261–1264 CAS.
- G. Vicente, M. Martínez, J. Aracil and A. Esteban, Ind. Eng. Chem. Res., 2005, 44, 5447–5454 CrossRef CAS.
- N. Hoda, Energy Sources, Part A, 2010, 32, 434–441 CrossRef CAS.
- D. Edinger, H. Weber, E. Žagar, D. Pahovnik and C. Slugovc, ACS Appl. Polym. Mater., 2021, 3, 2018–2026 CrossRef CAS.
- J. Clayden, N. Greeves, S. Warren and P. Wothers, Organic chemistry, Oxford University Press, 2009 Search PubMed.
- K. Roos, M. Planes, C. Bakkali-Hassani, J. Mehats, A. Vax and S. Carlotti, Macromolecules, 2016, 49, 2039–2045 CrossRef CAS.
- P. Q. Huang, Y. H. Huang, H. Geng and J. L. Ye, Sci. Rep., 2016, 6, 1–10 CrossRef.
- S. Morita, T. Yoshimura and J. I. Matsuo, Green Chem., 2021, 23, 1160–1164 RSC.
- T. Iwamura, I. Tomita, M. Suzuki and T. Endo, ACS Appl. Polym. Mater., 2000, 38, 430–435 CAS.
- I. Tarnavchyk, A. Popadyuk, N. Popadyuk and A. Voronov, ACS Sustainable Chem. Eng., 2015, 3, 1618–1622 CrossRef CAS.
- Z. Demchuk, O. Shevchuk, I. Tarnavchyk, V. Kirianchuk, A. Kohut, S. Voronov and A. Voronov, ACS Sustainable Chem. Eng., 2016, 4, 6974–6980 CrossRef CAS.
- R. Naureen, M. Tariq, I. Yusoff, A. J. K. Chowdhury and M. A. Ashraf, Saudi J. Biol. Sci., 2015, 22, 332–339 CrossRef CAS.
- F. Jin, K. Kawasaki, H. Kishida, K. Tohji, T. Moriya and H. Enomoto, Fuel, 2007, 86, 1201–1207 CrossRef CAS.
- L. A. Anderson and A. K. Franz, Energy Fuels, 2012, 26, 6404–6410 CrossRef CAS.
- B. Nieva-Echevarría, E. Goicoechea, M. J. Manzanos and M. D. Guillén, Food Res. Int., 2014, 66, 379–387 Search PubMed.
- C. Fauhl, F. Reniero and C. Guillou, Magn. Reson. Chem., 2000, 38, 436–443 Search PubMed.
- L. L. Gur'eva, A. I. Tkachuk, Y. I. Estrin, B. A. Komarov, E. A. Dzhavadyan, G. A. Estrina, L. M. Bogdanova, N. F. Surkov and B. A. Rozenberg, Polym. Sci., Ser. A, 2008, 50, 283–290 Search PubMed.
- Y. Sun, L. Fu, M. Olszewski, K. Matyjaszewski, Y. Sun, L. Fu, M. Olszewski and K. Matyjaszewski, Macromol. Rapid Commun., 2019, 40, 1800877 Search PubMed.
- B. De Lambert, M. T. Charreyre, C. Chaix and C. Pichot, Polymer, 2005, 46, 623–637 CAS.
- Y. Cao, X. X. Zhu, J. Luo and H. Liu, Macromolecules, 2007, 40, 6481–6488 CAS.
- B. A. Abel and C. L. McCormick, Macromolecules, 2016, 49, 465–474 CAS.
- O. Creese, P. Adoni, G. Su, A. Romanyuk and P. Fernandez-Trillo, Polym. Chem., 2019, 10, 5645–5651 Search PubMed.
- N. J. Fox and G. W. Stachowiak, Tribol. Int., 2007, 40, 1035–1046 CAS.
- S. A. Harrison and D. H. Wheeler, J. Am. Chem. Soc., 1951, 73, 839–842 CAS.
- N. G. Gaylord, J. Polym. Sci., 1956, 22, 71–78 CrossRef CAS.
- D. J. Carlsson, K. U. Ingold, H. G. Kuivila, L. W. Menapace, C. R. Warner, J. Am, R. J. Strunk, O. Chem, C. Walling, J. H. Cooley, A. A. Ponaras, E. J. Racah, A. K. Sawyer and C. Ind, Tetrahedron Lett., 1968, 90, 2041 Search PubMed.
- D. B. Thomas, A. J. Convertine, L. J. Myrick, C. W. Scales, A. E. Smith, A. B. Lowe, Y. A. Vasilieva, N. Ayres and C. L. McCormick, Macromolecules, 2004, 37, 8941–8950 CrossRef CAS.
- D. J. Keddie, G. Moad, E. Rizzardo and S. H. Thang, Macromolecules, 2012, 45, 5321–5342 CrossRef CAS.
- S. Zhou, Y. Zhao, Y. Cai, Y. Zhou, D. Wang, C. C. Han and D. Xu, Polymer, 2004, 45, 6261–6268 CrossRef CAS.
- J. Cai, Z. Wei, W. Luo and W. Hu, Polymer, 2022, 252, 124922 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4py01100g |
| This journal is © The Royal Society of Chemistry 2025 |