
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Environmentally sustainable lithium-ion battery cathode binders based on cellulose nanocrystals†
Xingkang Huang‡
 *ab,
Haoyang You‡c,
Xiaoli Yan‡d,
Olaf J. Borkiewicze,
Kamila M. Wiadereke,
Janan Huif,
Mark C. Hersamfgh,
Santanu Chaudhuri*di,
Stuart J. Rowan*abc and
Junhong Chen*ab
*ab,
Haoyang You‡c,
Xiaoli Yan‡d,
Olaf J. Borkiewicze,
Kamila M. Wiadereke,
Janan Huif,
Mark C. Hersamfgh,
Santanu Chaudhuri*di,
Stuart J. Rowan*abc and
Junhong Chen*ab
aPritzker School of Molecular Engineering, The University of Chicago, 5640 S. Ellis Ave, Chicago, IL 60637, USA. E-mail: junhongchen@uchicago.edu; stuartrowan@uchicago.edu; xkhuang@uchicago.edu
bChemical Sciences and Engineering Division, Physical Sciences and Engineering Directorate, Argonne National Laboratory, Lemont, Illinois 60439, USA
cDepartment of Chemistry, The University of Chicago, Chicago, IL 60637, USA
dDepartment of Civil, Materials, and Environmental Engineering, University of Illinois, Chicago, Illinois 60439, USA. E-mail: santc@uic.edu
eX-ray Science Division, Advanced Photon Source, Argonne National Laboratory, Lemont, IL 60439, USA
fDepartment of Materials Science and Engineering, Northwestern University, Evanston, IL 60208, USA
gDepartment of Chemistry, Northwestern University, Evanston, IL 60208, USA
hDepartment of Electrical and Computer Engineering, Northwestern University, Evanston, IL 60208, USA
iApplied Materials Division, Advanced Energy Technologies Directorate, Argonne National Laboratory, Lemont, IL 60439, USA
First published on 16th August 2024
Abstract
Aqueous binders as environmentally sustainable alternatives to conventional polyvinylidene difluoride (PVDF) binders have not yet been successful for cathodes in lithium-ion batteries (LIBs). Here, carboxylic acid functionalized cellulose nanocrystals (CNC-COOHs) have been obtained from Miscanthus × giganteus (M×G) biomass and evaluated as aqueous binders for LIB cathodes.
Electrodes in lithium-ion batteries (LIBs) generally consist of an electroactive material, conductive carbon, and a polymer binder. Polymer binders play an important role in battery electrodes to aid the formation of a uniform slurry for film formation and to bind active materials and conductive carbon on the current collectors. Polyvinylidene difluoride (PVDF) is the most common commercially available binder for cathodes in LIBs. However, dissolution of PVDF requires strong solvents such as N-methyl-2-pyrrolidone (NMP), which is a costly solvent. In addition, there are toxicity issues on account of NMP's reproductive hazards.1 To avoid the use of NMP and utilize greener approaches to batteries, there have been significant efforts devoted to researching aqueous binders such as carboxymethyl cellulose (CMC),2,3 CMC/styrene butadiene rubber (SBR),4 alginate,5–7 polyacrylic latex (LA132),8,9 lignin,10 polyacrylic acid (PAA) and its salt,11,12 and chitosan.13 One of the major challenges for these aqueous binders to be used in cathodes of LIBs is the corrosion of aluminum current collectors.14 The native oxide protective layer on the aluminum surface is only stable at a pH value ranging from 4.5 to 8.5, whereas cathode materials in water typically display pH higher than 8.5.15 This high pH is the result of excessive lithium ions (e.g., Li2O, LiOH or Li2CO3) added during preparation of cathodes, which is intended to compensate for the loss of lithium at high temperatures; however, the residual Li2O or Li2CO3 may dissolve in the aqueous binder, leading to an increased pH value. In addition, when cathode materials are mixed with aqueous binders, Li+ in the cathode materials may exchange with H+ in the water, and the leached Li+ from cathode materials leads to LiOH that further increases the pH value. Addition of acid (such as H3PO4) has been reported as necessary to prevent Al foil corrosion in the cases of CMC/SBR as the binders.16 However, Bauer et al. reported significantly reduced adhesion strength of the cathode coating on the aluminum foil in the presence of most acids such as acetic, citric, oxalic, and phosphoric acids, with the exception of high molecular weight organic acids, such as polyacrylic acid (PAA).15
Therefore, it is promising to develop an aqueous binder with weak acidity but strong adhesion, and cellulose nanocrystals (CNCs) with carboxylic acid groups may meet this requirement and hold promise for aqueous binders in LIB cathodes. CNCs are highly crystalline rod-like nanomaterials that can be obtained from a wide range of biomass.15 Here, carboxylic acid functionalized CNCs (CNC-COOHs), obtained from Miscanthus × giganteus (M×G) stalks, are explored as a potential binder for LIB cathodes, such as spinel lithium manganese oxide (LiMn2O4 or LMO) and LiCoO2 (LCO) since they are less sensitive to water compared with other cathode materials such as Ni-rich cathodes.17 It has been shown in the literature18–22 that CNCs possess amphiphilic characteristics as they have both hydrophilic (110 and 1![[1 with combining macron]](https://www.rsc.org/images/entities/char_0031_0304.gif) 0) and hydrophobic (200) crystal faces. As such it is hypothesized that the CNC-COOH have the potential to be an effective binder for both LMO and carbon black (CB) and result in the formation of homogenous slurries. Our results indicate that the cathodes prepared using CNC-COOH binders exhibit improved electrochemical performances, especially the rate capability, compared with the cathodes using the incumbent PVDF binder. As CNC-COOHs are biodegradable and renewable, using them as an aqueous-based binder would enhance sustainability and reduce environmental concerns caused by traditional PVDF binders. In addition, the use of aqueous CNC-COOH binder could benefit recycling of spent cathode materials, compared with PVDF binders. To recover cathode materials from spent LIBs, strong solvents such as NMP are widely used to dissolve PVDF; alternatively, pyrometallurgy is another popular approach to remove PVDF, which releases hazardous byproducts.23 In contrast, removal of CNC-COOH binders from spent cathodes is much easier by using water without introducing extra hazard. After being washed with water 5 times, the content of CNC-COOH in an LMO electrode with CNC-COOH binder significantly decreased, as suggested by the decreased C/Mn atomic ratio from 0.46 to 0.08 (Fig. S1, Tables S1 and S2†).
0) and hydrophobic (200) crystal faces. As such it is hypothesized that the CNC-COOH have the potential to be an effective binder for both LMO and carbon black (CB) and result in the formation of homogenous slurries. Our results indicate that the cathodes prepared using CNC-COOH binders exhibit improved electrochemical performances, especially the rate capability, compared with the cathodes using the incumbent PVDF binder. As CNC-COOHs are biodegradable and renewable, using them as an aqueous-based binder would enhance sustainability and reduce environmental concerns caused by traditional PVDF binders. In addition, the use of aqueous CNC-COOH binder could benefit recycling of spent cathode materials, compared with PVDF binders. To recover cathode materials from spent LIBs, strong solvents such as NMP are widely used to dissolve PVDF; alternatively, pyrometallurgy is another popular approach to remove PVDF, which releases hazardous byproducts.23 In contrast, removal of CNC-COOH binders from spent cathodes is much easier by using water without introducing extra hazard. After being washed with water 5 times, the content of CNC-COOH in an LMO electrode with CNC-COOH binder significantly decreased, as suggested by the decreased C/Mn atomic ratio from 0.46 to 0.08 (Fig. S1, Tables S1 and S2†).
The preparation of CNC-COOHs is based on a previous report24 with some slight modifications. In short, CNCs were isolated from M×G stalks by NaOH washing, NaClO2 bleaching, and HCl hydrolysis, followed by oxidation using 2,2,6,6-tetramethylpiperidinyloxy (TEMPO) to obtain the CNC-COOHs. The CNC-COOHs utilized in this work have a height of 2.6 ± 0.8 nm and a length of 183 ± 91 nm, as measured by atomic force microscopy (Fig. 1a). The crystallinity index of the CNC-COOH is approximately 85%, determined by peak deconvolution of wide-angle X-ray scattering (WAXS) data25 (Fig. S2a†). The content of the carboxylic acid group in the CNC-COOH was determined to be approximately 2.4 mol kg−1 by conductometric titration (Fig. S2b†). CNC-COOH suspension was prepared by dispersing CNC-COOHs in deionized water (2–4 wt%) with the assistance of probe sonication. The pH value of the 2 wt% CNC-COOH dispersion is approximately 5.7; this weak acidity helps limit the corrosion of the Al current collector by the residual LiOH or Li2CO3 in cathodes.
 | ||
| Fig. 1 (a) AFM height images of M×G-CNC-COOH, (b) SEM image of LMO, and (c) SEM image of LMO with 10 wt% CNC, in which 2.5 nm of Au was sputtered to improve the electronic conductivity to observe CNCs, (d) PMF values of PVDF and CNC-COOH's different surfaces during the binding process with spinel LMO, (e) molecular dynamics simulation snapshot of a CNC-COOH crystal approaching LMO. A pair of constant velocity moving spring forces are exerted on the centers of mass of both groups of atoms so that they can move toward each other, in which C, O, H, Li, and Mn are represented by grey, red, white, pink, and blue atoms, respectively, (f) observation of representative coordination structures of Mn and –COOH: hydrogen bonds and Mn–O bonds shown in the left and right panel, respectively, (g) a CNC-COOH unit with atoms labeled, and radial distribution functions (h) between O in LMO and H in CNC-COOH, (i) between Mn in LMO and O in CNC-COOH. | ||
The LMO has a bimodal distribution of particle sizes (ca. 5 μm and 30–100 nm, Fig. 1b); more SEM images of the LMO are shown in Fig. S3.† The LMO was mixed with carbon black (CB) and the CNC-COOHs by a weight ratio of 80![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :10:10 to make a homogenous slurry, which was cast onto an Al substrate and dried. The mass loading on the electrode was 3.5 mg cm−2, which is within the typical range used in most literature (e.g., 1.5–5 mg cm−2).26–28 As CB typically consists of particles approximately 50–100 nm, similar to the smaller particles of the LMO (Fig. 1b), it can be difficult to differentiate CNC-COOHs covered on LMO versus CB surfaces. Therefore, a film comprised of just LMO and CNC-COOH (90:10 by weight) was produced and as shown in the SEM image of Fig. 1c, the CNC-COOH appears as a mesh covering LMO particles.
:10:10 to make a homogenous slurry, which was cast onto an Al substrate and dried. The mass loading on the electrode was 3.5 mg cm−2, which is within the typical range used in most literature (e.g., 1.5–5 mg cm−2).26–28 As CB typically consists of particles approximately 50–100 nm, similar to the smaller particles of the LMO (Fig. 1b), it can be difficult to differentiate CNC-COOHs covered on LMO versus CB surfaces. Therefore, a film comprised of just LMO and CNC-COOH (90:10 by weight) was produced and as shown in the SEM image of Fig. 1c, the CNC-COOH appears as a mesh covering LMO particles.
To understand the binding mechanism between LMO and CNC-COOH, a series of reactive molecular dynamics (MD) simulations were conducted to provide mechanistic insights into the binding performance of CNC-COOH with LMO (Fig. 1d–f). The MD simulations utilize an existing ReaxFF force field for LIB systems.29 CNCs have hydrophilic faces (e.g., 110 and 10) that contain the polar surface –OH and –COOH moieties and hydrophobic faces (e.g., 200) that consist of non-polar surface C–H bonds. As shown in Fig. 1d, the minimum normalized potential of mean force (PMF) between the (10) facet of the CNC-COOH and the LMO was approximately −12.2 Cal Å−2 mol−1, smaller than that between the PVDF and the LMO (−0.75 Cal Å−2 mol−1), which suggests CNC-COOH has stronger binding with LMO than PVDF. PVDF binds LMO through the interaction between the F atoms in the PVDF and the Mn in the LMO (Fig. S4†), while CNC-COOH can bind the LMO more efficiently through hydroxyl and carboxyl groups on the (10) facet (Fig. 1e and f); specifically, both O and Mn atoms from the LMO form coordination interactions with H atoms and O atoms from the CNC's carboxyl/hydroxyl and carboxyl groups, respectively (Fig. 1f). To demonstrate the binding mechanism between the LMO and the CNC-COOH, a CNC-COOH repeat unit is shown in Fig. 1g with the atoms labeled. According to the radial distribution functions from MD simulations, the major hydrogen bonding interactions between the LMO and the CNC-COOH come from the H6′ in the carboxylic acid group on C6′ as opposed to the H2 and H3 in hydroxyl groups (Fig. 1h). In addition, the Mn atoms in the LMO predominantly interact with the O6′ and O6′′ in the carboxyl groups rather than the O2 and O3 in hydroxyl groups (Fig. 1i). The finding explains the important role that –COOH moieties on the surface of the CNCs play in the binding between the CNC-COOH and the LMO.
As might be expected, when the hydrophobic (200) facet of the CNC-COOH approaches the LMO, the PMF remains at near zero and starts to increase after their distance is closer than 6.5 Å. This means there is no binding between the LMO and the (200) facet of the CNC-COOHs. Given that carbon is non-polar and hydrophobic,30–32 CB is prone to aggregation in water. However, in the presence of CNC-COOHs, the non-polar (200) facet of the CNC-COOH can be attracted to the surface of CB, reducing the interfacial free energy between CB and water,33 and the charged hydrophilic (10) facet of the CNC-COOH helps stabilize the CB in water, achieving more uniform slurry and effective binding between cathode components.
The LMO/CNC-COOH electrode was assembled into coin cells to study its electrochemical performance. The LMO delivered an initial capacity of 113.7 mA h g−1 at 0.1C (1C = 110 mA g−1) with an initial coulombic efficiency (ICE) of 95.0%, in the presence of the CNC-COOH binder (10 wt%) (Fig. 2a); in contrast, the LMO electrode with the PVDF binder (10 wt%) had an initial capacity of 106 mA h g−1 with an ICE of 92.1%. After 100 cycles at 0.5C, the capacity retention was 93.8% and 86.5% for the CNC-COOH and the PVDF-containing electrodes, respectively (Fig. 2a and b). Furthermore, the LMO exhibited a better rate capability with the CNC-COOH binders (Fig. 2c and d). In the presence of the PVDF binder, the LMO delivered capacities of 108.2, 106.0, 101.5, 96.8, 91.6, 78.4, 46.9 mA h g−1 at 0.1, 0.2, 0.5, 1, 2, 5, 10C, respectively. In contrast, with the CNC-COOH, the LMO delivered 110.6, 109.0, 106.3, 102.2, 96.4, 78.7, 56.1 mA h g−1 at 0.1, 0.2, 0.5, 1, 2, 5, 10C, respectively. As a result, the LMO with the CNC-COOH binder showed better electrochemical performance than that with the PVDF binder in both cyclic performance and rate capability. Ryou et al. reported that oxygen-containing groups (such as carboxyl, hydroxyl, and ether groups) in an alginate binder might help alleviate Mn2+ dissolution, which improved the cyclic performance of LMO cathodes.34 While our CNC-COOH binder might share the similar mechanism improving the cyclic performance, the improvement on the rate capability of the LMO cathode is more remarkable. As indicated by the binding mechanism discussed above, the CNC-COOH possesses both hydrophilic (110 and 10) and hydrophobic (200) facets, which benefit binding both active material (LMO) and conductive CB, thereby resulting in a highly uniform slurry. In other words, with the CNC-COOH binders, the LMO may have opportunity to contact more CB, thereby exhibiting reduced polarization and improved rate capability, compared to that with the PVDF binders. This result is consistent with the findings by Guerfi et al.;35 they compared a water-soluble elastomer and PVDF as binders for LiFePO4 cathodes and concluded that the LiFePO4 cathode was less uniform while using the PVDF binder, thereby showing inferior performance than that with the aqueous elastomer binder.
 | ||
| Fig. 2 (a) Cyclic performance and (b) charge/discharge curves of the LMO with CNC-COOH binder (dashed lines) and PVDF binder (solid lines), (c) rate capability and (d) charge/discharge curves for the LMO at various current rates with CNC-COOH binder (dashed lines) and PVDF binder (solid lines) of half-cells; (e) cyclic performance and (f) charge/discharge curves of the LMO‖graphite full cells, in which the LMO cathode was prepared with CNC-COOH binder. | ||
To demonstrate the feasibility of the CNC-COOH as a binder in potential practical applications, we investigated the full-cell performance of the LMO cathode with the CNC-COOH binder, in which graphite was used as the anode. The full cell was designed with a capacity loading of 1 mA h cm−2 and an N/P ratio of 1.1. To achieve that goal, the slurry formula was LMO, conductive carbon, and binder with a mass ratio of 93:5:2, with the resulting active material loading on the electrode of approximately 9.2 mg cm−2. 2 wt% of CNC-COOH exhibited good binding performance, and the low electronic conductivity of LMO required the addition of conductive carbon.36,37 The conductive carbon content consisted of 4.7 wt% CB with 0.3 wt% graphene3 as an additive to improve the electronic conductivity of the LMO cathode (Fig. S5†).
The LMO/CNC-COOH‖graphite full cell showed a reversible capacity of approximately 80 mA h g−1 and retained 87.5% of the capacity after 200 cycles (Fig. 2e), which is better than the LMO/PVDF‖graphite full cell with the same anode (Fig. S6†). This cyclic performance of our LMO/CNC-COOH‖graphite full cell is also slightly better than previously reported LMO full cells with alginate and PVDF binders that retained 79.4% and 76.4% of the capacity, respectively, after 200 cycles.34 In addition, by comparing the charge/discharge curves at 0.1C and 0.5C between the half cell (Fig. S5a†) and the full cell (Fig. 2f), we can see the substantially lower polarization in the full cell than that in the half cell, presumably on account of a thinner SEI layer and thus lower resistance in the full cell.
Electrochemical impedance spectroscopy (EIS) was measured to explain the better rate capability of the LMO with the CNC-COOH binder than that with the PVDF. While the Nyquist plots at all states of charge (SOCs) are shown in Fig. S7† and 3 depicts the LMO with PVDF and CNC-COOH binders at SOCs of 10%, 50%, and 100%, for direct comparison. At low SOCs (e.g., 10%), the LMO with the CNC-COOH showed smaller complex impedance than that with the PVDF binder, mainly from the capacitance instead of resistance, which is interesting, but the mechanism is uncertain at this stage. Upon increasing SOCs such as at 50% and 100% SOCs, the impedance of the LMO electrodes decreased with both binders but decreased more in the presence of the CNC-COOH binder. The lower impedance of the LMO/CNC-COOH cathode explains its better rate capability than the LMO/PVDF cathode, as the amphiphilic property of the CNC-COOH leads to more uniformly distributed LMO and CB in the cathode; the better contact between the LMO and the CB likely help decrease charge transfer resistance (Fig. 3).
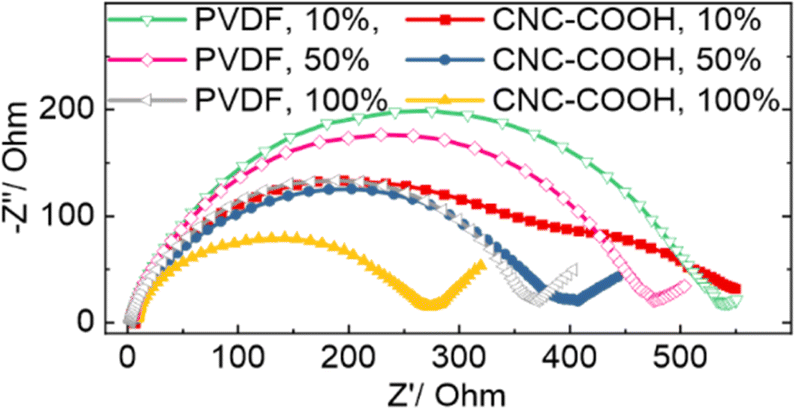 | ||
| Fig. 3 Nyquist plots of LMO electrodes with PVDF and CNC-COOH binders at three different states of charge (SOC), 10%, 50%, and 100%. | ||
The excellent cyclic performance of the LMO with CNC-COOH binder was also supported by operando synchrotron X-ray diffraction (XRD) characterization. A radially accessible tubular in situ X-ray (RATIX) cell38 was modified to carry out the operando XRD experiment, which allows the X-ray to penetrate only the electrode layer, besides the glass wall of the RATIX cell, thereby minimizing the interference from other components such as stainless steel, lithium metal, and Al substrate in the cell (Fig. 4a). Fig. 4b depicts the charge/discharge curves of the LMO electrode with the CNC-COOH binder, indicating reliable battery performance using the modified RATIX cell. As shown in Fig. 4c, most of the peaks shifted towards higher angles upon charging, which is consistent with the fact that crystal parameter a of the cubic LMO phase decreased along with de-intercalation of lithium ions from the LMO. To quantify the changes of a, these patterns were refined using GSAS II software.39 The initial a was 8.2228(2) Å and decreased to 8.0557(2) Å upon being fully charged, which recovered to 8.2234(2) Å at the fully discharged state (Fig. 4d). The recovered a helps explain the superlative cyclic performance of LMO with the CNC-COOH binder. Note that symbols * in Fig. 4c indicate the peaks from stainless steel (SS) and aluminum (Al), which were considered as Fe and Al phases instead of background during Rietveld refinement as shown in Fig. S8.† The observed residual errors (Rwp) were between 5.1% and 6.6% during the sequential refinement.
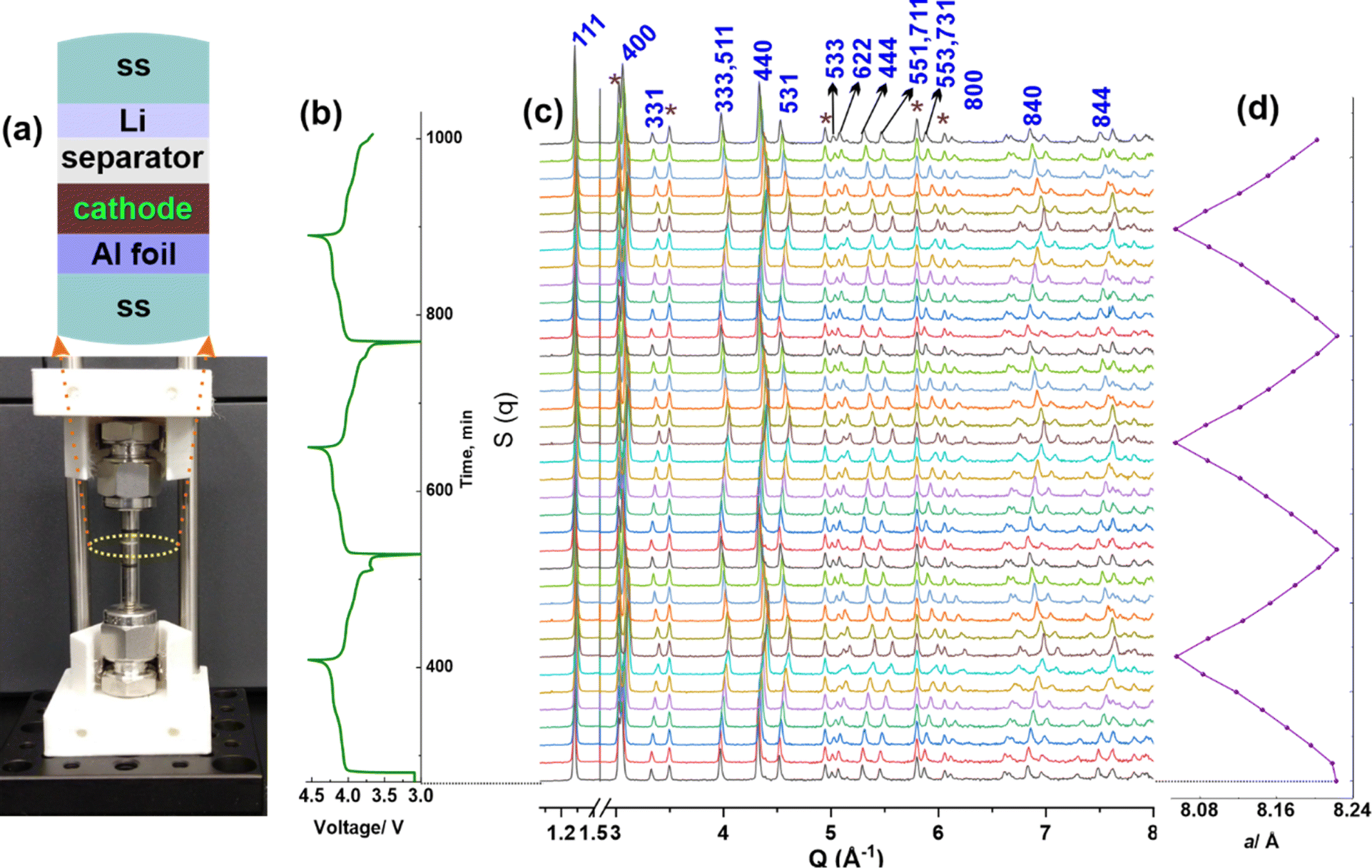 | ||
| Fig. 4 (a) Modified RATIX cell with 3D printed cell holder, (b) charge/discharge curves of the LMO, (c) operando synchrotron XRD of the LMO cycled at 0.5C for 3 cycles, and (d) refined crystal parameter a for LixMn2O4. | ||
The coin cell was also dissembled after cycling to examine the integrity of the LMO electrode. By comparing the cycled LMO electrode (Fig. S9†) with a fresh LMO electrode (Fig. S10†), it is clear that the cycled LMO electrode remained intact, without forming significant cracks or losing active materials on the electrode. Consequently, it can be concluded that the CNC-COOH functioned well as the binder for the LMO cathode upon cycling.
While demonstrating the great potential of the CNC-COOH as a binder for LMO cathode, we extended our effort to other cathodes such as LiCoO2 (LCO) and LiFePO4 (LFP). With the CNC-COOH binder, the LCO delivered a reversible capacity of 140 mA h g−1 with an ICE of 98.1% at 0.1C when the voltage was cut off at 4.2 V, which is very close to that of the LCO/PVDF electrode with a reversible capacity of 141 mA h g−1 and an ICE of 98.5% (Fig. S11†); at 0.5C, the LCO/CNC-COOH and the LCO/PVDF cathodes delivered a reversible capacity of 132 and 133 mA h g−1, respectively, and retained 121 and 125 mA h g−1, respectively, after 100 cycles. As the LCO cathode with CNC-COOH binder showed very comparable cyclic performance, we expanded the voltage upper limit upon charging to challenge its cyclic performance with higher capacities. As shown in Fig. 5a and b, both LCO/CNC-COOH and LCO/PVDF exhibited 190 mA h g−1 at 0.1C when they were charged to 4.5 V with similar ICEs of 98% while delivering 181 and 183 mA h g−1, respectively, at 0.5C. Although the LCO/CNC-COOH showed slightly faster decay at early stage, after 150 cycles, it retained a higher capacity (140 mA h g−1) than the LCO/PVDF cathode (133 mA h g−1). Note that after approximately 140 cycles, the LCO/PVDF cathode showed a faster capacity decay than the LCO/CNC-COOH cathode. This might be attributed to the decay of the anode, as the relatively high capacity-loading of approximately 2.1 mA h cm−2 might lead to the formation of dendrites on the lithium anode upon repeating charging/discharging cycles, which increases the impedance of the half cells.
 | ||
| Fig. 5 (a) Cyclic performance and (b) representative charge/discharge curves of LCO cathode with PVDF and CNC-COOH binders with a voltage upper limit of 4.5 V; (c and d) rate capability of the LCO cathodes with dashed lines for CNC-COOH and solid lines for PVDF in (d); 1C rate was defined as 180 mA g−1 for the LCO; (e) cyclic performance and (f) representative charge/discharge curves of LCO/CNC-COOH‖graphite full cell with a voltage upper limit of 4.4 V. | ||
While the LCO/CNC-COOH cathode showed comparable cyclic performance to the LCO/PVDF cathode, the former displayed a better rate capability, as shown in Fig. 5c and d. The LCO/CNC-COOH delivered a similar capacity at 0.1C but much higher capacities at high C-rates, compared with the LCO/PVDF cathode; for example, at 5C, the LCO/CNC-COOH delivered 67 mA h g−1, much higher than that of the LCO/PVDF cathode (42 mA h g−1), and the LCO/CNC-COOH cathode was able to deliver 11 mA h g−1 at 10C while the LCO/PVDF cathode failed in performing at such a high current density. Note that at some current rates (from 0.2C to 2C), the capacities of the LCO/CNC-COOH cathode were lower than those of the LCO/PVDF cathode (Fig. 5c), which is because the LCO/CNC-COOH exhibited a slightly faster capacity decay at early stage upon cycling (Fig. 5a). By looking at their charge/discharge curves, e.g., at 2C, we can identify the lower polarization (lower charge voltages and higher discharge voltages) of the LCO/CNC-COOH binders than that of the LCO/PVDF binders (Fig. 5d). The improved rate capability of the LCO/CNC-COOH is likely associated with the amphiphilic characteristics of the CNC-COOH, which helps obtain more uniform slurry, thereby leading to reduced charge transfer resistance, compared with the cathode with PVDF binders. The excellent performance of the LCO/CNC-COOH cathode encouraged us to investigate its performance in full cells. As shown in Fig. 5e and f, the LCO/CNC-COOH‖graphite full cell delivered an initial capacity of approximately 170.2 mA h g−1 at 19 mA g−1 and 154.7 mA h g−1 at 190 mA g−1, which retained approximately 105 mA h g−1 after 400 cycles. In addition, the LCO/CNC-COOH‖graphite full exhibited good rate capability, as it delivered 165.2, 160.5, 149.6, 138.7, 118.0, 48.6 mA h g−1 at 0.1, 0.2, 0.5, 1, 2, 5C, respectively (Fig. S12†), which means the full cell is capable of charging or discharging its approximately 71% capacity within 20 min.
In addition, the CNC-COOH was used as binder for LFP cathodes. As shown in Fig. S13,† the reversible capacities at 0.1C and 0.5C were approximately 151 and 143 mA h g−1, respectively, and no capacity decay was observed after 150 cycles.
In summary, CNC-COOH derived from M×G was demonstrated as a more sustainable binder for LMO cathodes. The LMO with CNC-COOH showed an improved electrochemical performance compared with the commonly used PVDF binder, which can be attributed to the superior binding property between the binder and the LMO/CB. The CNC-COOH contains both hydrophilic and hydrophobic crystal faces to bind with LMO and CB, respectively, which helps obtain a more uniform slurry. MD simulations indicate the effective binding between the LMO and the CNC-COOH is the result of O and Mn atoms from the LMO forming non-covalent interactions with H atoms and O atoms from the CNC-COOH's carboxyl groups, respectively. In addition, the CNC-COOH was also demonstrated as a suitable binder for LCO and LFP cathodes. Considering that CNC-COOH is made from widely available biomass, CNC-COOH is more environmentally friendly than conventional binders, thereby providing a sustainable alternative binder solution for LIB cathodes in practical applications.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
X. H., H. Y., S. R., and J. C. conceptualized the use of CNC-COOH as the binder for LIB cathode. X. Y. and S. C. conceptualized the MD study. H. Y. prepared and characterized the CNC-COOH. X. H. evaluated the cell performance. J. H. prepared graphene under the supervision of M. H. Operando XRD was conducted and analyzed by O. J. B., K. M. W., and X. H. The original manuscript was written by X. H., H. Y., X. Y. and J. H. All authors discussed and provided input on writing the original version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors acknowledge support from the National Science Foundation MADE-PUBLIC Future Manufacturing Research Grant Program (CMMI-2037026). SEM characterization was performed at the Center for Nanoscale Materials, a U.S. Department of Energy (DOE) Office of Science User Facility, supported by the U.S. DOE, Office of Basic Energy Sciences, under Contract No. DE-AC02-06CH11357. This research used resources of the Advanced Photon Source, a U.S. DOE Office of Science user facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357. The anodes used for full cells in this work are from Argonne's Cell Analysis, Modeling and Prototyping (CAMP) Facility, which is fully supported by the DOE Vehicle Technologies Office (VTO). This work also made use of the shared facilities at the University of Chicago Materials Research Science and Engineering Center, supported by National Science Foundation under award number DMR-2011854. Part of this work was carried out at the Soft Matter Characterization Facility of the University of Chicago. Part of the computation work was conducted with resources from the Delta supercomputer through allocation MAT220042 from the Advanced Cyberinfrastructure Coordination Ecosystem: Services & Support (ACCESS) program. Delta is a joint effort of the University of Illinois Urbana-Champaign and the National Center for Supercomputing Applications (NCSA). The computation works also used resources from the Extreme supercomputer at the Advanced Cyberinfrastructure for Education and Research (ACER) at the University of Illinois at Chicago. X. Y. thanks the support from Janki Brahmbhatt for the assistance on the simulations.Notes and references
- D. Bresser, D. Buchholz, A. Moretti, A. Varzi and S. Passerini, Energy Environ. Sci., 2018, 11, 3096–3127 RSC.
- Z. L. Wang, N. Dupre, A. C. Gaillot, B. Lestriez, J. F. Martin, L. Daniel, S. Patoux and D. Guyomard, Electrochim. Acta, 2012, 62, 77–83 CrossRef CAS.
- Z. Chen, G. T. Kim, D. L. Chao, N. Loeffler, M. Copley, J. Y. Lin, Z. X. Shen and S. Passerini, J. Power Sources, 2017, 372, 180–187 CrossRef CAS.
- C. C. Li and Y. W. Wang, J. Electrochem. Soc., 2011, 158, A1361–A1370 CrossRef CAS.
- J. T. Xu, S. L. Chou, Q. F. Gu, H. K. Liu and S. X. Dou, J. Power Sources, 2013, 225, 172–178 CrossRef CAS.
- T. L. Zhao, Y. Meng, R. X. Ji, F. Wu, L. Li and R. J. Chen, J. Alloys Compd., 2019, 811, 152060 CrossRef CAS.
- S. J. Zhang, Y. P. Deng, Q. H. Wu, Y. Zhou, J. T. Li, Z. Y. Wu, Z. W. Yin, Y. Q. Lu, C. H. Shen, L. Huang and S. G. Sun, ChemElectroChem, 2018, 5, 1321–1329 CrossRef CAS.
- H. X. Zhong, M. H. Sun, Y. Li, J. R. He, J. W. Yang and L. Z. Zhang, J. Solid State Electrochem., 2016, 20, 1–8 CrossRef CAS.
- J. T. Lee, Y. J. Chu, X. W. Peng, F. M. Wang, C. R. Yang and C. C. Li, J. Power Sources, 2007, 173, 985–989 CrossRef CAS.
- S. N. Bryntesen, I. Tolstorebrov, A. M. Svensson, P. Shearing, J. J. Lamb and O. S. Burheim, Mater. Adv., 2023, 4, 523–541 RSC.
- M. Yu, Y. Wang, Z. Y. Wang, Y. P. Fan, J. H. Song, D. F. Zhou, K. Wang, Q. S. Zhang, H. T. Gu and J. Y. Xie, J. Electrochem. Soc., 2019, 166, A4122–A4127 CrossRef CAS.
- Z. P. Cai, Y. Liang, W. S. Li, L. D. Xing and Y. H. Liao, J. Power Sources, 2009, 189, 547–551 CrossRef CAS.
- S. Kunne, F. Puttmann, M. Linhorst, B. M. Moerschbacher, M. Winter, J. Li and T. Placke, ChemElectroChem, 2022, 9, e202200600 CrossRef.
- I. Doberdo, N. Loffler, N. Laszczynski, D. Cericola, N. Penazzi, S. Bodoardo, G. T. Kim and S. Passerini, J. Power Sources, 2014, 248, 1000–1006 CrossRef CAS.
- W. Bauer, F. A. Cetinel, M. Muller and U. Kaufmann, Electrochim. Acta, 2019, 317, 112–119 CrossRef CAS.
- S. Radloff, R. G. Scurtu, M. Holzle and M. Wohlfahrt-Mehrens, J. Electrochem. Soc., 2021, 168, 100506 CrossRef CAS.
- A. M. Pillai, P. S. Salini, B. John and M. T. Devassy, Energy Fuels, 2022, 36, 5063–5087 CrossRef CAS.
- J. Lehtiö, J. Sugiyama, M. Gustavsson, L. Fransson, M. Linder and T. T. Teeri, Proc. Natl. Acad. Sci. U.S.A., 2003, 100, 484–489 CrossRef PubMed.
- I. Kalashnikova, H. Bizot, B. Cathala and I. Capron, Biomacromolecules, 2012, 13, 267–275 CrossRef CAS PubMed.
- K. Y. Lee, J. J. Blaker, R. Murakami, J. Y. Y. Heng and A. Bismarck, Langmuir, 2014, 30, 452–460 CrossRef CAS PubMed.
- F. Cherhal, F. Cousin and I. Capron, Biomacromolecules, 2016, 17, 496–502 CrossRef CAS PubMed.
- Y. F. Zhang, V. Karimkhani, B. T. Makowski, G. Samaranayake and S. J. Rowan, Macromolecules, 2017, 50, 6032–6042 CrossRef CAS.
- T. Raj, K. Chandrasekhar, A. N. Kumar, P. Sharma, A. Pandey, M. Jang, B. H. Jeon, S. Varjani and S. H. Kim, J. Hazard. Mater., 2022, 429, 128312 CrossRef CAS PubMed.
- E. Cudjoe, M. Hunsen, Z. J. Xue, A. E. Way, E. Barrios, R. A. Olson, M. J. A. Hore and S. J. Rowan, Carbohydr. Polym., 2017, 155, 230–241 CrossRef CAS PubMed.
- S. Park, J. O. Baker, M. E. Himmel, P. A. Parilla and D. K. Johnson, Biotechnol. Biofuels, 2010, 3, 10 CrossRef PubMed.
- M. J. Young, S. Letourneau, R. E. Warburton, W. M. Dose, C. Johnson, J. Greeley and J. W. Elam, J. Phys. Chem. C, 2019, 123, 23783–23790 CrossRef CAS.
- D. K. Kim, P. Muralidharan, H. W. Lee, R. Ruffo, Y. Yang, C. K. Chan, H. Peng, R. A. Huggins and Y. Cui, Nano Lett., 2008, 8, 3948–3952 CrossRef CAS PubMed.
- T. Huang, X. Zheng, G. Fang, Y. Pan, W. Wang and M. Wu, RSC Adv., 2018, 8, 38831–38835 RSC.
- V. Gomzi, I. M. Sapic and A. Vidak, J. Phys. Chem. A, 2021, 125, 10649–10656 CrossRef CAS PubMed.
- A. Kozbial, F. Zhou, Z. T. Li, H. T. Liu and L. Li, Acc. Chem. Res., 2016, 49, 2765–2773 CrossRef CAS PubMed.
- T. Takamura, in Encyclopedia of Electrochemical Power Sources, ed. J. Garche, Elsevier, Amsterdam, 2009, pp. 709–743, DOI:10.1016/B978-044452745-5.00836-4.
- M. Bogunia and M. Makowski, J. Phys. Chem. B, 2020, 124, 10326–10336 CrossRef CAS PubMed.
- C. M. Gonzalez-Garcia, M. L. Gonzalez-Martin, V. Gomez-Serrano, J. M. Bruque and L. Labajos-Broncano, Langmuir, 2000, 16, 3950–3956 CrossRef CAS.
- M. H. Ryou, S. Hong, M. Winter, H. Lee and J. W. Choi, J. Mater. Chem. A, 2013, 1, 15224–15229 RSC.
- A. Guerfi, M. Kaneko, M. Petitclerc, M. Mori and K. Zaghib, J. Power Sources, 2007, 163, 1047–1052 CrossRef CAS.
- J. Marzec, K. Swierczek, J. Przewoznik, J. Molenda, D. R. Simon, E. M. Kelder and J. Schoonman, Solid State Ionics, 2002, 146, 225–237 CrossRef CAS.
- B. N. Rao, O. Padmaraj, D. Narsimulu, M. Venkateswarlu and N. Satyanarayana, Ceram. Int., 2015, 41, 14070–14077 CrossRef.
- H. Liu, P. K. Allan, O. J. Borkiewicz, C. Kurtz, C. P. Grey, K. W. Chapman and P. J. Chupas, J. Appl. Crystallogr., 2016, 49, 1665–1673 CrossRef CAS.
- B. H. Toby and R. B. Von Dreele, J. Appl. Crystallogr., 2013, 46, 544–549 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ta03305a |
| ‡ These authors contributed equally. |
| This journal is © The Royal Society of Chemistry 2024 |