
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNanoassemblies of heptamethine cyanine dye-initiated poly(amino acid) enhance ROS generation for effective antitumour phototherapy†
Pengwen
Chen
a,
Shangwei
Li
a,
Zhining
Xu
bc and
Horacio
Cabral
 *a
*a
aDepartment of Bioengineering, Graduate School of Engineering, The University of Tokyo, Bunkyo-ku, Tokyo, 113-8656, Japan. E-mail: horacio@bmw.t.u-tokyo.ac.jp
bPolymer Chemistry and Physics Research Group, HUN-REN Research Centre for Natural Sciences, Budapest, H-1117, Hungary
cFaculty of Science, Eötvös Loránd University, Budapest, H-1117, Hungary
First published on 13th March 2024
Abstract
Phototherapy shows great potential for pinpoint tumour treatment. Heptamethine cyanine dyes like IR783 have high potential as agents for antitumour phototherapy due to their inherent tumour targeting ability, though their effectiveness in vivo is unsatisfactory for clinical translation. To overcome this limitation, we present an innovative strategy involving IR783-based polymeric nanoassemblies that improve the dye's performance as an antitumoural photosensitizer. In the formulation, IR783 is modified with cysteamine and used to initiate the ring-opening polymerization (ROP) of the N-carboxyanhydride of benzyl-L-aspartate (BLA), resulting in IR783-installed poly(BLA). Compared to free IR783, the IR783 dye in the polymer adopts a twisted molecular conformation and tuned electron orbital distribution, remarkably enhancing its optical properties. In aqueous environments, the polymers spontaneously assemble into nanostructures with 60 nm diameter, showcasing surface-exposed IR783 dyes that function as ligands for cancer cell and mitochondria targeting. Moreover, the nanoassemblies stabilized the dyes and enhanced the generation of reactive oxygen species (ROS) upon laser irradiation. Thus, in murine tumor models, a single injection of the nanoassemblies with laser irradiation significantly inhibits tumour growth with no detectable off-target toxicity. These findings highlight the potential for improving the performance of heptamethine cyanine dyes in antitumor phototherapy through nano-enabled strategies.
New conceptsOur research presents an innovative approach to antitumor phototherapy by enhancing the efficacy of heptamethine cyanine dyes, exemplified by IR783, through the use of nanoassemblies. The uniqueness of our concept is evident in several key aspects, as follows: (i) by polymerizing hydrophobic poly(amino acid)s from the meso-position of the dye, we significantly improved the stability and ROS generation capacity of the dyes. This was achieved through a unique process involving the twisting of the molecular conformation and altering the electron orbital distribution of the dyes without altering the chemical backbone of the dye; (ii) the resulting poly(amino acid)s self-assembled into nanostructures in water, presenting the negatively charged dyes on their surface. This design capitalizes on the dyes' inherent targeting capabilities toward cancer cells and their natural tropism to mitochondria for intracellular delivery. Thus, a single injection of the nanoassemblies is able to regress tumours after irradiation. |
Introduction
Phototherapy is an exceptional non-invasive treatment for tumours, offering precise control over cytotoxic effects by regulating external light irradiation.1,2 The success of phototherapy largely depends on photosensitizing molecules that convert light into reactive oxygen species (ROS) for photodynamic therapy (PDT) or heat for photothermal therapy (PTT).3–7 Currently, several photosensitizers have been approved for clinical treatment against different cancers.8–10 However, these photosensitizers present poor tumour selectivity and off-target dark toxicity.8–10 Heptamethine cyanine dyes are alternative photosensitizer candidates with advantageous properties including long absorbance wavelength (around the near-infrared (NIR) range), high molar extinction coefficient and low toxicity.11–14 Notably, certain heptamethine cyanine dyes, such as IR780, IR783 and IR808, exhibit intrinsic tumour targeting capability due to their affinity for organic anion transporting polypeptides (OATPs) overexpressed in various cancer cells,15–17 which makes them compelling candidates for tumour-targeted phototherapy. However, the heptamethine cyanine dyes have significant drawbacks hindering their clinical translation, including photobleaching upon irradiation due to the fragile tricarbocyanine backbones, and inadequate ROS generation or photothermal conversion to attain effective therapeutic efficacy.18,19 Furthermore, these dyes display poor pharmacokinetics with fast clearance from the body.20 Thus, solutions are highly demanded to aid the clinical translation of heptamethine cyanine dyes.A few strategies have been proposed for improving the performance of heptamethine cyanine dyes as photosensitizers for effective tumour phototherapy. For example, structural modifications of the dye backbone offered opportunities to enhance the photostability and improve the optical properties,21,22 and developing carrier systems loading the dyes helped to ameliorate the pharmacokinetics.23,24 However, altering the backbone structure has risks to diminish the intrinsic tumour-targeting ability of these dyes, since the structure–activity relationship of the tumour-targeting ability has not been clarified yet,25 and encapsulating the dyes into delivery systems would mask the dye-mediated tumour-targeting. Thus, strategies that can improve the optical properties and pharmacokinetics without diminishing the inherent tumour targeting capability of heptamethine cyanine dyes are required.
Herein, we devised an innovative approach employing IR783-based polymeric nanoassemblies to augment the PDT performance without altering the backbone structure of the dye molecule. In the polymer, the N-carboxy anhydride of benzyl-L-aspartate (BLA) was polymerized from the meso-chlorine position (Scheme 1A). The introduction of the poly(BLA) (pBLA) block twists the molecular conformation of IR783 and enhances the intramolecular charge transfer (ICT), as predicted in our in silico models, which can potentiate the ROS generation and enhance the photostability.22,26–28 Moreover, the hydrophobicity and helical structure of the pBLA block can enable the assembly of the polymers into nanostructures29,30 for further enhancing the non-radiative energy conversion and facilitate the uptake by tumour cells (Scheme 1B). Our results showed that the polymers assembled into nanoparticles (IR-PA) with around 60 nm diameter in aqueous conditions with the IR783 moieties exposed on the surface. The IR-PA achieved high and selective tumour accumulation upon systemic injection in a mouse model, and demonstrated effective PDT against CT26 tumours after a single intravenous (i.v.) injection and light irradiation. This formulation serves as a unique paradigm for improving the performance of heptamethine cyanine dyes as photosensitizers for antitumour phototherapy.
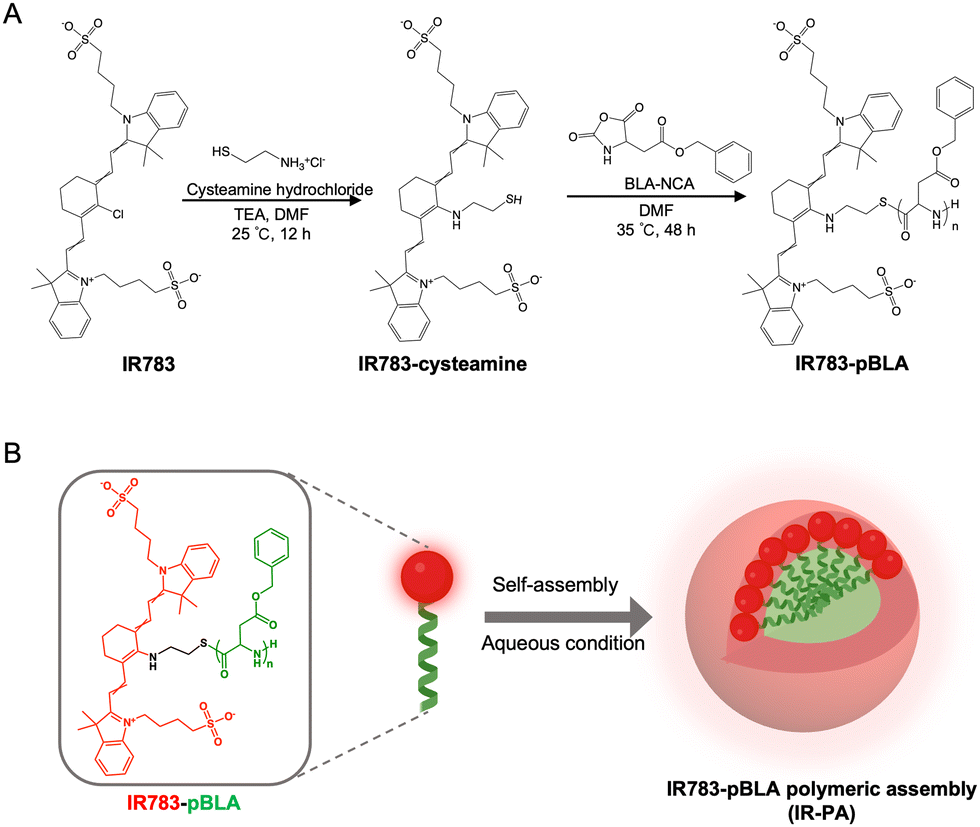 | ||
| Scheme 1 Schematic illustration of material synthesis and the formation of a polymeric nanoassembly. (A) Synthesis route of the materials. IR783 was firstly reacted with cysteamine to introduce the active thiol into the molecule. The IR783-cysteamine derivative then initiated the N-carboxyanhydride (NCA)-mediated ROP reaction to generate the polymer (IR783-pBLA). (B) The IR783-pBLA polymers self-assemble to form IR-PA nanoparticles in an aqueous environment by hydrophobic interactions, which are further stabilized by the negative surface charge and the helical structure of the pBLA block. | ||
Results and discussion
Synthesis, characterization, and theoretical computation of the materials
By mixing IR783 with cysteamine, the meso-chloride of IR783 was preferentially substituted by amines via nucleophilic substitution31–33 (Scheme 1A). Confirmation of the successful substitution was conducted by time-of-flight mass spectrometry (TOF-MS). The resulting product, namely IR783-cysteamine, exhibited a distinct peak at m/z = 766.3 (Fig. S1B, ESI†), contrasting with the unmodified IR783 molecule that displayed a peak at m/z = 725.3 (Fig. S1A, ESI†). Moreover, IR783-cysteamine showed an absorption peak around 650 nm (Fig. S2, ESI†), indicating the replacement of the meso-chloride by the amine of cysteamine.31–33 Subsequently, the thiol presented in IR783-cysteamine was used to initiate a ring-opening polymerization (ROP) reaction with N-carboxyanhydride of benzyl-L-aspartate (BLA-NCA) to generate the pBLA polymer.34–36 The final product, termed IR783-poly(β-benzyl-L-aspartate) (IR783-pBLA), was characterized by 1H-NMR (Fig. S3, ESI†). By comparing the areas of the peak from –CH– on the IR783 moiety (δ ≈ 7.9 ppm) and the peak from –CH– on the backbone of the pBLA block (δ ≈ 4.6 ppm), the average degree of polymerization (DP) of the pBLA block was calculated to be 17.Theoretical computations were performed to explore the conformational and electronic structures of the molecules under investigation. The thermodynamically optimal conformations of the molecules were determined (Fig. 1A). Notably, while IR783 exhibited a nearly planar structure, the introduction of cysteamine and the pBLA block altered the optimal conformation of the molecule backbone. In particular, in IR783-pBLA, the incorporation of the pBLA block induced a highly twisted molecule conformation. Such highly twisted conformation could reduce the π–π stacking between the molecules and elevate the excited-state intramolecular motion.22,26 Furthermore, the presence of the pBLA block significantly modulated the electron distribution in the molecule (Fig. S4, ESI†). Compared to IR783 and IR783-cysteamine, IR783-pBLA exhibited a smaller HOMO–LUMO gap, which can enhance the non-radiation decay rate.22 Moreover, HOMO and LUMO of IR783-pBLA showed heightened discrepancy in the spatial distribution, confirming the enhanced ICT effect, which can facilitate ROS generation.26–28
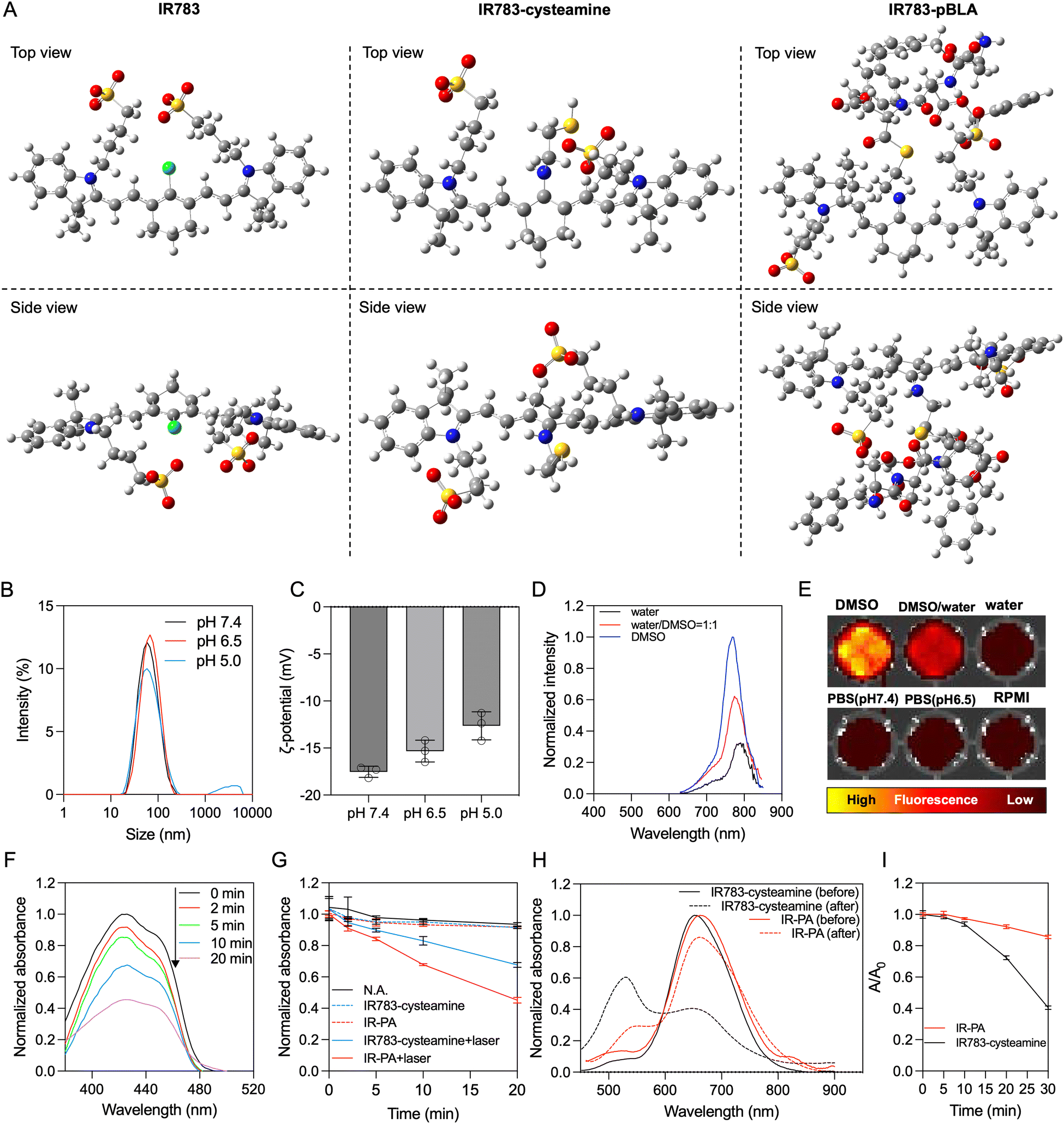 | ||
| Fig. 1 Characterization of IR-PA. (A) Thermodynamically optimal conformation of the IR783-derivative materials. (B) Representative histogram showing the hydrodynamic size distribution of IR-PA in aqueous solution. (C) ζ-Potential of IR-PA in aqueous buffer with different pH. Data shown as mean ± S.D., n = 3. (D) Representative fluorescence emission spectra of IR-PA in different solvents. The emission intensities are normalized to the maximal emission intensity of IR-PA in DMSO. (E) Representative fluorescence image of IR-PA samples dispersed in different solvents. (Excitation filter: 640 nm, emission filter: 780 nm). (F) Representative absorbance spectra of 1,3-diphenylisobenzofuran (DBPF) incubated with IR-PA after irradiation with a 633 nm laser for different times. (G) ROS generation efficiency from IR783-cysteamine or IR-PA reflected by the absorbance intensity of DBPF at 430 nm after different treatments. Data shown as mean ± S.D., n = 3. (H) Absorbance spectra of IR783-cysteamine (black lines) and IR-PA (red lines) before (solid lines) and after (dashed lines) 30 min irradiation with a 633 nm laser. (I) Relative absorbance at 660 nm of IR783-cysteamine or IR-PA after irradiation with a 633 nm laser for different times. Data shown as mean ± S.D., n = 3. | ||
Preparation and characterization of IR-PA
In the IR783-pBLA polymer, the IR783 moiety is hydrophilic and has negative charges from the sulfonates, while the pBLA block is hydrophobic with a helical structure. Thus, it was anticipated that the IR783-pBLA would undergo self-assembly in aqueous conditions (Scheme 1B). We firstly investigated the aggregation behaviour of the IR783-pBLA polymer in varying water/DMSO ratios. The sample containing IR783-pBLA exhibited increased light scattering intensity in dynamic laser scattering (DLS) measurement as the water fraction in the solvent increased (Fig. S5, ESI†), indicating the formation of aggregates. Subsequently, purified IR-PA nanoparticles were prepared by dialyzing IR783-pBLA DMSO solution against aqueous buffer for complete solvent exchange. The prepared IR-PA showed narrow size distribution, consistently maintaining an average hydrodynamic diameter of around 60 nm across different pHs, ranging from pH 5.0 to pH 7.4 (Fig. 1B). Furthermore, the nanoparticles displayed a negative surface charge (ζ-potential < −10 mV) (Fig. 1C), indicating the exposure of IR783 moieties on the particle surface. Such exposure could enhance the tumour-targeting capability through the OATP-mediated cellular uptake.15–17In aqueous conditions, the IR-PA manifested as a cyan-blue solution with a maximal absorption wavelength at 660 nm (Fig. S6, ESI†). The fluorescence emission peak of IR-PA centred around 770 nm, and its emission intensity demonstrated a significant correlation with the solvent (Fig. 1D and E). While strong fluorescence was observed from IR-PA in DMSO, a moderate emission was detected in water-based solvents, such as PBS buffer and RPMI cell culture medium (Fig. 1E). Moreover, the addition of surfactants or heating amplified the fluorescence emission of IR-PA in the aqueous system (Fig. S7, ESI†), confirming that the reduction of fluorescence emission intensity was due to intermolecular quenching. Moreover, the Stokes shift of IR-PA (≈110 nm) was notably larger than that of IR783-cysteamine (≈54 nm) and Cy5 (≈19 nm) (Table S1, ESI†). This discrepancy serves as evidence supporting the existence of an enhanced ICT mechanism in IR783-pBLA, which resulted from its twisted molecular structure and tuned molecular orbital.37–39 In comparison with the small molecular IR783-cysteamine, IR-PA exhibited a higher molar extinction coefficient and a lower fluorescence quantum yield (Table S1, ESI†), indicating a stronger non-radiative energy decay post light excitation. Subsequent evaluation aimed to specify the energy conversion pathway through assessing photothermal conversion and ROS generation upon irradiation of the samples. Surprisingly, neither IR783-cysteamine nor IR-PA demonstrated significant photothermal conversion under 633 nm laser irradiation, displaying similar temperature profiles to PBS even at a high concentration (20 μM) (Fig. S8, ESI†). Next, the ROS generation from the samples under 633 nm laser irradiation was evaluated by a 1,3-diphenylisobenzofuran (DBPF) probe. After incubation with IR-PA, the absorbance peak of DBPF at 430 nm significantly decreased (Fig. 1F), indicating the generation of ROS that oxidized DBPF to diminish its absorbance. Moreover, compared with IR783-cysteamine, IR-PA showed more effective ROS generation (Fig. 1G), indicated by the faster decay of the absorbance of DBPF at 430 nm. Finally, the photostability of the IR-PA and IR783-cysteamine was assessed. Upon irradiation by a 633 nm laser, IR783-cysteamine displayed a clear photobleaching, with a notable decrease in absorbance around 660 nm and a shift in maximal absorbance wavelength to 540 nm (Fig. 1H). Additionally, the colour of the IR783-cysteamine changed from cyan blue to pink (Fig. S9, ESI†). In contrast, IR-PA exhibited improved photostability. Even after 30 min irradiation by the laser, the absorbance at 660 nm (Abs660) of IR-PA only dropped to around 0.85 of the initial value, while the Abs660 of IR783-cysteamine dropped below 0.4 of the initial value (Fig. 1I). This enhanced photostability resulted from the formation of the nanoassembly, which may restrict the conformation change of the backbone of IR783 during potential oxidation.40 This hypothesis was confirmed by investigating the photostability of IR783-pBLA in DMSO, where it cannot form IR-PA. The results showed that the free polymers have comparable photostability to IR783-cysteamine (Fig. S10, ESI†). In summary, IR-PA demonstrated superior ROS generation ability and photostability compared to small molecule IR783-cysteamine, which could benefit the phototherapy performance.
In vitro performance of IR-PA
The cellular uptake behaviour of IR-PA was studied in CT26 cells. Compared to the small molecular IR783-cysteamine, IR-PA exhibited enhanced cellular uptake by CT26 cells (Fig. 2A). To elucidate the uptake mechanism of IR-PA, cells were tested with different treatments, including chlorpromazine (clathrin inhibitor), methyl-β-cyclodextrin (caveolae-mediated endocytosis inhibitor), amiloride (macropinocytosis inhibitor), free IR783 (competitive inhibition on OATPs), and low temperature (4 °C). As a result, methyl-β-cyclodextrin, free IR783 and low temperature significantly reduced the uptake of IR-PA by CT26 cells (Fig. 2B), confirming that the uptake of IR-PA relies on energy-dependent OTAPs-mediated transportation and caveolae-mediated endocytosis.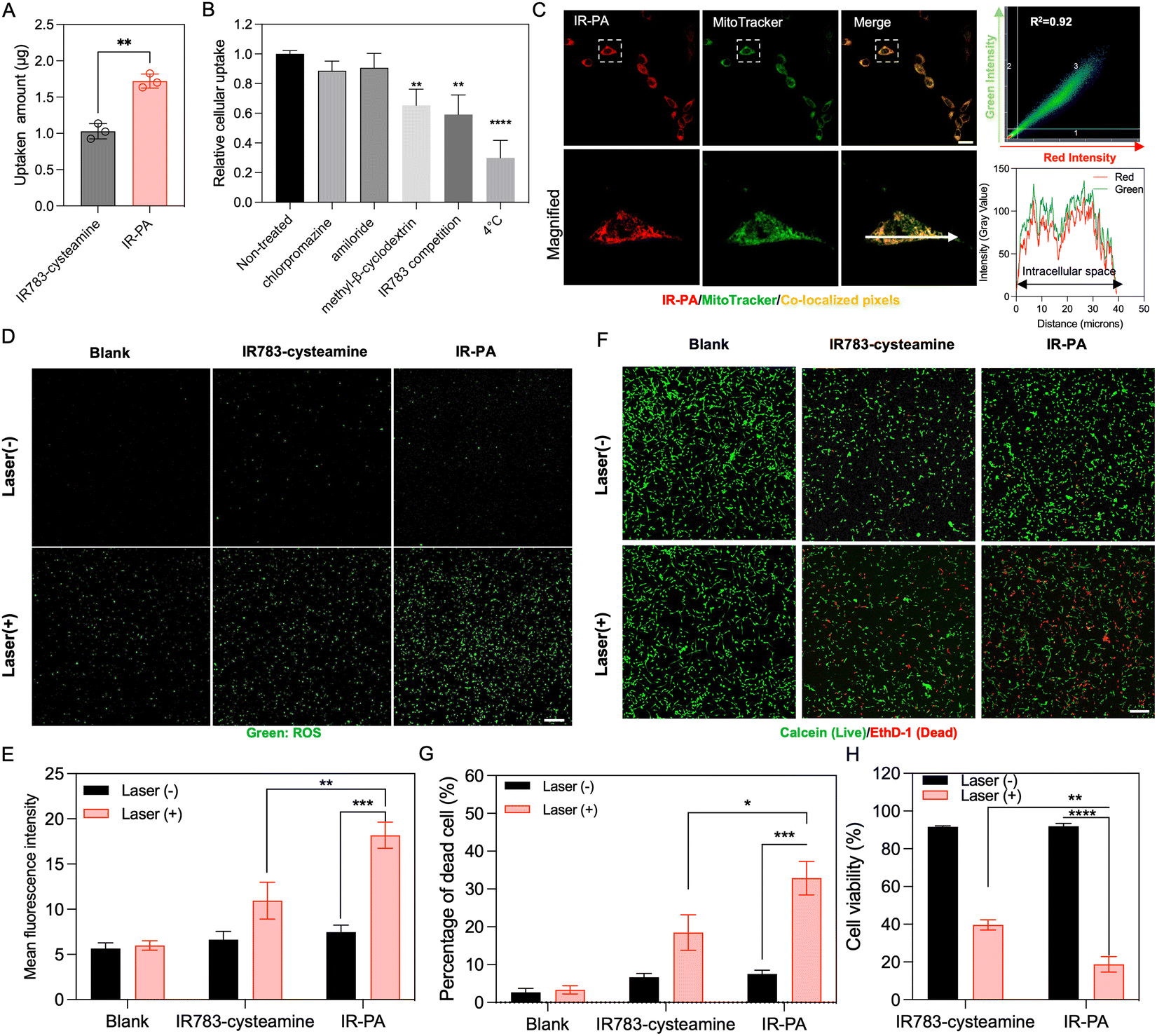 | ||
| Fig. 2 In vitro performance of IP-PA. (A) Cellular uptake of IR783-cysteamine and IR-PA after incubation with CT26 cells for 6 h. Data shown as mean ± S.D., n = 3. The values are compared via unpaired t-test. **p < 0.002. (B) Cellular uptake of IR-PA in CT26 cells after different treatments. Data shown as mean ± S.D., n = 3. The values are compared with the non-treated group via one-way ANOVA. **p < 0.002, *** p < 0.0002. (C) Representative fluorescence images of CT26 cells after incubating with IR-PA for 6 h (red: IR-PA, green: mitochondria stained by MitoTracker, yellow: pixels with co-localized red and green colours). Scale bar = 20 μm. Quantitative co-localization analysis of red and green signals is performed by correlation regression between red and green intensities shown in the scatterplot at the upper right corner. Qualitative co-localization analysis is visualized by the plot profile (lower right corner) along the direction of the white arrow in the microscopic image. (D) Representative fluorescence images of CT26 cells incubated with CM-H2DCFDA probes after different treatment (green: activated CM-H2DCFDA probes indicating generated ROS). Scale bar = 100 μm. (E) Mean green fluorescence intensity from the images in (D). Data shown as mean ± S.D., n = 3. The values are compared via unpaired t-test. **p < 0.002, *** p < 0.0002. (F) Live/dead (calcein/EthD-1) cytotoxic assay staining of CT26 cells after different treatments (green: calcein staining indicating living cells, red: EthD-1 staining indicating dead cells). Scale bar = 100 μm. (G) Quantified result showing the dead cell percentage from the experiment presented in Fig. 2D. Data shown as mean ± S.D., n = 4. The values are compared via unpaired t-test. *p < 0.03, *** p < 0.0002. (H) Cytotoxicity of different treatments measured by CCK-8 kit with CT26 cells. Data shown as mean ± S.D., n = 3. The values are compared via unpaired t-test, **p < 0.002, **** p < 0.0001. | ||
The subcellular distribution of IR-PA upon uptake was then investigated by microscopy observation (Fig. 2C). The fluorescence signal of internalized IR-PA (red pixels) displayed robust co-localization (R2 = 0.92) with the staining of MitoTracker Green (green pixels). This suggests the capability of IR-PA to target mitochondria, which is advantageous for photodynamic therapy due to the sensitivity of mitochondria to ROS.16,41,42 Subsequently, the intracellular ROS generation induced by IR-PA was evaluated. Upon treatment with IR-PA and subsequent laser irradiation, intense green fluorescence from the activated CM-H2DCFDA probes was detected in CT26 cells (Fig. 2D), confirming the efficient ROS generation inside the cells. Moreover, in comparison to IR783-cysteamine, the cells treated with IR-PA exhibited a higher ROS signal (Fig. 2E). This heightened signal can be ascribed to the stronger ROS generation capacity and augmented cellular uptake of IR-PA. Conversely, in all the groups without laser irradiation, no substantial ROS signal was detected, validating that ROS generation occurred solely through the photodynamic mechanism. Consequently, IR-PA showed potent PDT efficacy against CT26 cells, confirmed by viability staining (Calcein/EthD-1 staining). After incubation with IR-PA and subsequent laser irradiation, strong red fluorescence signals (EthD-1 staining) were observed, referring to dead cells (Fig. 2F and G).
While IR783-cysteamine treatment also induced cell death upon laser irradiation, the resulting red cells/green cells number ratio was lower than that of IR-PA + laser treatment, indicating a comparatively lower photodynamic therapy efficacy. For a more accurate quantification of the cytotoxicity, a CCK-8 kit was used to evaluate the cell viability after treatment (Fig. 2H). The result corroborated the superior cytotoxic effect of IR-PA over equivalent IR783-cysteamine upon laser irradiation. Furthermore, no cytotoxicity was observed in any group without laser irradiation, confirming the biocompatibility of the materials.
In vivo performance of IR-PA
We firstly evaluated the in vivo intratumoral accumulation after systemic (i.v.) injection of IR-PA in a subcutaneous (s.c.) CT26 tumour model. The imaging results revealed detectable fluorescence signals from the tumour area after injection, with an observed time-dependent increase in the intratumoral fluorescence intensity (Fig. 3A and B). This intratumoral accumulation of IR-PA was further confirmed by ex vivo fluorescence imaging of the excised tissue samples (Fig. 3C). IR-PA also exhibited retention in the liver, which is reasonable considering that 60 nm of IR-PA could lead to sequestration in the liver.43–45 On the other hand, IR-PA displayed a stronger fluorescence signal within the tumour in comparison to other major organs. These results confirmed the effective accumulation of IR-PA within the tumour, which is a characteristic necessary for enhancing the in vivo efficacy of PDT. As previous studies have shown that IR-783 can enhance the accumulation in tumours after binding with albumin,25 we checked if IR-PA can associate non-covalently with albumins for improving its pharmacokinetics and accumulation in tumours. The study was done by measuring the UV-vis absorbance spectra of IR783-cysteamine and IR-PA in aqueous buffer with or without BSA (Fig. S11, ESI†). The results showed that free IR783-cysteamine interacted with albumin and its spectrum showed a red-shift, whereas IR-PA did not show a significant change in its spectrum in the presence of albumin, suggesting that IR783 on IR-PA is not interacting with BSA. Thus, the interaction of IR-PA and albumin can be excluded as a mechanism of tumour accumulation. Considering that IR-PA has 60 nm diameter and presents the IR-783 dye on its surface, the effective accumulation of IR-PA in tumours could be attributed to both the enhanced permeability and retention (EPR) effect46–48 and the OTAP-mediated cellular uptake.15–17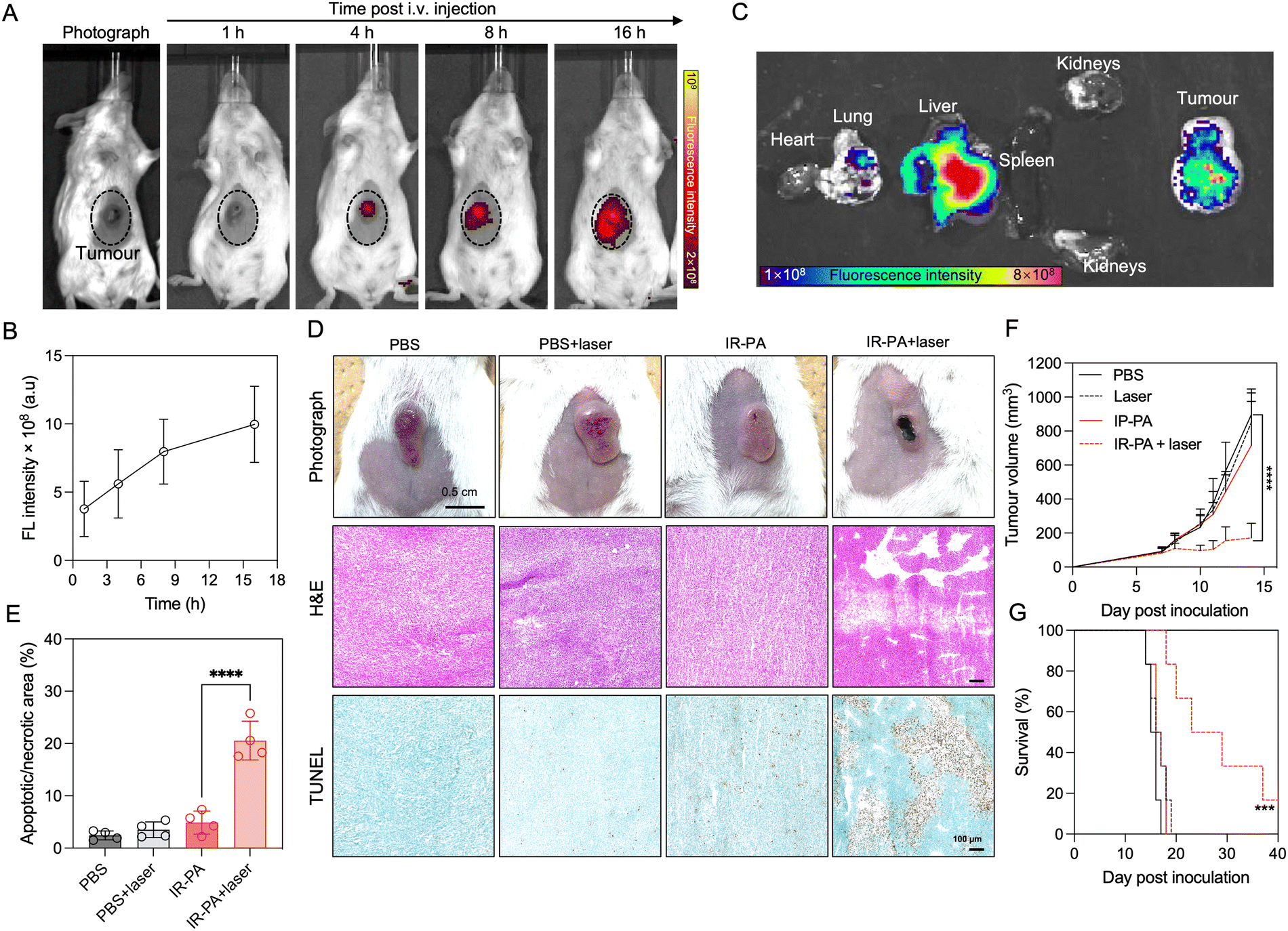 | ||
| Fig. 3 Tumour-targeted accumulation and PDT efficacy of IR-PA upon i.v. injection. (A) Representative in vivo fluorescence images of a mouse bearing a CT26 tumour after i.v. injection of IR-PA. The tumour area is marked by the dotted circle. (B) Quantified fluorescence intensities from the tumour area at a determined time after injection. Data are plotted as the mean ± S.D., n = 3. (C) Representative ex vivo fluorescence image of tissue samples (main organs and tumour) excised from mice 16 h post injection. (D) Representative photograph, H&E-stained sections, and TUNEL assay-stained sections of tumours on day 12 post inoculation. (E) Quantified percentage of the apoptotic/necrotic area in the TUNEL assay results. Data are plotted as the mean ± S.D., n = 4. Results in different groups were via one-way ANOVA analysis. **** p < 0.0001. (F) Average tumour volume after different treatments. Data are plotted as the mean ± SEM, n = 6. The curves stopped on day 14 post inoculation when the first tumour exceeding 1000 mm3 appeared. The average tumour volumes of different groups on day 14 were compared via one-way ANOVA analysis. **** p < 0.0001. (G) Survival curves of the animals during the experiment. Mice were euthanized when the tumour exceeded 1000 mm3 and counted as dead. The curves were compared via log-rank analysis. *** p < 0.0002. | ||
The therapeutic efficacy of IR-PA was subsequently assessed in the CT26 tumour model. In the treatment group, the mice received intravenous (i.v.) injections of IR-PA, followed by laser irradiation to the tumours 16 h post-injection. Clearly, the tumours in the treatment group exhibited strong necrosis (Fig. 3D), while there were no discernible changes in tumour morphology observed in the other groups. To further validate the impact of the treatment on the tumours, histological analysis was conducted. H&E staining of the tumour sections revealed large damage in the group treated with IR-PA and laser irradiation, a pattern not observed in other groups. Additionally, TUNEL assay results exhibited robust apoptosis/necrosis signals within tumours treated with IR-PA and laser irradiation (Fig. 3E), indicating the successful PDT within the tumours. Thus, IR-PA + laser irradiation treatment remarkably inhibited the tumour growth (Fig. 3F) and extended the survival time of the animals (Fig. 3G). Conversely, no significant differences were observed among the other groups.
No treatment-associated toxicity was detected throughout the experiment. Animals across all groups displayed a similar trend in body weight change (Fig. 4A). A more quantitative evaluation was performed by measuring the blood biomarkers associated with liver and kidney damage (Fig. 4B–E), but no significant difference could be determined between any two groups. Even in the liver, despite the high accumulation of IR-PA, no toxicity was observed in both blood tests (Fig. 4B and C) and histological analysis (Fig. 4F). Moreover, H&E staining images of all other organs displayed no pathological changes (Fig. S12, ESI†). Hence, IR-PA demonstrated efficient and safe photodynamic therapy against CT26 tumours, as evidenced by its notable therapeutic effects without inducing observable toxicity in the experimental animals.
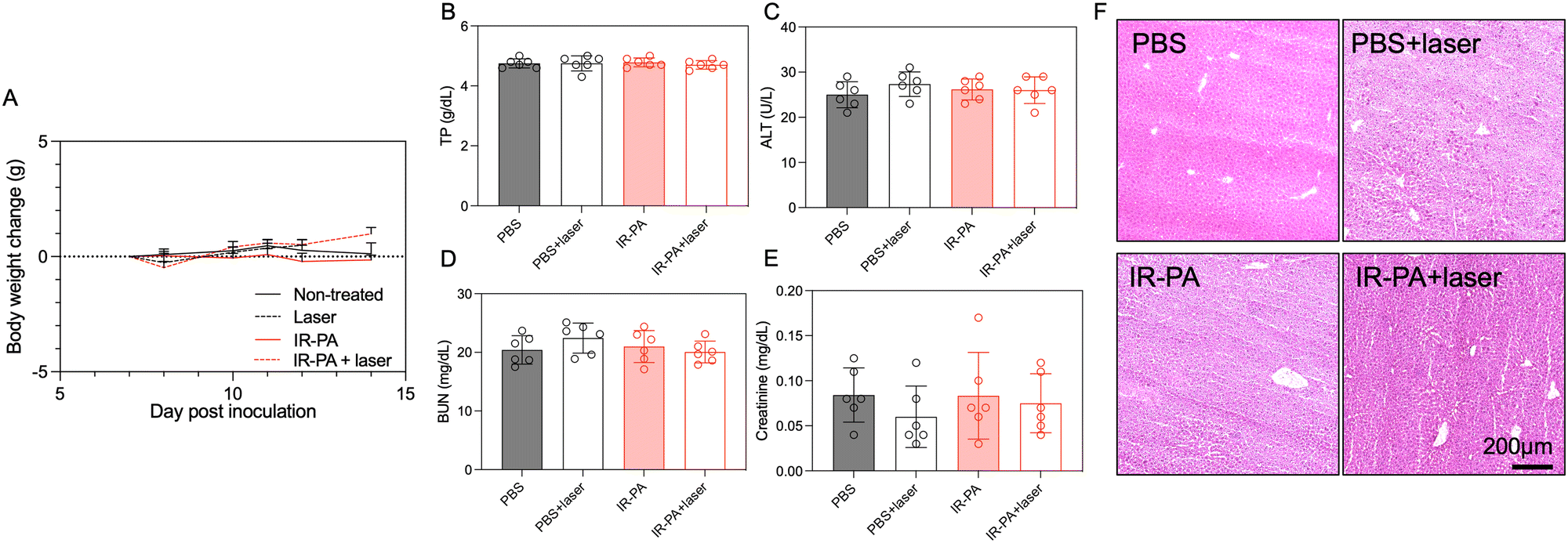 | ||
| Fig. 4 Toxicity analysis of IR-PA-based phototherapy. (A) Curves showing the average bodyweight change of animals in different groups during the phototherapy experiment. Data are plotted as the mean ± S.D., n = 6. The values on day 14 were compared via one-way ANOVA but no significance was found among any groups. (B)–(E) Blood test results detecting the corresponding organ damage-associated biomarkers. Blood samples were collected on day 12 post inoculation. Data are plotted as the mean ± S.D., n = 6. The values were compared via one-way ANOVA, but no significance was found among any groups. (F) Representative images of liver sections with H&E staining. The liver samples were collected on day 12 post inoculation. Scale bar = 200 μm. | ||
Conclusions
In this study, we introduced an innovative IR783-initiated polymer material capable of self-assembling into nanoparticles within an aqueous environment. These particles exhibited high efficacy in PDT and demonstrated remarkable photostability. In CT26 tumours, these nanoparticles efficiently accumulated in the tumour upon systemic administration. Following a single injection coupled with laser irradiation, the particles induced apoptosis/necrosis of the tumour cells and effectively impeded tumour growth. Overall, the formulation presented in this study holds promise as an advantageous photosensitizer with high potential for future translation and represents an innovative strategy to enhance heptamethine cyanine dyes for therapeutic application. Furthermore, this formulation can potentially serve as a tumour-targeted nanocarrier with drug payloads loaded inside to conduct combination therapy with PDT.Materials and methods
Materials
IR783 (sodium salt), triethylamine (TEA), ethyl acetate, and 1,3-diphenylisobenzofuran (DPBF) were purchased from Tokyo Chemical Industry Co., Ltd (Tokyo, Japan). N,N-Dimethylformamide (DMF), diethyl ether, dimethyl sulfoxide (DMSO), RPMI 1640 (RPMI) medium, fetal bovine serum (FBS), bovine serum albumin (BSA), penicillin–streptomycin (×100) and optimal cutting temperature compound (O.C.T compound) were purchased from Fujifilm Corporation (Tokyo, Japan). Cysteamine hydrochloride, chlorpromazine, amiloride, and methyl-β-cyclodextrin (MβCD) were purchased from Sigma-Aldrich (Burlington, USA). β-Benzyl-L-aspartic acid N-carboxyanhydride (BLA-NCA) was purchased from Chuo Kasei Co., Ltd (Osaka, Tokyo). RIPA cell lysis buffer, MitoTracker Green FM, the CM-H2DCFDA probe and the live/dead viability/cytotoxicity kit were purchased from Thermo Fisher Scientific Inc. (Waltham, USA). Cell Counting Kit-8 (CCK-8) was purchased from Dojindo Laboratories Co., Ltd (Kumamoto, Japan). The hematoxylin and eosin (H&E) staining kit and TUNEL assay kit were purchased from Abcam Inc. (Cambridge, UK).Cell lines and animals
Murine colon carcinoma cell line CT26 was obtained from RIKEN BioResourse Research Centre (Tsukuba, Japan). If not specified, the cells were cultured in RPMI containing 10% FBS and 1% penicillin–streptomycin with 5% CO2 at 37 °C. BALB/c mice were purchased from Charles River Laboratories Japan, Inc. (Yokohama, Japan). All animal experiments in this study were approved by the Animal Ethics Committee of The University of Tokyo and conducted following the Guidelines for the Care and Use of Laboratory Animals of The University of Tokyo and the Law for the Humane Treatment and Management of Animals of Japan.Synthesis and characterization of materials
The synthesis route of the materials is shown in Scheme 1A. IR783 was firstly reacted with cysteamine through nucleophilic substitution of the meso-chlorine on IR783 by the amines of cysteamine.31–33 Briefly, IR783 (0.75 g, 1 mmol) and cysteamine hydrochloride (0.017 g, 1.5 mmol) were dissolved in DMF (10 mL). TEA (0.4 mL, 3 mmol) was added to the mixture solution. The solution was kept stirring under 25 °C for 12 h and precipitated against ethyl acetate (200 mL). The product, IR783-cysteamine, was collected by filtration. The IR783-cysteamine was characterized by time-of-flight mass spectroscopy (TOF-MS) with an ESI source (ESI-TOF MS micro TOF; Bruker, USA).Next, NCA ring-opening-polymerization (ROP) was performed by using IR783-cysteamine as the initiator.34–36 IR783 (0.77 g, 1 mmol) and BLA-NCA (5 g, 20 mmol) were dissolved in anhydrous DMF (10 mL). The solution was kept reacting under 35 °C for 48 h in an Ar atmosphere and then precipitated against diethyl ether. The product, namely IR783-poly(β-benzyl-L-aspartate) (IR783-pBLA), was collected by filtration. The IR783-pBLA was characterized by 1H-NMR in d6-DMSO under 80 °C (NMR ECS-400; JEOL, Japan).
Theoretical computation was performed to investigate the molecular structures of the materials by Gaussian software. In the case of IR-pBLA, simplification was conducted by shortening the pBLA block into 3 BLA units to facilitate the computation. All the molecular structures were optimized by the Gaussian 16 QM modelling package with the B3LYP/6-31G(d,p) density functional theory (DFT) method.
Preparation and characterization of IR-PA
The aggregation behaviour of IR783-pBLA under different DMSO/H2O ratios was studied to confirm the self-assembly of IR783-pBLA in an aqueous environment. IR783-pBLA was firstly dissolved in DMSO at a concentration of 1 mg mL−1 to prepare the stock solution. Subsequently, the stock solution was 10× diluted into DMSO/water mixtures with different water volume fractions and vigorously stirred under 25 °C for 1 h. The well-mixed samples were measured by dynamic light scattering (DLS) using a 532 nm laser with 173° backward detection angle (Zetasizer Nano-ZS; Malvern, UK). The derivative count rate of each sample was recorded.To prepare IR-PA, the IR783-pBLA polymer was dissolved in DMSO (10 mg mL−1) and further 10× diluted into pure water with vigorous stirring. The diluted solution was then dialyzed against water or 10 mM phosphate buffer with different pHs to remove the remaining DMSO. DLS measurement was conducted to determine the size distribution of the samples. The surface charge of the IR-PA was determined by ζ-potential measurement (Zetasizer Nano-ZS; Malvern, UK). Absorbance and emission spectra of the samples were determined by a microplate reader (Spark; Tecan, Switzerland) with a quartz plate. To investigate the fluorescence quenching mechanism, the IR-PA water solution was added with 10% Triton X-100 or heated up to 80 °C before measurement. For taking the fluorescence images of IR783-pBLA in different solvents, the samples were added to a 96-well plate and imaged by an IVIS Spectrum imaging system (SP-BFM-T1, PerkinElmer, USA) (excitation filter: 640 nm, emission filter: 780 nm). The molar extinction coefficients of IR783-cysteamine and IR-PA at 650 nm in water solution were detected by comparing the absorbance of the samples at 650 nm with equivalent Cy5 as a standard.49 The molar extinction coefficients of the samples were calculated by the following equation.
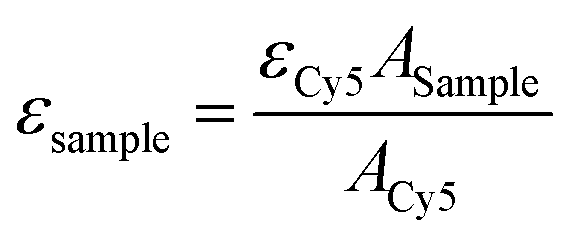
The fluorescence quantum yield of IR783-cysteamine and IR-PA in water solution was detected by comparing the fluorescence emission spectra of the samples with equivalent Cy5 as a standard.50 The samples were excited by 620 nm light and the emission spectra were recorded from 640 nm to 850 nm. The fluorescence quantum yield of the samples was calculated by the following equation.22
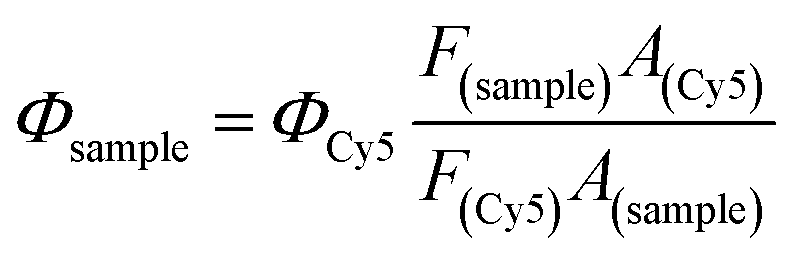
The photothermal conversion efficiency of IR-PA and IR783-cysteamine was evaluated in PBS. The samples (20 μM IR783 eq., 200 μL) were added into a 96-well plate and continuously irradiated by 633 nm laser (300 mW cm−2). The temperature of the sample was tracked by an electronic thermometer (Lutron TM-947SD; Lutron, Taiwan). Pure PBS was used for comparison. The ROS generation efficiency of IR-PA and IR783-cysteamine was evaluated by a DPBF probe. IR-PA or IR783-cysteamine (5 μM IR783 eq. in water) was incubated with DPBF (20 μM). Upon irradiation by a 633 nm laser (300 mW cm−2) for the determined time, the absorbance spectrum (350–500 nm) of the sample was measured by a microplate reader. The absorbance at 430 nm was recorded for indicating the ROS generation.
For evaluating their photostability, IR-PA or IR783-cysteamine water or DMSO solutions (5 μM IR783 eq.) were irradiated by a 633 nm laser (300 mW cm−2) for the determined time. The absorbance spectrum (450–900 nm) of the samples was measured by a microplate reader and the absorbance values at 660 nm were recorded to indicate the photostability.
To test the possible interaction between IR-PA and albumin, the absorbance spectra (550–900 nm) of IR783-cysteamine or IR-PA (20 μM) were measured in aqueous buffer with or without BSA (20 μM).
In vitro performance of IR-PA
Cellular uptake of IR-PA was evaluated in CT26 cells. CT26 cells were seeded into a 6-well plate (106 cells per well) and left for overnight culture before the experiment. IR-PA (20 μM) was then added to the culture medium. In comparison groups, equivalent IR783-cysteamine was added. After 6 h incubation, the culture supernatants were discarded, and the cells were washed with PBS 3 times. RIPA cell lysis buffer was then added into the wells (2 mL per well) and the cells were collected by a scraper. The harvested cell lysates were sonicated for 1 h, then centrifuged (10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000g × 10 min) to collect the supernatants. The absorbance of the supernatants at 650 nm was detected by a microplate reader to indicate the cellular uptake. To determine the uptake mechanism, different treatments were applied. In one experiment, cells were added with different endocytosis inhibitors (20 μM chlorpromazine, 5 mM MβCD, 2 mM amiloride) and incubated for 1 h under 37 °C before incubating with IR-PA (20 μM). In another experiment, the cells were incubated in IR-PA together with 20 μM IR783 for competitive inhibition. Additionally, the cells were also incubated with IR-PA under 4 °C. After 6 h, the cells were harvested and measured with the same protocol described above to determine the cellular uptake.
000g × 10 min) to collect the supernatants. The absorbance of the supernatants at 650 nm was detected by a microplate reader to indicate the cellular uptake. To determine the uptake mechanism, different treatments were applied. In one experiment, cells were added with different endocytosis inhibitors (20 μM chlorpromazine, 5 mM MβCD, 2 mM amiloride) and incubated for 1 h under 37 °C before incubating with IR-PA (20 μM). In another experiment, the cells were incubated in IR-PA together with 20 μM IR783 for competitive inhibition. Additionally, the cells were also incubated with IR-PA under 4 °C. After 6 h, the cells were harvested and measured with the same protocol described above to determine the cellular uptake.
The subcellular distribution of IR-PA upon internalization was then evaluated. CT26 cells were seeded into an 8-well chamber slide (5 × 103 cells per well) and left for overnight culture before the experiment. IR-PA (5 μM) was added into the culture medium. After 12 h incubation, the cells were washed by PBS 3 times and stained by MitoTracker with the protocol provided by the manufacturer. The cells were then imaged by a confocal laser scanning microscope (LSM780; Zeiss, Germany) (excitation laser: 488 and 633 nm; detection range: 495–520 nm and 680–780 nm, respectively). Scatterplot-based co-localization analysis of the two fluorescence channels was performed by Zen software. Plot profile analysis was performed by Image J software.
The CM-H2DCFDA probe was used to evaluate the in vitro ROS generation from IR-PA. CT26 cells were seeded into a 48-well plate (104 cells per well) and left for overnight culture before the experiment. IR-PA or equivalent IR783-cystamine (10 μM) was added into the culture medium. After 6 h incubation, the culture supernatants were discarded and changed to PBS containing 10 μM CM-H2DCFDA. After more 30 min incubation, the culture supernatants were again changed to clean PBS and cells in the irradiation groups were irradiated with a 633 nm laser (300 mW cm−2) for 15 min. Finally, the cells were observed by a confocal laser scanning microscope (excitation laser: 488 nm; detection range: 495–520 nm).
The cytotoxicity of IR-PA was firstly evaluated by live/dead viability/cytotoxicity staining. CT26 cells were seeded into a 48-well plate (104 cells per well) and left for overnight culture before the experiment. IR-PA or equivalent IR783-cystamine (10 μM) was added into the culture medium. After 6 h incubation, the culture supernatants were discarded and changed to fresh medium. In the irradiation groups, cells were irradiated with a 633 nm laser (300 mW cm−2) for 15 min. After more 24 h incubation, the cells were stained with a live/dead viability/cytotoxicity kit with the protocol provided by the manufacturer, then observed by a confocal laser scanning microscope (excitation laser: 488 and 514 nm; detection range: 495–510 nm and 600–620 nm, respectively). The images were analysed by Image J software to quantify the number of cells stained with red fluorescence and green fluorescence. A more quantitative cytotoxicity evaluation was performed by using CCK-8 kit. CT26 cells were seeded into a 96-well plate (5 × 103 cells per well) and left for overnight culture before the experiment. IR-PA or equivalent IR783-cystamine (10 μM) was added into the culture medium. After 6 h incubation, the culture supernatants were discarded and changed to fresh medium. In the irradiation groups, cells were irradiated with a 633 nm laser (300 mW cm−2) for 15 min. After more 24 h incubation, the cell viability was measured by CCK-8 kit with the protocol provided by the manufacturer.
Pharmacokinetics of IR-PA
BALB/c mice (female, 6 weeks old) were subcutaneously (s.c.) inoculated with CT26 cells (107 cells suspended in 100 μL RPMI) at the lower abdomen. The experiment was conducted on day 15 post inoculation. IR-PA (100 μg dispersed in 200 μL PBS) was intravenously (i.v.) injected into mice via the tail vein. After determined time points, the mice were anesthetized by 2.5% isoflurane air flow and imaged by an IVIS spectrum imaging system (excitation filter: 640 nm, emission filter: 780 nm). The fluorescence emission intensity from the tumour area was recorded. After 16 h upon injection, the mice were sacrificed, and tissue samples (organs and tumours) were collected for ex vivo imaging by the IVIS spectrum imaging system.Therapeutical efficacy and toxicity evaluation of IR-PA
BALB/c mice (female, 6 weeks old) were s.c. inoculated with CT26 cells with the same protocol described in Section 2.5. On day 7 post inoculation, the mice were randomized into 4 groups. IR-PA (100 μg dispersed in 200 μL PBS) or PBS (200 μL) were intravenously (i.v.) injected into the mice of the corresponding groups via the tail vein. For the irradiation groups, the mice were anesthetized by 2.5% isoflurane air flow 16 h post injection and the tumours were irradiated by a 633 nm laser (1000 mW cm−2) for 20 min. The tumour size of each individual mouse was tracked by calliper measurement. The tumour volume was calculated with the following formula.L and W are the length and width of the tumour, respectively. The bodyweight of each individual mouse was also recorded during the experiment. The mice were euthanized after the tumour volume exceeded 1000 mm3.
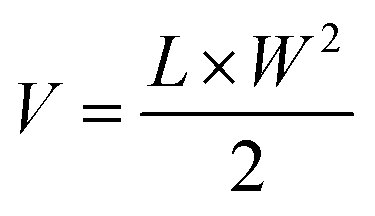
Representative tumour samples and organs were harvested from the mice on day 12 post inoculation. The tissues were embedded in O.C.T. compound and frozen in hexane under −90 °C, then sliced by a cryostat (CM1950; Leica Biosystems, Germany) into 10 μm thick sections. The tumour sections were stained by a H&E staining kit and TUNEL assay kit with the protocol provided by the manufacturer. The organ sections were stained by a H&E staining kit. The stained sections were observed by a brightness field microscope (BZ-X; Keyence Corporation, USA). To quantify the TUNEL assay results, microscopy pictures were analysed by Image J software with an IHC profiler plugin to specify the apoptotic/necrotic area and calculate the area percentage of the apoptotic/necrotic part in each picture.51 For each treatment group, four independent pictures were analysed. Blood samples (150 μL) were collected from all mice on day 12 by capillary tubes via the orbital vein. The samples were coagulated by placing under 25 °C for 20 min, then centrifuged (10000g × 10 min) to collect the serum. Toxicity markers (total protein, TP; alanine transaminase, ALT; blood urea nitrogen, BUN; creatinine, CRE) in the serum samples were measured by a blood chemistry analyser (DRI-CHEM NX700; Fujifilm, Japan).
Statistical analysis
All the statistical analyses in this study were performed by GraphPad Prism software (Version 9.5.0). Analysis methods were described in the corresponding content. Results were defined as statistically significant when *p < 0.03, **p < 0.002 and *** p < 0.0002 and **** p < 0.0001.Author contributions
P. C. performed the experiments and wrote the manuscript. S. L. assisted with the experiments. Z. X. conducted the theoretical computation. H. C. conceived the study, supervised the project, and edited the manuscript.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
This work was supported by the Project for Cancer Research and Therapeutic Evolution (P-CREATE) (H.C.) from the Japan Agency for Medical Research and Development (AMED). The work was also supported by the AMED Seeds A grant (203; H.C.). The study was partially supported by Grants-in-Aid for Scientific Research A (23H00546; H.C.), Grants-in-Aid for Exploratory Research (22K19541; H.C.), and the Fund for the Promotion of Joint International Research (Fostering Joint International Research (B), 21KK0197; H.C.), from the Japan Society for the Promotion of Science (JSPS). The study was also partially supported by Support for Pioneering Research Initiated by the Next Generation (SPRING) (JPMJSP2108; P.C.) from Japan Science and Technology Agency (JST).References
- M. Overchuk, R. A. Weersink, B. C. Wilson and G. Zheng, ACS Nano, 2023, 17(9), 7979–8003 CrossRef CAS.
- Z. Zhang, W. Xu, M. Kang, H. Wen, H. Guo, P. Zhang, L. Xi, K. Li, L. Wang, D. Wang, B. Zhong Tang, Z. Zhang, M. Kang, H. Wen, L. Wang, D. Wang, W. Xu, B. Z. Tang, H. Guo, L. Xi, K. Li and P. Zhang, Adv. Mater., 2020, 32(36), 2003210 CrossRef CAS PubMed.
- W. Li, J. Yang, L. Luo, M. Jiang, B. Qin, H. Yin, C. Zhu, X. Yuan, J. Zhang, Z. Luo, Y. Du, Q. Li, Y. Lou, Y. Qiu and J. You, Nat. Commun., 2019, 10(1), 3349 CrossRef PubMed.
- J. Li and K. Pu, Chem. Soc. Rev., 2019, 48(1), 38–71 RSC.
- X. Li, J. F. Lovell, J. Yoon and X. Chen, Nat. Rev. Clin. Oncol., 2020, 17(11), 657–674 CrossRef PubMed.
- X. Li, X. H. Peng, B. D. Zheng, J. Tang, Y. Zhao, B. Y. Zheng, M. R. Ke and J. D. Huang, Chem. Sci., 2018, 9(8), 2098–2104 RSC.
- K. Bilici, S. Cetin, E. Aydındogan, H. Yagci Acar and S. Kolemen, Front. Chem., 2021, 9(June), 1–15 Search PubMed.
- R. Baskaran, J. Lee and S. G. Yang, Biomater. Res., 2018, 22, 25 CrossRef PubMed.
- A. Hak, M. S. Ali, S. A. Sankaranarayanan, V. R. Shinde and A. K. Rengan, ACS Appl. Bio Mater., 2023, 6(2), 349–364 CrossRef CAS PubMed.
- C.-H. Su, M. Nowakowska, T. E. Kim and J.-E. Chang, Pharm., 2023, 15(9), 2257 Search PubMed.
- L. Zhang, H. Jia, X. Liu, Y. Zou, J. Sun, M. Liu, S. Jia, N. Liu, Y. Li and Q. Wang, Front. Pharmacol., 2022, 12, 764654 CrossRef PubMed.
- N. Lange, W. Szlasa, J. Saczko, A. Chwiłkowska, M. Wojcik, E. Preis and G. Litscher, Pharm., 2021, 13(6), 818 CAS.
- Y. Wang, J. Weng, J. Lin, D. Ye and Y. Zhang, J. Am. Chem. Soc., 2020, 142(6), 2787–2794 CrossRef CAS PubMed.
- D. H. Li, C. L. Schreiber and B. D. Smith, Angew. Chem., Int. Ed., 2020, 59(29), 12154–12161 CrossRef CAS PubMed.
- E. Zhang, S. Luo, X. Tan and C. Shi, Biomaterials, 2014, 35(2), 771–778 CrossRef CAS PubMed.
- Y. Wang, X. Liao, J. Sun, B. Yi, S. Luo, T. Liu, X. Tan, D. Liu, Z. Chen, X. Wang and C. Shi, Adv. Ther., 2018, 1(7), 1800069 CrossRef.
- Y. Wang, X. Liao, J. Sun, B. Yi, S. Luo, T. Liu, X. Tan, D. Liu, Z. Chen, X. Wang and C. Shi, Adv. Sci., 2018, 5(3), 1700392 CrossRef PubMed.
- T. Chen, Y. Zheng, Y. Gao and H. Chen, Bioorg. Chem., 2022, 126, 105903 CrossRef CAS PubMed.
- S. Luo, Y. Wang, S. Shen, P. Tang, Z. Liu, S. Zhang, D. Wu, S. Luo, Y. Wang, S. Shen, P. Tang, Z. Liu, S. Zhang and D. Wu, Adv. Funct. Mater., 2021, 31(24), 2100954 CrossRef CAS.
- R. Tian, Q. Zeng, S. Zhu, J. Lau, S. Chandra, R. Ertsey, K. S. Hettie, T. Teraphongphom, Z. Hu, G. Niu, D. O. Kiesewetter, H. Sun, X. Zhang, A. L. Antaris, B. R. Brooks and X. Chen, Sci. Adv., 2019, 5(9), 672–685 Search PubMed.
- Y. Xu, J. Yu, J. Hu, K. Sun, W. Lu, F. Zeng, J. Chen, M. Liu, Z. Cai, X. He, W. Wei and B. Sun, Adv. Healthcare Mater., 2023, 12(15), 2203080 CrossRef CAS.
- X. Zhao, H. Zhao, S. Wang, Z. Fan, Y. Ma, Y. Yin, W. Wang, R. Xi and M. Meng, J. Am. Chem. Soc., 2021, 143(49), 20828–20836 CrossRef CAS PubMed.
- J. Lv, H. Li, M. Yang, X. Li, J. Gao and Z. Yuan, ChemistrySelect, 2022, 7(36), e202202560 CrossRef CAS.
- Z. Hu, R. Li, X. Cui and Z. Chen, ACS Appl. Mater. Interfaces, 2023, 15(28), 33890–33902 CrossRef CAS.
- S. M. Usama, C. M. Lin and K. Burgess, Bioconjugate Chem., 2018, 29(11), 3886–3895 CrossRef CAS PubMed.
- Z. Zhao, C. Chen, W. Wu, F. Wang, L. Du, X. Zhang, Y. Xiong, X. He, Y. Cai, R. T. K. Kwok, J. W. Y. Lam, X. Gao, P. Sun, D. L. Phillips, D. Ding and B. Z. Tang, Nat. Commun., 2019, 10, 768 CrossRef CAS PubMed.
- R. Singh, D. G. Chen, C. H. Wang, C. C. Wu, C. H. Hsu, C. H. Wu, T. Y. Lai, P. T. Chou and C. T. Chen, J. Mater. Chem. B, 2022, 10(32), 6228–6236 RSC.
- J. Ni, Y. Wang, H. Zhang, J. Z. Sun and B. Z. Tang, Mol, 2021, 26(2), 268 CrossRef CAS PubMed.
- Y. Mochida, H. Cabral, Y. Miura, F. Albertini, S. Fukushima, K. Osada, N. Nishiyama and K. Kataoka, ACS Nano, 2014, 8(7), 6724–6738 CAS.
- R. H. Karlson, K. S. Norland, G. D. Fasman and E. R. Blout, J. Am. Chem. Soc., 1960, 82(9), 2268–2275 CAS.
- L. Niu, H. Zheng, Y. Chen, L. Wu, C. Tung and Q. Zhang, Analyst, 2014, 139, 1389–1395 RSC.
- H. Gorris, S. Saleh, D. Groegel, S. Ernst, K. Reiner, H. Mustroph and O. Wolfbeis, Bioconjugate Chem., 2011, 22(7), 1433–1437 CrossRef CAS.
- C. Lin, S. Usama and K. Burgess, Molecules, 2018, 23(11), 2900 CrossRef PubMed.
- X. Zhang, M. Oddon, O. Giani, S. Monge and J. Robin, Macromolecules, 2010, 43(6), 2654–2656 CrossRef CAS.
- X. Zhang, S. Monge, M. In, O. Giani and J. Robin, Soft Matter, 2013, 9, 1301–1309 RSC.
- Z. Tian, Z. Zhang, S. Wang and H. Lu, Nat. Commun., 2021, 12, 5810 CrossRef CAS PubMed.
- X. Peng, F. Song, E. Lu, Y. Wang, W. Zhou, J. Fan and Y. Gao, J. Am. Chem. Soc., 2005, 127(12), 4170–4171 CrossRef CAS PubMed.
- Y. Wu, Y. Chen, G. Gou, W. Mu, X. Lv, M. Du and W. Fu, Org. Lett., 2012, 14(20), 5226–5229 CrossRef CAS PubMed.
- C. S. Abeywickrama, Chem. Commun., 2022, 58, 9855–9869 RSC.
- B. Jana, A. P. Thomas, S. Kim, I. S. Lee, H. Choi, S. Jin, S. A. Park, S. K. Min, C. Kim and J.-H. Ryu, Chem. – Eur. J., 2020, 26, 10695 CrossRef CAS PubMed.
- M. D. Yaqoob, L. Xu, C. Li, M. M. L. Leong and D. D. Xu, Photodyn. Ther., 2022, 38, 102830 CrossRef CAS.
- A. P. Thomas, L. Palanikumar, M. T. Jeena, K. Kim and J. H. Ryu, Chem. Sci., 2017, 8(12), 8351–8356 RSC.
- M. Longmire, P. L. Choyke and H. Kobayashi, Nanomedicien, 2008, 3(5), 703–717 CrossRef CAS PubMed.
- N. Hoshyar, S. Gray, H. Han and G. Bao, Nanomedicien, 2016, 11(6), 673–692 CrossRef CAS PubMed.
- H. Cabral, J. Li, K. Miyata and K. Kataoka, Nat. Rev. Bioeng., 2024, 2, 214–232 CrossRef.
- H. Cabral, Y. Matsumoto, K. Mizuno, Q. Chen, M. Murakami, M. Kimura, Y. Terada, M. R. Kano, K. Miyazono, M. Uesaka, N. Nishiyama and K. Kataoka, Nat. Nanotechnol., 2021, 6, 815 CrossRef PubMed.
- D. Rosenblum, N. Jpshi, W. Tao, J. M. Karp and D. Peer, Nat. Commun., 2018, 9, 1410 CrossRef PubMed.
- J. Shi, P. W. Kantoff, R. Wooster and O. C. Farokhzad, Nat. Rev. Cancer, 2017, 17, 20 CrossRef CAS PubMed.
- J. Kang, O. Kaczmarek, J. Liebscher and L. Dähne, Int. J. Polym. Sci., 2010, 2010, 264781 Search PubMed.
- N. A. Pace, S. P. Hennelly and P. M. Goodwin, J. Phys. Chem. Lett., 2021, 12(37), 8963–8971 CrossRef CAS PubMed.
- F. Varghese, A. B. Bukhari, R. Malhotra and A. De, PLoS One, 2014, 9(5), e96801 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d3nh00584d |
| This journal is © The Royal Society of Chemistry 2024 |