
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
DNA origami: thinking ‘outside the fold’ for direct integrity testing of membranes for virus removal in potable reuse applications†
Hannah
Ray
 a,
Katerina
Papp
a,
Leopold
Green
b,
Boo Shan
Tseng
c,
Eric
Dickenson
a and
Daniel
Gerrity
*a
a,
Katerina
Papp
a,
Leopold
Green
b,
Boo Shan
Tseng
c,
Eric
Dickenson
a and
Daniel
Gerrity
*a
aSouthern Nevada Water Authority, P.O. Box 99954, Las Vegas, NV 89193, USA. E-mail: Daniel.Gerrity@snwa.com
bPurdue University, 610 Purdue Mall, West Lafayette, Indiana 47907, USA
cUniversity of Nevada Las Vegas, 4700 S. Maryland Parkway, Las Vegas, NV 89119, USA
First published on 19th July 2024
Abstract
Increasing water scarcity and water quality impairment have led to broader implementation of potable reuse throughout the world. High pressure membranes, including nanofiltration (NF) and reverse osmosis (RO), play a critical role in many potable reuse treatment trains because they are robust barriers against chemical and microbiological constituents. Despite achieving high pathogen log reduction values (LRVs) in practice (e.g., LRV > 5), high pressure membranes are often credited for only a fraction of observed LRVs (e.g., LRV < 3), which results in an LRV ‘gap’. This is because commonly used bulk water quality surrogates, namely total organic carbon (TOC) and electrical conductivity (EC), lack the resolution or analytical dynamic range to justify higher credit. The industry is now evaluating alternative surrogates (e.g., sucralose, sulfate, and strontium) that are both discrete and abundant in wastewater to narrow this regulatory ‘gap’. DNA origami technology can synthesize DNA nanostructures that mimic the size and morphology of viruses, potentially offering another novel surrogate for direct integrity testing. This study simultaneously evaluated pilot-scale NF and RO rejection of spiked MS2 bacteriophage (culture and qPCR), spiked DNA nanostructures (qPCR), and the aforementioned water quality surrogates. RO and NF achieved LRVs of ∼5 for culturable MS2 and censored LRVs of >4 for MS2 RNA. For RO, DNA nanostructure LRVs (up to ∼3) were comparable to the more advanced surrogates (e.g., sucralose, sulfate, and strontium), while DNA nanostructure LRVs for the NF membranes were generally <1 and consistent with EC and strontium. This study demonstrates that DNA nanostructures may have future value for potable reuse as they can be directly quantified via qPCR (without nucleic extraction) and can provide tailored structures that target various pathogens of interest. However, this study also highlights knowledge gaps that require further study, including the potential adsorption of DNA nanostructures to membrane surfaces and their ability to retain three-dimensional morphology in non-ideal wastewater matrices. Beyond the potential use of DNA origami technology, this study also highlights the value of rapid molecular methods in complementing, or even replacing, traditional culture methods when quantifying targets in membrane challenge tests.
Water impactPotable reuse is necessary to meet demands in water scarce or impaired areas. High pressure membranes, such as reverse osmosis and nanofiltration, are robust processes that remove chemical and microbial contaminants, ensuring high quality drinking water. Novel surrogates can be used to validate the performance of these membranes, award log reduction value credits, and ensure adequate public health protection. |
1 Introduction
Water reuse can be an effective strategy to mitigate uncertainty surrounding more conventional water supplies—either from a quality or quantity perspective. To ensure reliable public health protection at all times, potable water reuse necessitates robust, redundant, and resilient advanced treatment.1 In the United States (U.S.), design and operational criteria for indirect potable reuse (IPR) and direct potable reuse (DPR) treatment trains are established by state regulations or guidelines.2 Particularly for DPR, in which the role of the environmental buffer is eliminated or significantly reduced, these frameworks compensate for short response retention times (RRTs) by mandating more extensive treatment and/or on-line monitoring to identify, respond, and buffer against off-specification conditions.1Analytical methods are sufficiently sensitive to detect and quantify nearly all chemicals of concern down to concentrations that are relevant to public health. Coupled with the fact that many chemicals pose a chronic rather than acute risk, demonstrating compliance with concentration-based regulations through intermittent monitoring is generally suitable for chemical control. In contrast, the concentrations at which pathogens might be found in advanced potable reuse treatment trains—and the concentrations at which they pose an acute risk to public health—are often so low they are beyond the practical limits of existing microbiological methods.
As a result—and consistent with the approach for conventional drinking water—potable reuse regulations often rely on a log reduction value (LRV) framework3 that specifies overall LRV targets for critical pathogen groups4 and approaches for awarding these LRVs.5 This treatment-centric approach is heavily dependent on challenge testing and/or routine monitoring of surrogate microorganisms6 or other water quality parameters that can be correlated with process performance.7,8 As an artifact of regulatory conservatism, there is often a substantial discrepancy, or ‘gap’, between credited LRVs and the LRVs measured during normal operation, challenge tests, or research studies. This is particularly true for high pressure membranes, including nanofiltration (NF) and reverse osmosis (RO).9 This undercrediting can potentially lead to risk overestimation, overly complex treatment train designs, and unnecessarily high capital and operations and maintenance (O&M) costs for full-scale potable reuse systems.
RO membranes are frequently included in potable reuse treatment trains due to their effective removal of particulates, dissolved constituents, and pathogens.3,10 NF, while not currently used for potable reuse, has potential to be a key treatment process in future applications, specifically where rejection of divalent ions, per- and polyfluoroalkyl substances (PFAS), trace organic compounds (TOrCs), and/or disinfection byproduct (DBP) precursors is more critical than rejection of monovalent ions.11–16 NF membranes have higher flux capabilities and lower operating pressures, thereby reducing energy demand and associated costs.17 In the U.S., Texas does not routinely award pathogen LRV credits for RO or NF,18 but credits can be sought in other jurisdictions assuming consistent validation of membrane integrity, specifically by measuring native wastewater constituents and their removals.9 Commonly used wastewater constituents include total dissolved solids (TDS), specifically via electrical conductivity (EC), and total organic carbon (TOC), although these constituents generally yield LRVs of <2 due to their limited dynamic range. Recent monitoring advancements have expanded the surrogate parameter list to fluorescent dyes, divalent ions (e.g., sulfate and strontium), organic compounds (e.g., sucralose), and a variety of microbiological targets.8,19,20 However, these alternatives are not always amenable to on-line monitoring and may still not achieve the high LRVs demonstrated for RO,9 thereby highlighting the potential for further technological advancements.
Direct integrity testing with spiked bacteriophages (e.g., MS2) can demonstrate LRVs >6, but this approach is limited by the labor and time associated with culture-based methods. Specifically, 18–24 hours of incubation may not be consistent with the RRT requirements of a potable reuse system, with some studies suggesting monitoring should be conducted every 15 minutes.21–23 Molecular testing, including quantitative polymerase chain reaction (qPCR) or digital PCR (dPCR), is more conducive to automation and potentially has shorter turnaround times, but ambient concentrations of viral genomes may still be too low to justify higher credits for RO.24 However, combining molecular testing with a spiked viral surrogate that satisfies the U.S. Environmental Protection Agency's ‘discrete particle’ requirement25 may be a viable approach for maximizing LRV credits.
One potential approach involves DNA origami, which has been applied in medical settings for drug delivery, cellular repair, and gene therapy.26,27 DNA origami leverages the inherent physicochemical properties of nucleic acids to induce spontaneous conformational changes. With an appropriate DNA ‘scaffold’ and carefully designed ‘staple’ sequences, it is possible to synthesize nano-scale DNA structures of desired size, shape, and configuration,28,29 including structures resembling viruses. Because the DNA sequences used as building blocks do not pose a concern for public or environmental health, DNA nanostructures could be spiked into feed water matrices and their removal measured via molecular methods to justify LRV credits for membrane treatment. This is predicated on the assumption that DNA nanostructures behave similarly to target viruses despite the fact that they lack a protein capsid (and other viral structures), potentially carry different surface charges and isoelectric points, and have uncertain stability in complex environmental matrices (e.g., wastewater). Future applications may be able to further optimize this technology by developing hybrid protein–DNA nanostructures that are increasingly similar to target viruses.30
Therefore, the goal of this study was to evaluate the potential use of DNA nanostructures as viral surrogates in membrane-based potable reuse applications. The main objectives were to (1) establish baseline LRVs for NF and RO using spiked MS2 bacteriophage analyzed by culture and molecular methods, (2) assess the stability and fate of spiked DNA nanostructures in the NF/RO system based on molecular methods, and (3) compare MS2 and DNA nanostructure LRVs against conventional (i.e., EC and TOC) and emerging (i.e., sucralose, sulfate, and strontium) surrogates. The results from this study can be used to assess the suitability of DNA nanostructures for potable reuse applications and identify research gaps that can be addressed to potentially increase adoption of DNA origami in the water sector.
2 Materials & methods
2.1 Single-element membrane system
All experiments were performed using a single-element membrane system (Fig. S1†) operated in a batch configuration under the following operational conditions: flow rate of ∼38 liters per minute (∼10 gallons per minute), recovery of ∼11%, flux of ∼30 liters per square meter per hour (∼16–18 gfd), and feed pressures ranging from 345–951 kPa (50–138 psi) depending on the membrane being tested. Separate experiments were conducted with an ESPA2-LD-4040 membrane (“RO”, Hydranautics, Oceanside, CA, USA), a CSM NE4040-40 membrane (“NF1”, Toray Advanced Materials Korea), and a Filmtec NF270 membrane (“NF2”, DuPont Water Solutions, Wilmington, DE, USA). Membrane-specific operational conditions are summarized in Table S2.† Prior to testing, each membrane was flushed with 90 L of deionized water. Next, the integrity of each membrane was confirmed through operation at the conditions listed in their respective membrane data sheets (e.g., salt concentration, pressure, recovery) to ensure the minimum salt rejection was achieved.The feed water to the system consisted of tertiary effluent obtained from a full-scale wastewater treatment plant in Las Vegas, NV, USA. The full-scale treatment train consists of primary clarification, activated sludge (full nitrification, partial denitrification, and biological phosphorus removal), secondary clarification, and ultrafiltration (UF); representative feed water quality is summarized in Table S1.† For each of three spiking experiments, UF filtrate was added to a 90 L aluminum tank, the pH of the UF filtrate was adjusted from 7.3 to 6.9 using sulfuric acid, aliquots of each spiking stock (described later) were added to the water, and the water was manually mixed. At this point, a 50 mL sample was collected into a conical tube to represent the combined feed water in the tank at time zero. After starting the feed pump, the system was allowed to stabilize for 30 min prior to sample collection, at which point 50 mL feed and permeate samples were collected every 5 (experiment 1) or 15 min (experiments 2 and 3) (N = 3 per sample type). With the batch configuration, permeate and concentrate flows were continuously recombined in the 90 L aluminum tank, which was housed within a larger plastic tank fed with single-pass cooling water (Fig. S1†). This was intended to maintain a stable feed water temperature during recirculation. After 60 min of continuous operation, a different membrane was installed, and the process was repeated. The same feed water was used for the three different membranes (RO, NF1, NF2) within spiking experiment 1 and two membranes (RO, NF1) within spiking experiments 2 and 3. New feed water was prepared for each of the three spiking experiments (summarized in Table S3†), and the system was flushed with deionized water between spiking experiments.
2.2 MS2 bacteriophage spiking stock and assays
MS2 bacteriophage stock with a reported titer of ∼5 × 1011 plaque-forming units (PFU) per mL was purchased from GAP EnviroMicrobial Services Ltd. (Ontario, Canada). Approximately 5 mL of the MS2 stock was diluted in 90 L of UF filtrate to achieve a theoretical starting concentration of ∼3 × 107 PFU mL−1. For culture-based MS2 analyses, samples were shipped overnight on ice to GAP and enumerated within 48 hours of sample collection using the double agar layer method (triplicate plates) with E. coli Famp (ATCC 700891) as the bacterial host. All method blanks were negative for the culture-based MS2 assay.MS2 was also quantified by the Southern Nevada Water Authority (SNWA) using two-step reverse transcription qPCR (RT-qPCR). Nucleic acids were directly extracted from 350 μL samples using a PureLink Viral RNA/DNA Mini Kit (ThermoFisher Scientific, Waltham, MA, USA) according to manufacturer's instructions, with the exception that carrier RNA (yeast transfer RNA) was omitted from the procedure. Extracts were eluted into 60 μL of nuclease-free water. A negative control consisting of Milli-Q water was included in each extraction. Complementary DNA (cDNA) was synthesized using a Maxima First Stand cDNA Synthesis Kit (ThermoFisher Scientific) in 20 μL reactions: 5 μL of nucleic acids, 9 μL of nuclease-free water, 4 μL of 5× Reaction Mix, and 2 μL of Maxima Enzyme Mix. Reactions were incubated in an Applied Biosystems 2720 Thermal Cycler (Applied Biosystems, Waltham, MA, USA) or a Bio-Rad T100 Thermal Cycler (Bio-Rad Laboratories, Hercules, CA, USA) at 25 °C for 10 minutes, followed by RT-enzyme activation at 50 °C for 30 minutes, inactivation at 85 °C for 5 minutes, and a final hold at 4 °C.
The cDNA was then analyzed using a published qPCR assay for MS2.31 Triplicate qPCR reactions were run on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad Laboratories) in 10 μL of total volume: 1 μL of cDNA, 5 μL of 2× iTaq Universal Probes Supermix (Bio-Rad Laboratories), 0.25 μL of each primer (0.5 μM final concentration), 0.1 μL of probe (0.2 μM final concentration), and 3.4 μL of Milli-Q water. The probe and primers were purchased from Integrated DNA Technologies (IDT, Coralville, IA, USA), and the sequences are provided in Table 1. Cycling conditions started with an initial denaturation step at 95 °C for 2 min, followed by 45 cycles of denaturation at 95 °C for 5 s and a combined annealing/extension at 60 °C for 30 s with signal acquisition.
| Assay | Primer/probe | Sequence | Ref. |
|---|---|---|---|
| a Nanostructures were quantified using a SYBR-based qPCR assay (i.e., no probe). b NS assay 1 was ultimately selected for quantifying DNA nanostructures in experimental samples. | |||
| MS2 | MS2_F | 5′-GTCCATACCTTAGATGCGTTAGC-3′ | Gendron et al. (2010)31 |
| MS2_R | 5′-CCGTTAGCGAAGTTGCTTGG-3′ | ||
| MS2_Pb | 5′-/56-FAM/ACGTCGCCAGTTCCGCCATTGTCG-3′/BHQ1 | ||
| NS assay 1a,b | P8064_F1 | 5′-ACT CGT TCT GGT GTT TCT CG-3′ | Okholm et al. (2014)32 |
| P8064_R1 | 5′-TGA AAG AGG ACA GAT GAA CGG-3′ | ||
| NS assay 2a | P8064_F2 | 5′-CTG GCT CGA AAA TGC CTC T-3′ | |
| P8064_R2 | 5′-ACC AGT ATA AAG CCA ACG CT-3′ | ||
Each qPCR plate included no-template controls (NTCs) as well as a freshly made, serially diluted gBlock Gene Fragment standard (Table S4†) to serve as a calibration curve ranging from 107 to 101 gene copies (gc) per μL. Assay efficiency was >96%. Starting quantities were converted to reaction-specific concentrations (in gc/reaction) using the standard curve, and then sample-specific concentrations were calculated based on the equivalent sample volume (ESV) analyzed in each qPCR reaction,33 which was ∼0.0015 mL for the MS2 assay (see eqn (S1)† for derivation). The limit of quantification (LoQ) for the MS2 assay was determined to be 6.17 × 103 gc per mL of sample (Table S5†); this LoQ was imputed for any left-censored permeate samples (i.e., non-detect or <LoQ). Additional QA/QC details are provided in Text S1.†
2.3 DNA nanostructure spiking stocks and assays
Three formulations of pre-synthesized “Gear v2.0” DNA nanostructures were purchased from tilibit nanosystems (Munich, Germany), as this prefabricated formulation was most similar to MS2 bacteriophage in terms of size and shape despite not being icosahedral34,35 (Fig. S2†). The “Gear v2.0” DNA nanostructures have dimensions of 41 nm × 31 nm × 33 nm and are synthesized using a single-stranded circular DNA scaffold (p8064) from the M13mp18 bacteriophage. The nanostructures were prepared in 1 mL of 1× Tris-EDTA (TE) buffer theoretically containing 50 pmol of “Gears”, which equates to a concentration of 50 nM or ∼3 × 1013 nanostructures per mL of stock. The buffer was supplemented with 5 mM MgCl2 and 5 mM NaCl to prevent nanostructure ‘melting’ (i.e., loss of three-dimensional morphology). Successful synthesis was confirmed by tilibit nanosystems using gel electrophoresis and transmission electron microscopy (Fig. S2†).The first spiking experiment was conducted with gel-purified, non-labeled (i.e., no fluorophore addition), non-stabilized DNA nanostructures purchased approximately one year prior to the membrane experiment; the DNA nanostructure stock was stored at 4 °C. The second and third spiking experiments were conducted with gel-purified, non-labeled DNA nanostructures immediately upon receipt, with these experiments differentiated based on whether the DNA nanostructures were oligolysine-stabilized. Researchers have previously demonstrated that oligolysine coatings may confer resistance to degradation under non-ideal conditions (e.g., low concentrations of divalent cations).36 For each spiking experiment, ∼0.5 mL of DNA nanostructure stock was added to 90 L of UF filtrate, resulting in a theoretical diluted concentration of ∼2 × 108 nanostructures per mL of sample. The experimental conditions for the three spiking experiments are summarized in Table S3.†
For the DNA nanostructures, samples were analyzed by qPCR with direct quantification (i.e., no extraction) or after direct extraction of nucleic acids with a PureLink Viral RNA/DNA Mini Kit (ThermoFisher Scientific), as described earlier. Table 1 summarizes the two SYBR-based qPCR assays that were initially evaluated for their ability to target the p8064 DNA scaffold from the M13mp18 bacteriophage;32 nanostructure assay 1 was ultimately selected for analysis of experimental samples due to lower background signal (Text S1†). All qPCR reactions were run in triplicate on a CFX384 Touch Real-Time PCR Detection System (Bio-Rad Laboratories) in 10 μL of total volume: 1 μL of template DNA, 5 μL of 5X iTaq Universal SYBR Green Supermix (Bio-Rad Laboratories), 0.1 μL of each primer (0.2 μM final concentration), and 3.8 μL of Milli-Q water. Cycling conditions started with a denaturation step at 95 °C for 5 min, followed by 40 cycles of denaturation at 95 °C for 10 s, annealing at 56 °C for 15 s, and extension at 72 °C for 20 s. The run was completed with a melt curve from 65 °C to 95 °C in 0.5 °C increments for 5 seconds. Efficiencies were 95.5% for the first assay and 97.1% for the second assay (Fig. S3†). Starting quantities were converted to reaction-specific concentrations (in gc/reaction) using the standard curve ranging from 108 to 102 gc per μL, and then sample-specific concentrations were calculated based on the ESV analyzed in each qPCR reaction. For the DNA nanostructure assays, the ESVs were ∼0.0058 mL with nucleic acid extraction and 0.0010 mL with direct quantification (see eqn (S2) and (S3)† for derivation). Because of the difference in ESV, the LoQs for DNA nanostructure assay 1 ranged from 1.06 × 104 to 6.20 × 104 gc per mL of sample with and without nucleic acid extraction, respectively (Table S5†). These LoQs were imputed for any left-censored samples (i.e., non-detect or <LoQ). Additional QA/QC details are provided in Text S1.†
2.4 Endonuclease treatments
Two enzymatic treatments were evaluated to assess the impact of free RNA when quantifying MS2 (i.e., RNase A treatment) and DNA nanostructure stability (i.e., DNase I treatment). For MS2, the intent of the RNase A treatment was to assess the relative proportion of free RNA vs. capsid-encapsulated RNA, or seemingly intact MS2 virions, in feed and permeate samples. For the DNA nanostructures, the intent of the DNase I treatment was to assess their stability in the non-ideal water matrices, specifically due to the relatively low concentrations of divalent cations, and also to assess the efficacy of oligolysine stabilization. 350 μL samples were incubated with 35 μL of RNase A (ThermoFisher Scientific, Waltham, MA) at 37 °C for 30 min; paired control samples were not subjected to RNase A digestion. 310 μL samples were incubated with 35 μL of 10× Reaction Buffer with MgCl2 and 5 μL DNase I (ThermoFisher Scientific) at 37 °C for 30 min, followed by inactivation at 65 °C for 10 min; paired control samples were not subjected to DNase I digestion.2.5 Analytical methods for surrogate analytes
In addition to the spiked MS2 bacteriophage and DNA nanostructures, a single set of samples was collected at the end of each membrane test and analyzed for several native wastewater constituents: sucralose, sulfate, strontium, TOC, and EC. Sucralose was measured by SNWA using automated solid phase extraction (ASPE) and liquid chromatography tandem mass spectrometry (LC–MS/MS) with isotope dilution according to previously published methods.37 Briefly, methanol extracts were processed with a CTC Autosampler (CTC Analytics, Zwingen, Switzerland) and an Agilent 1260 LC Binary Pump (Palo Alto, CA, USA) and analyzed with a SCIEX API 4000-series mass spectrometer (Redwood City, CA). Data were collected in multiple reaction monitoring (MRM) [Q1: 395(397) m/z; Q3: 35 m/z] and negative electrospray ionization (ESI) modes for sucralose and its isotopically-labeled analog (d6-sucralose). The sucralose method reporting limit (MRL) was 25 ng L−1.Samples were shipped to Eurofins (Lancaster, PA, USA) for sulfate and strontium analyses; sulfate required no preservation and strontium required nitric acid preservation. Sulfate was measured by ion chromatography (IC) using EPA Method 300.0, which achieved an MRL of 0.25 mg L−1 and method detection limit (MDL) of 0.06 mg L−1. Strontium was measured by inductively coupled plasma mass spectrometry (ICP-MS) using EPA Method 200.8, which achieved an MRL of 0.01 mg L−1 and MDL of 0.002 mg L−1.
After acidification to pH < 2, total organic carbon (TOC) was measured by SNWA as non-purgeable organic carbon according to Standard Method 5310B using a Shimadzu TOC-L Analyzer (Kyoto, Japan); the MRL was 0.5 mg-C/L. Electrical conductivity (EC) was measured with a handheld 4360 Traceable expanded range conductivity meter.
3 Results and discussion
3.1 MS2 feed concentrations
Based on spiking experiment 1, culture-based MS2 concentrations in each tank sample (i.e., time 0 for each membrane) were within a factor of 2 relative to the corresponding membrane feed concentrations (Table S6†). However, the overall average MS2 concentration for all tank/feed samples was (1.17 ± 0.43) × 106 PFU mL−1, which is 1.41 logs lower than the expected concentration of 3 × 107 PFU mL−1 (Fig. 1). This may have been due to initial die-off following propagation by the commercial laboratory, as MS2 concentrations remained consistent throughout the three spiking experiments. For experiment 1, in which three different membranes were evaluated using the same recirculated feed water (i.e., recombined concentrate and permeate), MS2 concentrations declined slightly between membrane 1 (RO) and membrane 3 (NF2). This effect was less pronounced for spiking experiments 2 and 3, in which only two membranes were tested with the same feed water (RO and NF1).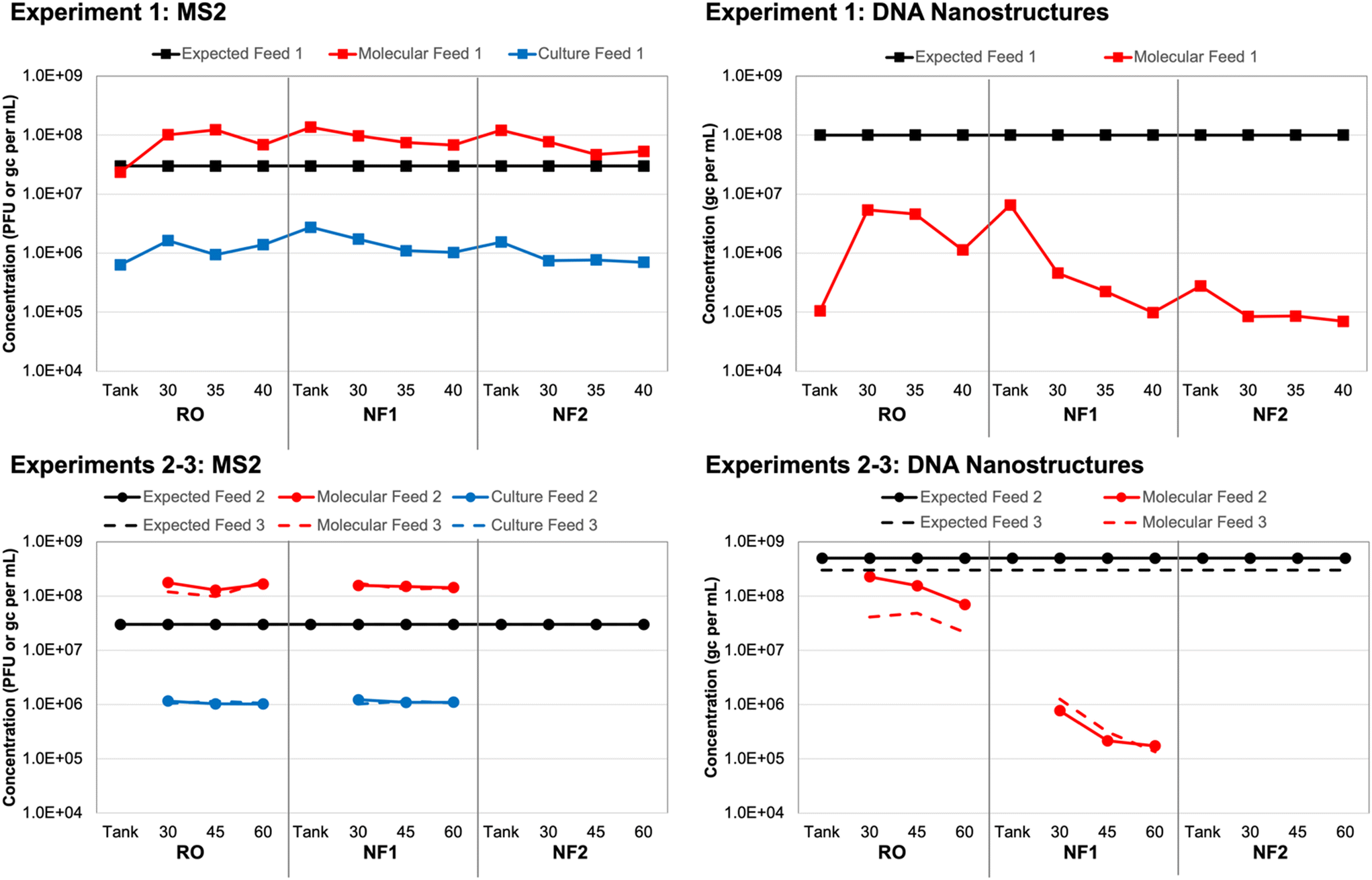 | ||
| Fig. 1 Expected and observed membrane feed concentrations of (left) MS2 bacteriophage and (right) DNA nanostructures in three spiking experiments. New feed water was prepared for each of three independent spiking experiments, with (top) experiment 1 comparing three different membranes and (bottom) experiments 2 and 3 comparing only one reverse osmosis (RO) and one nanofiltration (NF) membrane. Samples were collected in the spiking tank (experiment 1 only) and membrane feeds at multiple time points (experiment 1: 30, 35, 40 min; experiments 2–3: 30, 45, 60 min) and analyzed using culture and molecular methods for MS2 and molecular methods only for the DNA nanostructures. | ||
Again, based on spiking experiment 1, molecular MS2 concentrations in the NF tank samples (i.e., time 0) were within a factor of 2 relative to the corresponding NF feed concentrations (Table S6†). The RO tank sample was approximately 4 times lower than the corresponding RO feeds, which suggests that recirculation within the system improved upon the initial manual mixing of the tank. This was also observed for the culture-based RO tank vs. feed concentrations, although those differed by a factor of only 2 instead of 4 (Fig. 1). The molecular MS2 concentration across all tank and feed samples was relatively consistent at an overall average of (1.15 ± 0.44) × 108 gc per mL. Still, similar to the culture-based data, there was a slight decline in membrane feed concentration between the RO, NF1, and NF2 membranes, which suggests a small degree of target degradation over the ∼3 hour duration of the test and/or membrane adsorption.
The trends in MS2 feed concentrations were nearly identical, but the molecular MS2 concentrations were consistently 2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logs higher than the corresponding culture-based concentrations (Fig. 1), which suggests that a large fraction of the stock was composed of inactivated MS2 virions and/or free MS2 RNA. Pretreating the same samples with RNase prior to nucleic acid extraction eliminated only about 30% of the MS2 signal on average (Table S6†); thus, free RNA did not fully account for the 2log difference between the culture-based and molecular feed concentrations. In a separate control experiment with extracted nucleic acids, the same RNase treatment completely eliminated the MS2 signal (data not shown), which confirmed that the RNase was functioning as expected. Therefore, a majority of the difference between the culture-based and molecular feed concentrations could be attributed to inactivated MS2 virions, albeit with sufficient structural integrity to protect the nucleic acid from RNase (i.e., not entirely free RNA).
logs higher than the corresponding culture-based concentrations (Fig. 1), which suggests that a large fraction of the stock was composed of inactivated MS2 virions and/or free MS2 RNA. Pretreating the same samples with RNase prior to nucleic acid extraction eliminated only about 30% of the MS2 signal on average (Table S6†); thus, free RNA did not fully account for the 2log difference between the culture-based and molecular feed concentrations. In a separate control experiment with extracted nucleic acids, the same RNase treatment completely eliminated the MS2 signal (data not shown), which confirmed that the RNase was functioning as expected. Therefore, a majority of the difference between the culture-based and molecular feed concentrations could be attributed to inactivated MS2 virions, albeit with sufficient structural integrity to protect the nucleic acid from RNase (i.e., not entirely free RNA).
3.2 DNA nanostructure feed concentrations
With respect to the DNA nanostructure stocks provided by the commercial supplier, both qPCR assays yielded concentrations that were consistent with the expected concentration of ∼3 × 1013 gc per mL (Table S7†). The first gel-purified, non-stabilized stock had a concentration of ∼2 × 1013 gc per mL, while the second and third batches purchased approximately one year later had concentrations of ∼9 × 1013 gc per mL without oligolysine stabilization and ∼6 × 1013 gc per mL with oligolysine stabilization. Thus, stabilization appeared to result in a slight decrease in nanostructure yield. Based on the similar results for both assays and its lower background signal (Text S1†), qPCR assay 1 (Table 1) was used for subsequent analysis of all experimental samples.After adjusting the observed stock concentrations for dilution, the tank/feed concentrations were expected to be ∼1 × 108 gc per mL for experiment 1, ∼5 × 108 gc per mL for experiment 2, and ∼3 × 108 gc per mL for experiment 3. For experiment 1, the DNA nanostructure concentration increased by nearly two orders of magnitude between the RO tank and the 30 min RO feed sample (Table S8†). Similar to the MS2 data, this suggests inadequate manual mixing of the tank, but this was resolved to some degree during the 30 min recirculation period prior to sampling. However, the observed RO feed concentrations were still approximately 1.5logs lower than expectations and decreased to as low as ∼8 × 104 gc per mL for the second NF membrane (Fig. 1). A decrease in feed concentration was also observed within each membrane test. These discrepancies were the driver for acquiring new DNA nanostructure stocks, specifically to account for potential instability during year-long storage (experiment 1 vs. experiments 2 and 3) and to evaluate potential instability upon spiking into the wastewater matrix (non-stabilized vs. oligolysine-stabilized). The RO feed concentrations in spiking experiments 2 and 3 were closer to expectations, but still differed by 0.5–1.0logs and further decreased between sample collections and between the RO and NF membrane tests (Fig. 1).
Ponnuswamy et al. (2017) noted that DNA nanostructures are highly susceptible to ‘melting’ in aqueous environments with low concentrations of divalent cations.36 However, they successfully demonstrated that DNA nanostructures retain their morphologies after oligolysine stabilization, even when magnesium concentrations were lowered to 0.6 mM (or 15 mg L−1). For comparison, the DNA nanostructure buffer and UF filtrate had a total hardness of ∼4–5 mM, but due to high calcium and magnesium rejection, the NF and RO permeates had a total hardness <0.6 mM. In the current study, oligolysine stabilization of the DNA nanostructures (i.e., experiment 3) did not eliminate the concentration discrepancy (Fig. 1). That being said, nanostructure ‘melting’ results only in loss of three-dimensional structure, with the scaffold theoretically remaining intact, which would still allow for quantification by qPCR. In the current study, subsequent control experiments with DNA nanostructures spiked into smaller volumes of Milli-Q water and UF filtrate yielded expected concentrations (data not shown). Thus, matrix effects were not entirely responsible for the concentration discrepancies, which suggests that adsorption to the membranes during recirculation of the batch system may have been a more significant factor.
Ponnuswamy et al. (2017) further modified their nanostructures by conjugating the oligolysine to polyethylene glycol (PEG) to also protect against nucleases found in physiological fluids.36 The DNA nanostructures in the current study included oligolysine stabilization to protect their structural integrity but did not contain PEG to protect against nuclease activity. Deliberate exposure to DNase had an adverse but inconsistent impact on DNA nanostructure concentrations (Table S8†), so DNase activity in the wastewater matrix also does not fully explain the concentration discrepancies. Collectively, these data suggest that long-term storage time (i.e., stock age) may be a more significant factor than ‘melting’ or nuclease-based degradation, at least from a concentration perspective. Membrane adsorption appears to be the most significant factor—and more so for the DNA nanostructures than spiked MS2—but this may be an artifact of this study's batch configuration. Adsorption may be less of a factor for a traditional flow-through membrane system, especially after the membrane establishes some form of equilibrium with the feed water. ‘Melting’ and loss of three-dimensional morphology may still be important in the context of membrane rejection and is discussed later.
Finally, nucleic acid extraction prior to qPCR analysis of the DNA nanostructures resulted in signal loss in the membrane feeds—by 73% on average relative to non-extracted samples (Table S8†). The concentrations for non-extracted vs. extracted samples were highly correlated (Fig. 2), so both approaches appear to be valid, but signal loss due to extraction increases the likelihood of left-censored data and decreases method sensitivity, which is particularly important for permeate samples. Initially, it was unclear whether oligolysine stabilization would adversely affect the efficiency of the qPCR assay. Direct quantification was similarly effective for non-stabilized and oligolysine-stabilized nanostructures, and extraction resulted in similar signal loss for both formulations. Thus, direct quantification (i.e., without nucleic acid extraction) proved to be the preferred approach for the DNA nanostructures, which is fortuitous because it makes this technology more amenable to online monitoring in the future.
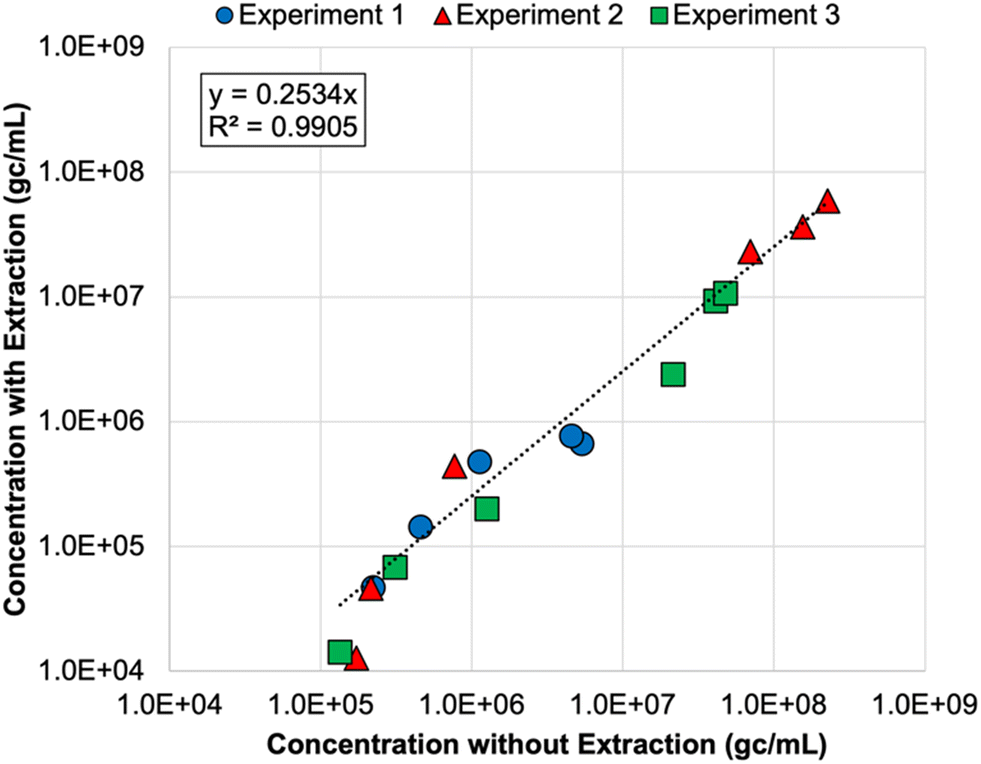 | ||
| Fig. 2 Correlation between DNA nanostructure concentrations determined with direct quantification (i.e., without nucleic acid extraction) and with direct extraction of nucleic acids prior to qPCR analysis. Colors and shapes differentiate the membrane feed concentrations for each spiking experiment, but no distinction is made for reverse osmosis (RO) vs. nanofiltration feeds. Only samples with extracted and non-extracted concentrations above the limit of quantification (LoQ) are included in this figure. Experiments 1 and 2 included non-stabilized DNA nanostructures, and experiment 3 included oligolysine-stabilized DNA nanostructures. See Table S8† for raw data. | ||
3.3 MS2 membrane log reduction values (LRVs)
As noted earlier, culture and molecular MS2 feed concentrations were relatively consistent across time points and membrane tests, particularly in comparison to the DNA nanostructures. Thus, differences in rejection (i.e., LRV) were primarily driven by MS2 permeate concentrations (Table S6†). All culture feed, culture permeate, and molecular feed concentrations were >LoQ, but only two molecular permeate concentrations were >LoQ (experiment 2: RO sample 2 and NF1 sample 1), resulting in censored LRVs for nearly all molecular data. However, the molecular feed concentrations were sufficiently high to demonstrate LRVs of approximately 4.0 for all membranes and across nearly all experiments. The one exception was the aforementioned NF1 ‘outlier’ (LRVculture = 2.45 and LRVmolecular = 3.34); the permeate concentration for the RO ‘outlier’ was still low enough to yield an LRV of 4.29 (Table S6†). On average, the censored molecular LRVs were more conservative than the corresponding culture-based LRVs for all membranes and experiments (Fig. 3). Again, the exception was the NF1 ‘outlier’ sample, for which the molecular data proved to be slightly less conservative. However, the culture and molecular data (with and without RNase pre-digestion) all highlighted that sample as having a high permeate concentration (Table S6†). The fact that RNase was unable to digest the target RNA is consistent with the detection of culturable MS2 in that NF1 sample.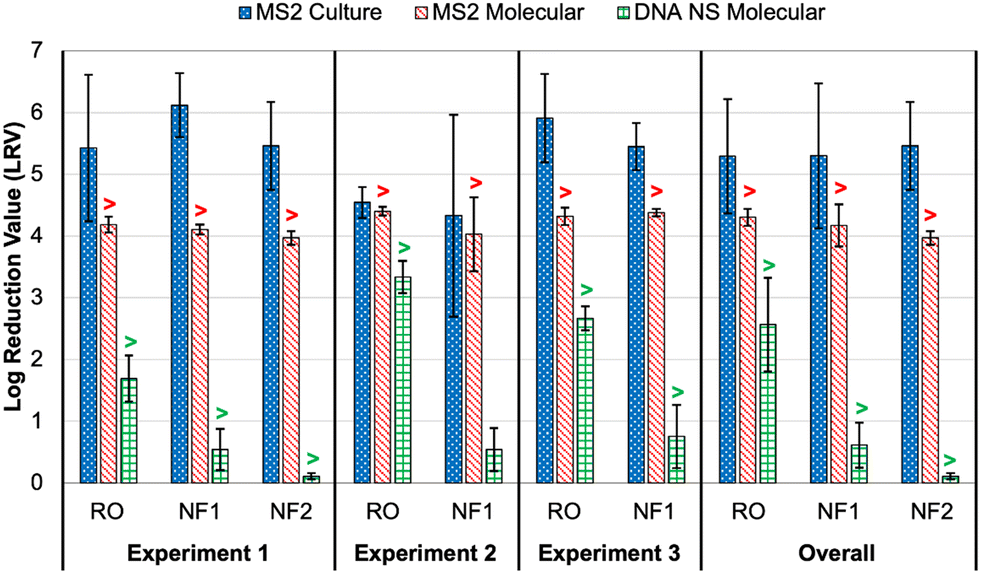 | ||
| Fig. 3 Membrane log reduction values (LRVs) for spiked MS2 bacteriophage (culture and molecular) and DNA nanostructures (molecular with direct quantification). Columns represent averages ±1 standard deviation across three or more time points. The overall averages include data from all time points and experiments for each membrane. The “>” symbols for the molecular data indicate that one or more LRVs were calculated after imputing the limit of quantification (LoQ) for left-censored permeate concentrations; this affected the molecular LRVs for all conditions except DNA nanostructure rejection by nanofiltration membrane 1 (NF1) in experiment 2. See Tables S6 and S8† for sample-specific LRVs. | ||
In experiment 1, the average culture-based LRVs were 5.43 ± 1.19 for the RO membrane, 5.46 ± 0.71 for the NF2 membrane, and 6.12 ± 0.52 for the NF1 membrane (Fig. 3). The slightly inferior performance of the RO membrane was driven by high variability between the 5 min time points (Table S6†). In experiment 2, RO performance was more consistent across samples, but the LRVs were ∼1log lower on average. As noted above, NF1 rejection in experiment 2 was adversely impacted by the first permeate sample, which contained relatively high concentrations of MS2 (4.40 × 103 PFU mL−1 and 7.24 × 104 gc per mL), but NF1 performed similarly to experiment 1 for the second and third time points. Finally, RO was slightly superior to the NF1 membrane in experiment 3, although both membranes consistently achieved LRVs >5.0. Based on the overall average LRVs, MS2 rejection was similar for RO and NF. MS2 rejection is dominated by steric hindrance (i.e., size exclusion) and Donnan exclusion (i.e., like-charge repulsion). The MS2 genome alone has a molecular weight of ∼1 × 106 g mol−1,38 which is equivalent to 1000 kDa. This is significantly larger than the molecular weight cut-off (MWCO) for dissolved organic compounds for NF and RO membranes (Table S2†), which is on the order of 1 kDa and 0.1 kDa, respectively,39 so MS2 rejection by size exclusion is expected. MS2 has an isoelectric point of 3.5 causing the virion to be negatively charged in potable reuse applications.40 Generally, thin-film composite membranes are negatively charged at neutral pH, and thus like-charge repulsion between MS2 and the membrane surface would further aid in MS2 rejection. It should be noted that adsorption of spiked MS2 to the membrane surface, particularly for aged membranes with increased hydrophobic interactions,41 may at least initially aid in removal, but adsorption may have less of an impact over time as the system reaches equilibrium.
The molecular data were generally more conservative because of the more limited dynamic range. The dynamic range could be expanded through sample concentration prior to analysis, but this increases the complexity of the method, lengthens the time to result, and also introduces uncertainty due to incomplete virus recovery. Despite being more conservative overall, it is somewhat problematic that the molecular method yielded a higher LRV than the culture method for the NF1 ‘outlier’ sample. With the culture data assumed to be more representative of actual public health risk, it is preferred for the surrogate LRV—based on molecular data in this case—to be lower, or more conservative, from a regulatory crediting perspective.
As described earlier, pre-digestion with RNase eliminated a portion of the feed MS2 signal, but the majority of the signal was associated with intact MS2 virions, or damaged virions that were sufficiently intact to protect the nucleic acid. Free RNA may pass through membranes, particularly fragmented molecules with molecular weights less than the MWCO of the membranes. For this reason, permeate samples were also subjected to RNase digestion to help differentiate passage of intact virions vs. free RNA. The majority of the permeate samples were <LoQ by the molecular method so this proved to be inconsequential; additionally, all permeate samples contained detectable MS2 by the culture method (Table S6†). Therefore, for this particular set of experiments, it was clear that intact virions were passing through the RO and NF membranes.
As noted above, MS2 is much larger than the MWCO for the RO and NF membranes, so MS2 virion passage would not be expected based on size alone. However, in contrast with low pressure membranes for which rejection is dominated by size exclusion, rejection by high pressure NF and RO membranes is also affected by concentration gradients and diffusion, among other physicochemical properties of the solute. Thus, it is possible for intact MS2 virions to pass across NF and RO membranes, even when membrane integrity is not compromised, and this is supported by the fact that MS2 was detected by culture and molecular methods (with and without RNase digestion) in the membrane permeates in the current study. Furthermore, Ray et al. (2024) and Vickers et al. (2019) both evaluated RO membrane integrity through MS2 testing using non-damaged and damaged membranes.8,42 Using culture methods, each study reported detection of MS2 in the permeate from intact, non-damaged membranes.
3.4 DNA nanostructure membrane log reduction values (LRVs)
With the exception of the first time point in experiment 3, all DNA nanostructure concentrations in the RO permeates were <LoQ, leading to censored LRVs that were driven by the feed concentrations (Table S8†). Experiment 2 had the highest membrane feed concentrations, resulting in an average LRV of >3.34 for the RO membrane. Experiment 3 with the oligolysine-stabilized nanostructures had an average LRV of >2.67, and experiment 1 with the older, non-stabilized nanostructure stock had an average LRV of >1.69 (Fig. 3). The one RO permeate with a quantifiable DNA nanostructure concentration yielded a true LRV of 2.58, which was comparable to the two censored LRVs for that experiment (Table S8†), so that particular sample was not necessarily an outlier. Therefore, the DNA nanostructures appeared to be useful in demonstrating rejection by the RO membranes, but their practical utility in terms of maximizing LRV credits appears to be highly dependent on the feed concentration, with lower concentrations yielding observed LRVs that are comparable to traditional water quality surrogates. Similar to molecular quantification of MS2, the dynamic range of the DNA nanostructure assay could be expanded by concentrating the permeate samples, but uncertainty related to potential signal loss during sample processing might even be greater for these novel targets. Additionally, sample processing negates the full benefit of direct quantification and hinders future online monitoring opportunities. In other words, future efforts to use DNA nanostructures as viral surrogates should focus on maximizing feed concentrations. Eliminating the background signal of the qPCR assay would also reduce the LoQ and increase the dynamic range, and this could perhaps be accomplished by choosing an alternative DNA scaffold for which there is no master mix cross-reactivity.For nanofiltration, NF2 appeared to be highly impacted by its low feed concentration in experiment 1, leading to an average censored LRV of only 0.11 ± 0.05 (Table S8†). While this is likely not representative of its true potential, particularly since the corresponding MS2 LRVs were >4 for culture and molecular methods (Fig. 3), NF1 also exhibited relatively low rejection, including three samples with quantifiable DNA nanostructure concentrations during experiment 2 (LRV = 0.54 ± 0.35). Therefore, the low LRVs for nanofiltration were not entirely driven by membrane feed concentration. Moreover, rejection of a particular constituent by RO or NF is governed by diffusion and concentration gradients,43 which means that higher feed concentrations are expected to yield higher permeate concentrations and vice versa. Lower NF feed concentrations should theoretically have yielded fully censored permeate concentrations, and since they did not, there appears to be a true difference in nanostructure rejection between RO (>90–99.9%) and NF (as low as 50%).
Rejection of DNA nanostructures is potentially dependent on the processing steps used during synthesis. In general, non-stabilized DNA nanostructures have a negative charge due to the sugar–phosphate backbone of the underlying scaffold and staples, and this is confirmed by DNA nanostructure migration during gel electrophoresis.44 However, the oligolysine-stabilized DNA nanostructures are presumed to have an overall neutral charge due to the addition of cationic oligolysine molecules.44 Therefore, rejection of non-stabilized DNA nanostructures would be characterized by both steric hindrance (size exclusion) and like-charge repulsion (Donnan exclusion), which aligns with the rejection mechanisms of MS2. The neutral, stabilized DNA nanostructures would be predominantly rejected by steric hindrance. Additional testing would be needed to determine whether surface charges of non-stabilized vs. stabilized DNA nanostructures significantly affected their rejection.
Loss of three-dimensional structure, or ‘melting’, may have been an increasingly important factor over the duration of each experiment (i.e., after longer storage in UF filtrate and membrane permeate). This might disproportionately affect NF membrane performance due to its larger pore structure, while tighter RO membranes might still be expected to reject free DNA. Even with the oligolysine-stabilized DNA nanostructures in experiment 3, NF rejection decreased from >1.31 to 0.65 to 0.30 over 30 minutes of sampling (Table S8†). Although ‘melting’ may have been a contributing factor, the signal from those permeate samples was resistant to DNase digestion so some level of protection (e.g., three-dimensional morphology) remained.
Unfortunately, it was not possible to verify the integrity of the DNA nanostructures during the membrane experiments, but future work should consider post-treatment verification by gel electrophoresis and transmission electron microscopy. Future studies should also consider membrane autopsies to assess DNA nanostructure adsorption. DNA adsorption might be more extensive for RO membranes due to their generally greater surface roughness, which is known to increase adsorption and membrane fouling.45 DNA nanostructure ‘loss’ was greater for the RO membrane when assessed based on absolute terms, but the effect was more pronounced for the NF membranes based on relative changes in concentration (i.e., log differences) (Fig. 1).
3.5 General water quality surrogate membrane log reduction values (LRVs)
The rejection of native wastewater constituents, specifically sucralose, sulfate, strontium, TOC, and EC, was relatively consistent with previous research.8,46,47 RO achieved LRVs of 3.5–3.7 for sucralose and sulfate, 3.0 for strontium, 2.1–2.3 for EC, and >1.1 for TOC, which were greater than the LRVs for the NF membranes for all constituents except TOC (limited by analytical sensitivity) (Table 2). While the NF membranes achieved >2log rejection of sucralose and sulfate (2.3–2.7), they achieved <1log rejection of strontium (0.7–0.9) and EC (0.3–0.4). This is somewhat expected because, while sucralose is neutral in charge, it is a larger compound (398 g mol−1) with a molecular weight approximately five times greater than strontium, and NF membranes are generally able to reject compounds with molecular weights >300 g mol−1.48 Therefore, high sucralose rejection is expected for NF membranes due to size exclusion. The disparity in rejection between sulfate (96 g mol−1) and strontium (88 g mol−1) can likely be explained by Donnan exclusion. As noted previously, thin-film composite membranes are, in general, negatively charged at neutral pH, which would increase sulfate rejection due to like-charge repulsion (Donnan exclusion). Conversely, this would decrease strontium rejection due to membrane attraction, promoting diffusion into the permeate.49 In fact, Cai et al. (2020) found that in groundwater at a pH 6–7, RO achieved nearly 100% strontium rejection, while NF achieved only ∼55%.50
| Experiment | Membrane | Sucralose | Sulfate | Strontium | TOCa | ECa |
|---|---|---|---|---|---|---|
| a NM = not measured; TOC = total organic carbon; EC = electrical conductivity. | ||||||
| 1 | RO | 3.7 | 3.6 | 3.0 | >1.1 | 2.3 |
| NF1 | 2.4 | 2.3 | 0.9 | >1.1 | 0.4 | |
| NF2 | 2.3 | 2.3 | 0.7 | >1.1 | 0.3 | |
| 2 | RO | 3.5 | 3.5 | NMa | >1.1 | 2.1 |
| NF1 | 2.4 | 2.6 | NMa | >1.1 | 0.4 | |
| 3 | RO | 3.6 | 3.5 | NMa | >1.1 | 2.2 |
| NF1 | 2.4 | 2.7 | NMa | >1.1 | 0.4 | |
In order to maximize LRV credit for RO membranes, the data from this study suggest the following sequence: sucralose ≈ sulfate > strontium > EC > TOC. However, there are also considerations for amenability to online monitoring, which might make sulfate the preferred option over sucralose, and the suitability of TOC would depend on method/sensor sensitivity. As expected, these common water quality surrogates were more conservative than the spiked MS2, regardless of whether MS2 was analyzed by culture or molecular methods, and this is consistent with previous studies.8,51 In contrast, the DNA nanostructure LRVs were comparable to those of the surrogates. For RO, this was in part due to the DNA nanostructure LRVs being censored (i.e., limited by method sensitivity), but for NF, even the non-censored LRVs were comparable to the low LRVs observed for strontium and EC. Collectively, these data suggest that the DNA nanostructures behaved more like dissolved organic/inorganic constituents rather than a more complex MS2 virion.
3.6 Practical considerations for the use of DNA nanostructures as viral surrogates
Protocols for DNA nanostructure synthesis are not particularly complex, as they involve straightforward incubation of nanostructure building blocks (i.e., scaffold and staple strands), leading to self-assembly. However, the process requires substantial expertise in identifying the staple strand sequences to generate sizes and morphologies of interest. Expertise is also needed if it is desired for the DNA nanostructures to have additional features, including purification, stabilization, and/or fluorophore labeling. Therefore, it is possible for ‘in-house’ DNA nanostructure synthesis, but some applications may have to rely on commercial suppliers that can provide end users with fit-for-purpose formulations. Ideally, these fit-for-purpose formulations could target the exact size and morphology of a target virus, and fluorophore labeling could even facilitate on-line monitoring via flow cytometry (FCM). However, it is currently unclear whether FCM has sufficient sensitivity to reliably quantify DNA nanostructures.52The apparent advantage of using this technology for direct integrity testing of membranes is its rapid turnaround time when using molecular methods. This is because major time-consuming steps can be omitted, specifically nucleic acid extraction and cDNA synthesis. As noted earlier, this even lends itself to the possibility of on-line monitoring in the future. At this time, DNA nanotechnology is yet to be cost competitive with the more common approach of using spiked bacteriophages (e.g., MS2) in challenge tests. For example, each experiment described in the current study would cost on the order of several hundred to several thousand U.S. dollars to acquire a stock with sufficient concentration to achieve desired membrane feed concentrations. Also, these experiments were performed at a flow rate of 38 liters per minute (or 10 gallons per minute), so higher flow rates would pose greater challenges when trying to maximize feed concentrations. That being said, the industry is already making advances in reducing cost, increasing yields, and expanding commercial and industrial applicability.53 By coupling these technological advancements with a better mechanistic understanding of DNA nanostructure fate, DNA origami may play an increasingly important role in verifying treatment process integrity and protecting public health in potable reuse applications.
4 Conclusion
This study simultaneously used MS2 bacteriophage, DNA nanostructures, and water quality surrogates to evaluate the performance of NF and RO membranes in the context of potable reuse. This study introduces the water sector to DNA origami technology, while highlighting important advantages and disadvantages based on the current state of the technology. Even today, DNA origami possesses a high technology readiness level (TRL) for potable reuse applications due to its commercially availability, but there are several issues that require further study or improvement before DNA origami is a viable alternative, particularly when verifying membrane integrity in large-scale systems. In terms of increasing LRV credits for membranes, DNA nanostructures appear to be superior to total organic carbon and electrical conductivity and are comparable to more advanced surrogates, such as sucralose, sulfate, strontium. As state regulations and guidelines for potable reuse are implemented, stringent virus LRV requirements, such as California's 20log virus reduction requirement, incentivize utilities to look to more advanced surrogates to achieve compliance. A notable advantage of DNA nanostructures is the potentially fast analysis time, as they can be directly quantified via qPCR without the need for nucleic acid extraction. Additionally, the inherent flexibility of DNA origami allows for tailored structures of virtually any size and shape, with additional possibilities for chemical modification (e.g., hybrid protein–DNA nanostructures or the addition of thiols, amines, or fluorescent probes). However, a better understanding of membrane adsorption potential is needed to determine whether DNA nanostructures behave like viruses. Their post-treatment stability should also be confirmed via transmission electron microscopy to determine if passage across the membrane is driven by a loss of three-dimensional morphology. Beyond the potential use of DNA origami technology, this study also highlighted the value of molecular methods in complementing, or even replacing, traditional culture methods when quantifying MS2 in membrane challenge tests. Molecular quantification of MS2 was generally still more conservative than the culture approach but demonstrated higher LRVs than the common water quality surrogates. Thus, molecular methods for bacteriophages and DNA nanostructures may ultimately facilitate automated qPCR-based monitoring in potable reuse applications, particularly with wastewater-based epidemiology (WBE) driving wider adoption of PCR-based platforms.54
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This publication was supported through funding from the Water Research Foundation as part of WRF Project 5104. This publication has not been formally reviewed by WRF so the views expressed here are solely those of the authors. We would like to acknowledge Harry Zhang (WRF Research Program Manager), as well as members of the WRF Project Advisory Committee: Katherine Alfredo, Heather Bischel, Blythe Layton, and Rich Sustich. We would also like to acknowledge staff from the Southern Nevada Water Authority (Katherine Crank, Janie Holady, Christina Morrison, Oscar Quinones, Brittney Stipanov, Brett Vanderford, Eric Wert), Purdue University (Sang Hoon Um), Clark County Water Reclamation District, and tilibit nanosystems (Jean-Philippe Sobczak) for their contributions to the study.References
- B. M. Pecson, R. S. Trussell, A. N. Pisarenko and R. R. Trussell, Achieving Reliability in Potable Reuse: The Four Rs, J. - Am. Water Works Assoc., 2015, 107, 48–58 CrossRef.
- USEPA, Guidelines for Water Reuse, Washington, D.C., 2012 Search PubMed.
- P. Jeffrey, Z. Yang and S. J. Judd, The status of potable water reuse implementation, Water Res., 2022, 214, 118198 CrossRef CAS PubMed.
- D. Gerrity, K. Crank, E. Steinle-Darling and B. M. Pecson, Establishing pathogen log reduction value targets for direct potable reuse in the United States, AWWA Water Sci., 2023, 5, e1353 CrossRef CAS.
- M. J. Adelman, P. Oberoi, J. Oppenheimer, L. Furatian, S. Arabi, C. M. Glover and J. G. Jacangelo, Aligning monitoring of virus barriers in potable reuse with unit process treatment mechanisms and time scales: A critical review and research needs, AWWA Water Sci., 2024, 6, e1368 CrossRef.
- J. Heffron, M. Samsami, S. Juedemann, J. Lavin, S. T. Nick, B. A. Kieke and B. K. Mayer, Mitigation of viruses of concern and bacteriophage surrogates via common unit processes for water reuse: A meta-analysis, Water Res., 2024, 252, 121242 CrossRef CAS PubMed.
- D. Sano, M. Amarasiri, A. Hata, T. Watanabe and H. Katayama, Risk management of viral infectious diseases in wastewater reclamation and reuse: Review, Environ. Int., 2016, 91, 220–229 CrossRef CAS PubMed.
- H. Ray, D. Hannoun, J. Assouline, J. Vickers and E. Dickenson, Pilot evaluation of the potential log removal credit using chemical markers for reverse osmosis and nanofiltration, Desalination, 2024, 572, 117148 CrossRef CAS.
- L. M. Hornstra, T. R. da Silva, B. Blankert, L. Heijnen, E. Beerendonk, E. R. Cornelissen and G. Medema, Monitoring the integrity of reverse osmosis membranes using novel indigenous freshwater viruses and bacteriophages, Environ. Sci.: Water Res. Technol., 2019, 5, 1535–1544 RSC.
- D. Gerrity, B. Pecson, R. S. Trussell and R. R. Trussell, Potable reuse treatment trains throughout the world, J. Water Supply: Res. Technol.--AQUA, 2013, 62, 321–338 CrossRef CAS.
- Y. Zhi, X. Zhao, S. Qian, A. F. Faria, X. Lu, X. Wang, W. Li, L. Han, Z. Tao, Q. He, J. Ma and C. Liu, Removing emerging perfluoroalkyl ether acids and fluorotelomer sulfonates from water by nanofiltration membranes: Insights into performance and underlying mechanisms, Sep. Purif. Technol., 2022, 298, 121648 CrossRef CAS.
- X. Wang, B. Li, T. Zhang and X. Li, Performance of nanofiltration membrane in rejecting trace organic compounds: Experiment and model prediction, Desalination, 2015, 370, 7–16 CrossRef CAS.
- V. Yangali-Quintanilla, S. K. Maeng, T. Fujioka, M. Kennedy and G. Amy, Proposing nanofiltration as acceptable barrier for organic contaminants in water reuse, J. Membr. Sci., 2010, 362, 334–345 CrossRef CAS.
- M. Itoh, S. Kunikane and Y. Magara, Evaluation of nanofiltration for disinfection by-products control in drinking water treatment, Water Supply, 2001, 1, 233–243 CrossRef CAS.
- J. Radjenović, M. Petrović, F. Ventura and D. Barceló, Rejection of pharmaceuticals in nanofiltration and reverse osmosis membrane drinking water treatment, Water Res., 2008, 42, 3601–3610 CrossRef PubMed.
- L. Yi-Li, Effects of Physicochemical Properties of Nanofiltration Membranes on the Rejection of Small Organic DBP Precursors, J. Environ. Eng., 2013, 139, 127–136 CrossRef.
- A. W. Mohammad, Y. H. Teow, W. L. Ang, Y. T. Chung, D. L. Oatley-Radcliffe and N. Hilal, Nanofiltration membranes review: Recent advances and future prospects, Desalination, 2015, 356, 226–254 CrossRef CAS.
- TWDB, Direct potable reuse resource document, 2015 Search PubMed.
- T. Fujioka, T. Ueyama, F. Mingliang and M. Leddy, Online assessment of sand filter performance for bacterial removal in a full-scale drinking water treatment plant, Chemosphere, 2019, 229, 509–514 CrossRef CAS PubMed.
- É. Sylvestre, M. Prévost, J.-B. Burnet, P. Smeets, G. Medema, M. Hachad and S. Dorner, Using surrogate data to assess risks associated with microbial peak events in source water at drinking water treatment plants, Water Res., 2021, 200, 117296 CrossRef PubMed.
- B. M. Pecson, S. C. Triolo, S. Olivieri, E. C. Chen, A. N. Pisarenko, C.-C. Yang, A. Olivieri, C. N. Haas, R. S. Trussell and R. R. Trussell, Reliability of pathogen control in direct potable reuse: Performance evaluation and QMRA of a full-scale 1 MGD advanced treatment train, Water Res., 2017, 122, 258–268 CrossRef CAS PubMed.
- J. A. Soller, A. M. Parker and A. Salveson, Public Health Implications of Short Duration, Off-Specification Conditions at Potable Reuse Water Treatment Facilities, Environ. Sci. Technol. Lett., 2018, 5, 675–680 CrossRef CAS.
- É. Sylvestre, E. Reynaert and T. R. Julian, Defining Risk-Based Monitoring Frequencies to Verify the Performance of Water Treatment Barriers, Environ. Sci. Technol. Lett., 2023, 10, 379–384 CrossRef PubMed.
- M. Yasui, H. Iso, S. Torii, Y. Matsui and H. Katayama, Applicability of pepper mild mottle virus and cucumber green mottle mosaic virus as process indicators of enteric virus removal by membrane processes at a potable reuse facility, Water Res., 2021, 206, 117735 CrossRef CAS PubMed.
- USEPA, Membrane Filtration Guidance Manual, Cincinnati, OH, 2005 Search PubMed.
- Y. Zhang, X. Tian, Z. Wang, H. Wang, F. Liu, Q. Long and S. Jiang, Advanced applications of DNA nanostructures dominated by DNA origami in antitumor drug delivery, Front. Mol. Biosci., 2023, 10, 1239952 CrossRef CAS PubMed.
- J. A. Kretzmann, A. Liedl, A. Monferrer, V. Mykhailiuk, S. Beerkens and H. Dietz, Gene-encoding DNA origami for mammalian cell expression, Nat. Commun., 2023, 14, 1017 CrossRef CAS PubMed.
- B. Saccà and C. M. Niemeyer, DNA origami: the art of folding DNA, Angew. Chem., Int. Ed., 2012, 51, 58–66 CrossRef PubMed.
- D. Han, S. Pal, J. Nangreave, Z. Deng, Y. Liu and H. Yan, DNA Origami with Complex Curvatures in Three-Dimensional Space, Science, 2011, 332, 342–346 CrossRef CAS PubMed.
- A. Hernandez-Garcia, Strategies to Build Hybrid Protein–DNA Nanostructures, Nanomaterials, 2021, 11 Search PubMed.
- L. Gendron, D. Verreault, M. Veillette, S. Moineau and C. Duchaine, Evaluation of Filters for the Sampling and Quantification of RNA Phage Aerosols, Aerosol Sci. Technol., 2010, 44, 893–901 CrossRef CAS.
- A. H. Okholm, J. S. Nielsen, M. Vinther, R. S. Sørensen, D. Schaffert and J. Kjems, Quantification of cellular uptake of DNA nanostructures by qPCR, Methods, 2014, 67, 193–197 CrossRef CAS PubMed.
- K. Crank, K. Papp, C. Barber, P. Wang, A. Bivins and D. Gerrity, Environ. Sci. Technol., 2023, 57, 20448–20449 CrossRef CAS PubMed.
- G. Herath, K. Yamamoto and T. Urase, Removal of viruses by microfiltration membranes at different solution environments, Water Sci. Technol., 1999, 40, 331–338 CrossRef CAS.
- G. Pierre, C. Causserand, C. Roques and P. Aimar, Adsorption of MS2 bacteriophage on ultrafiltration membrane laboratory equipments, Desalination, 2010, 250, 762–766 CrossRef CAS.
- N. Ponnuswamy, M. M. C. Bastings, B. Nathwani, J. H. Ryu, L. Y. T. Chou, M. Vinther, W. A. Li, F. M. Anastassacos, D. J. Mooney and W. M. Shih, Oligolysine-based coating protects DNA nanostructures from low-salt denaturation and nuclease degradation, Nat. Commun., 2017, 8, 15654 CrossRef CAS PubMed.
- D. B. Mawhinney, R. B. Young, B. J. Vanderford, T. Borch and S. A. Snyder, Artificial sweetener sucralose in U.S. drinking water systems, Environ. Sci. Technol., 2011, 45, 8716–8722 CrossRef CAS PubMed.
- D. A. Kuzmanovic, I. Elashvili, C. Wick, C. O'Connell and S. Krueger, Bacteriophage MS2: Molecular Weight and Spatial Distribution of the Protein and RNA Components by Small-Angle Neutron Scattering and Virus Counting, Structure, 2003, 11, 1339–1348 CrossRef CAS PubMed.
- I. Koyuncu, R. Sengur, T. Turken, S. Guclu and M. E. Pasaoglu, in Advances in Membrane Technologies for Water Treatment: Materials, Processes and Applications, ed. A. Basile, A. Cassano and N. K. Rastogi, Woodhead Publishing, Oxford, 2015, pp. 83–128 Search PubMed.
- A. M. ElHadidy, S. Peldszus and M. I. Van Dyke, An evaluation of virus removal mechanisms by ultrafiltration membranes using MS2 and φX174 bacteriophage, Sep. Purif. Technol., 2013, 120, 215–223 CrossRef CAS.
- M.-L. Pype, B. C. Donose, L. Martí, D. Patureau, N. Wery and W. Gernjak, Virus removal and integrity in aged RO membranes, Water Res., 2016, 90, 167–175 CrossRef CAS PubMed.
- J. C. Vickers, M. Dummer, T. Le and J. B. Zoba, Removal of MS-2 coliphage in full-scale reverse osmosis systems, AWWA Water Sci., 2019, 1, e1158 CrossRef CAS.
- J. G. Wijmans and R. W. Baker, The solution-diffusion model: a review, J. Membr. Sci., 1995, 107, 1–21 CrossRef CAS.
- E. Bertosin, P. Stömmer, E. Feigl, M. Wenig, M. N. Honemann and H. Dietz, Cryo-Electron Microscopy and Mass Analysis of Oligolysine-Coated DNA Nanostructures, ACS Nano, 2021, 15, 9391–9403 CrossRef CAS PubMed.
- E. M. Vrijenhoek, S. Hong and M. Elimelech, Influence of membrane surface properties on initial rate of colloidal fouling of reverse osmosis and nanofiltration membranes, J. Membr. Sci., 2001, 188, 115–128 CrossRef CAS.
- R. S. Trussell, R. A. Tackaert, A. N. Pisarenko, E. Y. Idica and R. R. Trussell, Realizing Reverse Osmosis Potential for Potable Reuse: Demonstrating Enhanced Pathogen Removal, Denver, CO, 2017 Search PubMed.
- M.-L. Pype, E. A. de Eulate, A. Antony, D. Arrigan, F. Busetti, P. Le-Clech and W. Gernjak, National Validation Guidelines for Water Recycling: Reverse Osmosis Membranes, Brisbane, Queensland, 2015 Search PubMed.
- M. Nyström, L. Kaipia and S. Luque, Fouling and retention of nanofiltration membranes, J. Membr. Sci., 1995, 98, 249–262 CrossRef.
- K. Sharma, N. Akther, Y. Choo, P. Zhang, H. Matsuyama, H. K. Shon and G. Naidu, Positively charged nanofiltration membranes for enhancing magnesium separation from seawater, Desalination, 2023, 568, 117026 CrossRef CAS.
- Y.-H. Cai, X. J. Yang and A. I. Schäfer, Removal of Naturally Occurring Strontium by Nanofiltration/Reverse Osmosis from Groundwater, Membranes, 2010, 10(11), 321 CrossRef PubMed.
- J. Polanco, J. Safarik and M. H. Plumlee, Demonstrating Virus Log Removal Credit for Wastewater Treatment and Reverse osmosis for Potable Reuse at OCWD, Alexandria, VA, 2022 Search PubMed.
- D. Gerrity, E. R. V. Dickenson, B. S. Tseng, E. C. Wert, L. Green, C. M. Morrison, K. Papp and H. Ray, DNA Nanostructures as Viral Surrogates in Potable Reuse Applications, Alexandria, VA, 2023 Search PubMed.
- P. D. Halley, R. A. Patton, A. Chowdhury, J. C. Byrd and C. E. Castro, Low-cost, simple, and scalable self-assembly of DNA origami nanostructures, Nano Res., 2019, 12, 1207–1215 CrossRef CAS.
- A. Ness, A. Zarei-Baygi and D. Macnevin, Measure Viruses in the Field to Demonstrate Water Reuse Safety, Opflow, 2023, 49, 24–27 CrossRef.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ew00285g |
| This journal is © The Royal Society of Chemistry 2024 |