
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpacts of trace level chromium on formation of superoxide within uranyl triperoxide complexes†
Sarah K. Scherrera,
Harindu Rajapaksha a,
Dmytro V. Kravchuka,
Sara E. Masonb and
Tori Z. Forbes*a
a,
Dmytro V. Kravchuka,
Sara E. Masonb and
Tori Z. Forbes*a
aDepartment of Chemistry, University of Iowa, Iowa City, Iowa 52242, USA. E-mail: tori-forbes@uiowa.edu
bCenter for Functional Nanomaterials, Brookhaven National Laboratory, Upton, New York 11973, USA
First published on 28th August 2024
Abstract
U(VI) peroxides are important within the nuclear fuel cycle, but reactive oxygen species (ROS) can form upon oxidation. Herein, we identified the spectral signatures of a U(VI) diperoxosuperoxide complex (KUPS-1) and observed that the transformation of U(VI) triperoxide (KUT-1) to superoxide forms occurred with trace-level Cr. U(VI) superoxide complexes were identified in EPR solution spectra without the use of spin-traps.
Uranyl peroxides are important corrosion phases that form on the surface of spent nuclear fuel due to alpha-radiolysis of water.1–4 H2O molecules interact with the inherent radiation field around nuclear fuel, starting with ionization and excitation events and then propagating through ion–molecule reactions, dissociative relaxation, and solvation of electrons to form free radical species (i.e. OH˙, H˙).5,6 In the presence of dissolved molecular oxygen (O2), these free radicals can generate reactive oxygen species (ROS), such as superoxide (O2˙−), hydroperoxyl radical (OOH˙), hydroperoxyl anion (OOH−) and hydrogen peroxide (H2O2).7,8 The peroxide anion is considered to be the longer-lived form of such ROS and readily complexes with U(VI) to form the uranyl peroxide solids.9,10 These solids, particularly studtite ((UO2)(O2)(H2O)2 ˙ 2 H2O), are stable for years and can be observed in natural ore deposits;11,12 thus, the peroxide anion is the only ROS that has typically been considered for long term stability. However, Scherrer et al. recently demonstrated that the amount of O2˙− present in the studtite lattice increases with specific activity of the U present in the solid and age of the sample.13 This suggests peroxide oxidation and some level of superoxide ingrowth needs to be considered within the material.
In the current study, we evaluated the presence of O2˙− in fresh and aged samples of a potassium uranyl triperoxide material (KUT-1: K4[UO2(O22−)3](H2O2)4(H2O)4) by electron paramagnetic resonance (EPR) spectroscopy in both the solution and solid state. Initial materials were characterized for purity via X-ray diffraction and Raman spectroscopy (Fig. S1–S9, ESI†). The related potassium uranyl diperoxosuperoxide solid (KUPS-1: K4[UO2(O22−/˙−)3](OOH−/H2O2)4(H2O)4) was previously isolated and characterized with EPR,14 but the nature of the free radical signatures were not clear. We have now completed density functional theory (DFT) calculations to provide an in-depth understanding of the spin states and contribution of the free radical species. These computational studies were used to understand spectral signatures during aging of KUT-1 and the spin density within the molecule. In addition, we explore the behaviour of the KUT-1 compound upon dissolution in water and observe surprising in-growth and stability of the free radical species.
Prior structural characterization of the KUPS-1 compound indicated the presence of the uranyl (UO22+) cation with one O2˙− and two O22− anions bound in the equatorial plane.14 Potassium cations are located around the uranyl complex and are coordinated to both ligated water and H2O2 molecules. The previously reported EPR spectra of solid-state KUPS-1 that was stored under Ar for 3.5 weeks identified five g-factors at 1.951, 1.983, 1.994, 2.017, 2.05 (Fig. 1A).14 Presence of the superoxide ligand in the complex lowers the point group symmetry from the D3h expected for the uranyl triperoxide [(UO2)(O22−)3]4− down to C2v for the [(UO2)(O22−)2(O2˙−)]3−, technically resulting in a rhombic magnetic moment. The ambiguities in bond lengths and close structural relationship between the superoxide and peroxide anions will likely result in an axial signal observed experimentally. Still the additional features in the EPR spectrum suggested the presence of other radical species present in the system.
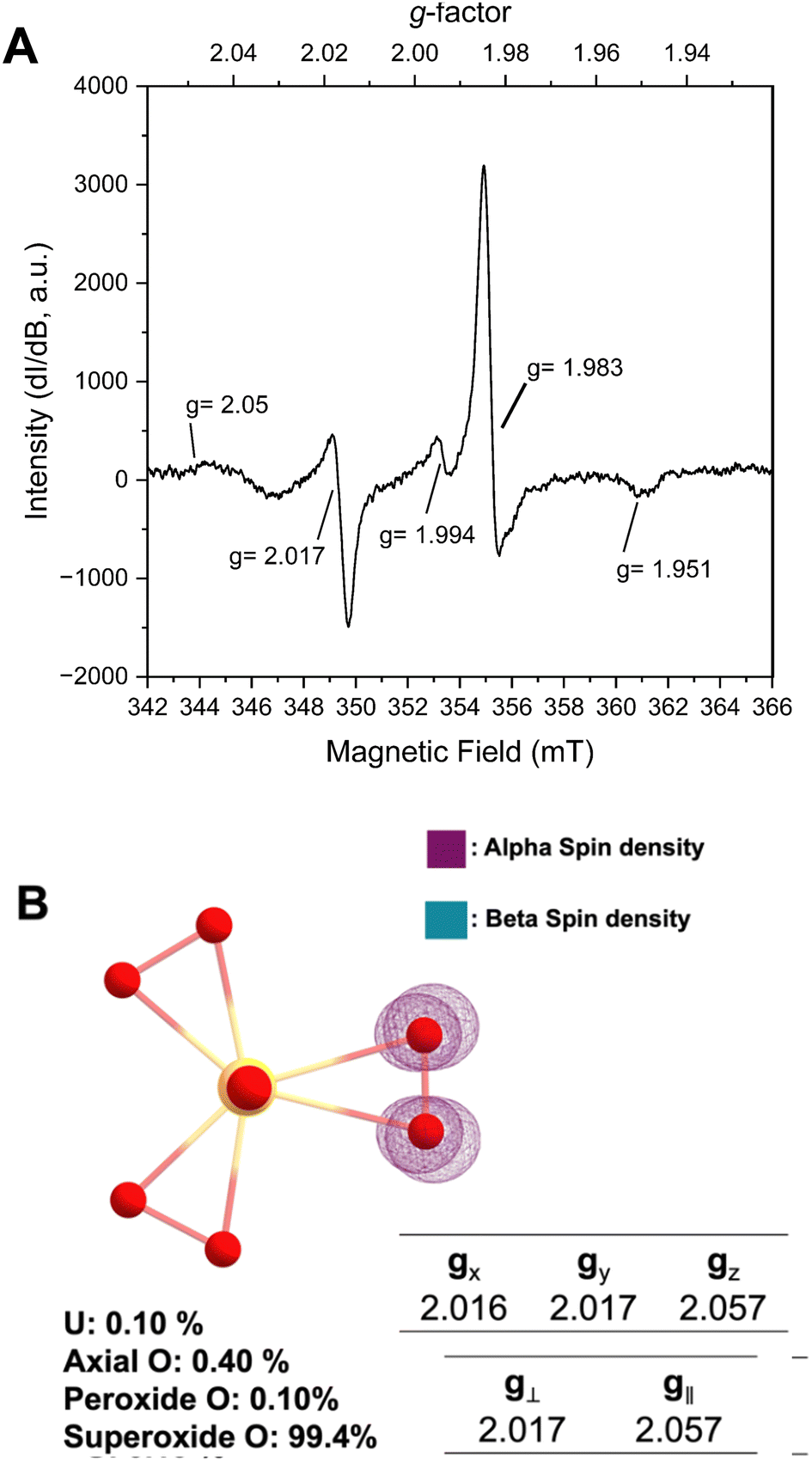 | ||
| Fig. 1 (A) Solid-state EPR of KUPS-1. (B) DFT simulation of the uranyl diperoxosuperoxide complex with spin density, % orbital contribution (localization of unpaired e−), and calculated g-tensors. Isosurfaces of spin densities have iso values of 0.0012 e− Bohr−3. | ||
DFT studies were performed to predict the EPR signatures for the KUPS-1 complex (two peroxide and one superoxide ligand) and provide additional insights into the nature of the molecular species. Predicted bond distances for the U![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O axial, U–Operoxide and U–Osuperoxide equatorial bond distances associated with [(UO2)(O22−)2(O2˙−)]3− are 1.868, 2.265, and 2.531 Å (Fig. S10, ESI†) respectively, which match well with the experimentally derived values.14 We note that the calculated values associated with the superoxide anion are shorter than what is observed (CCDC 135984†)14 in the structural data due to the average bond distance obtained from the single-crystal X-ray diffraction data representing the average of that site and likely contains contributions from geometries with multiple reactive oxygen species (i.e. O22−, O2˙−). Spin density maps of the molecular complex indicate that 99.4% of the unpaired electron is localized on two equatorial O atoms; thus, the spin solely remains on the ligand with no additional spin noted on the metal centre (Fig. 1B). This confirms that the U(VI) is not redox active even in the presence of the superoxide anion and we should not expect any contributions from U(V). Predicted spectral signals for the KUPS-1 complex are observed at gx = 2.016, gy = 2.017, and gz = 2.057 (giso = 2.032). These values suggest the likely presence of an axial signal that matches two of the experimentally observed values (g‖ = 2.05, g⊥ = 2.017) for the KUPS-1 compound. These g-factors are different from calculated and known superoxide phases (Tables S2 and S3, ESI†),15,16 including KO2, indicating that it is not a signature from the K+/H2O2 network.
O axial, U–Operoxide and U–Osuperoxide equatorial bond distances associated with [(UO2)(O22−)2(O2˙−)]3− are 1.868, 2.265, and 2.531 Å (Fig. S10, ESI†) respectively, which match well with the experimentally derived values.14 We note that the calculated values associated with the superoxide anion are shorter than what is observed (CCDC 135984†)14 in the structural data due to the average bond distance obtained from the single-crystal X-ray diffraction data representing the average of that site and likely contains contributions from geometries with multiple reactive oxygen species (i.e. O22−, O2˙−). Spin density maps of the molecular complex indicate that 99.4% of the unpaired electron is localized on two equatorial O atoms; thus, the spin solely remains on the ligand with no additional spin noted on the metal centre (Fig. 1B). This confirms that the U(VI) is not redox active even in the presence of the superoxide anion and we should not expect any contributions from U(V). Predicted spectral signals for the KUPS-1 complex are observed at gx = 2.016, gy = 2.017, and gz = 2.057 (giso = 2.032). These values suggest the likely presence of an axial signal that matches two of the experimentally observed values (g‖ = 2.05, g⊥ = 2.017) for the KUPS-1 compound. These g-factors are different from calculated and known superoxide phases (Tables S2 and S3, ESI†),15,16 including KO2, indicating that it is not a signature from the K+/H2O2 network.
While we can account for EPR signals at g‖ = 2.05 and g⊥ = 2.017 as arising from the coordinated O2˙−, the nature of g-factors at 1.951, 1.983, 1.994 remained ambiguous, especially since O-centred radicals are unlikely to appear in that range. Interestingly, we were able to match the resonance at g = 1.983 to the paramagnetic chromium(V) peroxo complex [Cr(V)(O)(O2)(OH2)x]+, with features at g = 1.951 and 1.994 matching well with hyperfine coupling signals for this form.17 We used inductively coupled plasma mass spectrometry (ICP-MS) to confirm the presence of trace chromium (ppb) across the stock solutions used in the KUPS-1 synthesis (Table S4, ESI†). This contaminant was not observed in the case of studtite,13 but the nature of the KUPS-1 lattice and the alkaline conditions used in the synthesis of the material likely enables the Cr(VI) and Cr(V) to incorporate into the solid phase.17 In the stock solutions, it is expected that Cr(VI) is present, but this metal cation will react with H2O2 to form a variety of species, including [Cr(VI)O(O2)2OH]−, four different Cr(V) peroxo complexes, and even Cr(III) peroxo intermediates. The Cr(VI)/H2O2 reaction is complicated by catalytic decomposition of H2O2 and specific reaction conditions (i.e. pH, H2O2 concentration).17 Given the prevalence for trace-level transition metals in uranium materials, this finding potentially has implications for the chemistry of reactive oxygen species within the uranyl peroxide systems, and may serve as a forensic marker during processing of uranium based fuel.
KUT-1 is synthesized in the same manner as KUPS-1 and when the EPR spectra of the KUT-1 compound was collected 24 h after synthesis of the solid (Fig. 2A), the only g-factors present in the spectrum were at g = 1.983 and 1.973. These features are assigned to Cr-centred [Cr(V)(O)(O2)(OH2)x]+ and [Cr(V)(O2)4]3− species3−,17 with no evident resonances associated with the O2˙− and confirming that the solid is a triperoxide phase. Interestingly, one of the K+ sites within the crystal structure of KUT-1 is exclusively coordinated by four side-on η2-O22− ligands, which appears to be the most likely substitutional site for the [Cr(V)(O2)4]3− species. After aging for 3 weeks under Ar, the signature at g = 1.983 grows in while the feature at g = 1.973 decreases, indicating partial transformation of [Cr(V)(O2)4]3− into [Cr(V)(O)(O2)(OH2)x]+. A new resonance at g⊥ = 2.016 is also present, consistent with the uranyl superoxide, however the g‖ = 2.05 is less resolved. To remove the trace contaminant metals from our uranyl nitrate stock solution and minimize the Cr(V) signature, we performed a liquid–liquid extraction using tributylphosphate.18 In this case, the fresh material contained no EPR signatures, but after three weeks weak features at g = 1.973, 1.983, 1.990 and 2.016 appeared in the spectrum (Fig. 2B). This indicates that the sub-ppb level Cr(VI) in the material reduced to Cr(V) over time.17
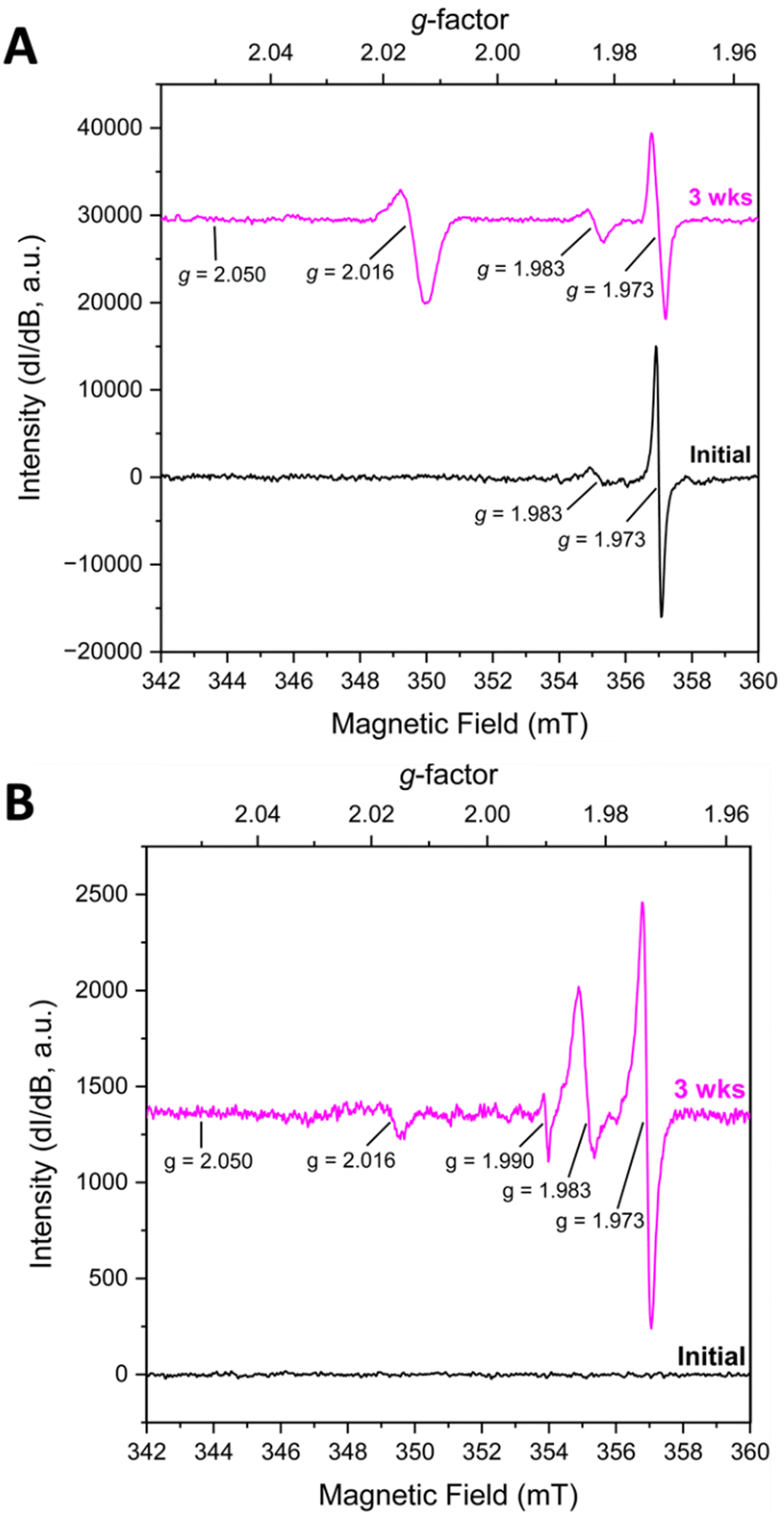 | ||
| Fig. 2 (A) Solid-state EPR of KUT-1 (∼24 h) and aged for 3 weeks indicates the presence of Cr(V). (B) Purification of the uranyl nitrate reactant resulted in no evidence of Cr(V) in the initial KUT-1 sample (∼24 h) but the Cr(V) and U(VI) superoxide signatures appear after 3 weeks of aging. | ||
From these results, we believe the changes in the K/Cr peroxide framework around the uranyl triperoxide monomer in KUT-1 induces the in situ formation of uranyl superoxide species (KUPS-1). Interestingly, the synthesis of a similar potassium uranyl triperoxide K4[UO2(O2)3]·4H2O was reported by Dembowski et al.19 who noted a formation of a transient “bright-yellow microcrystalline solid” with unexpected spectroscopic signatures that converted into the desired K4[UO2(O2)3]·4H2O phase within one week under ambient conditions. The physical properties (i.e. color and morphology) of this transient phase resemble the KUPS-1 compound, but this would suggest that the superoxide phase converts into the triperoxide form. However, this particular system has not been characterized by EPR spectroscopy, so it is unclear what ROS is present in the material.
It is also important to note that redox chemistry of coordinated peroxide ligands appears to be linked to trace-level Cr in the sample that serves as an electron sink. This is supported by the fact that the superoxide signature at g‖ = 2.05, g⊥ = 2.016 is more intense when more Cr(V/VI) is present in the material. In addition, previous work by Orhanovic and Wilkins20 demonstrated that Cr(VI) will reduce to Cr(V) peroxides in acidic solutions. This suggests that both Cr(VI) and Cr(V) may be present in the KUPS-1 compound but Cr(VI) will be EPR silent. Presence of trace transition metals with uranium solids is expected, including in UO2 fuel pellets.21 This adds an additional layer of complexity to the ingrowth and fate of ROS species within these materials and related solutions.
We also explored the change in spectral signals for KUT-1 in solution. In this case, fresh, purified KUT-1 was dissolved in water (30 mM), which resulted in a solution pH of 12. Initially the only EPR signature (Fig. 3) was located at g = 1.973, corresponding to the presence of the residual [Cr(V)(O2)4]3− complex upon dissolution.17 After 24 h, the signal associated with the Cr(V) peroxo moiety disappeared, likely due to oxidation to Cr(VI). After 48 h, a persistent new signature is observed at g = 2.031 and the intensity continues to increase with 72 h of aging under ambient conditions. This g-factor matches well with the calculated giso value (2.032) for the U(VI) superoxide anion and is shifted from the expected value for a free superoxide (g = 2.062) anion calculated using DFT (Table S2, ESI†). This result indicates that the uranyl triperoxide species alters to some amount of the U(VI) superoxide form in solution.
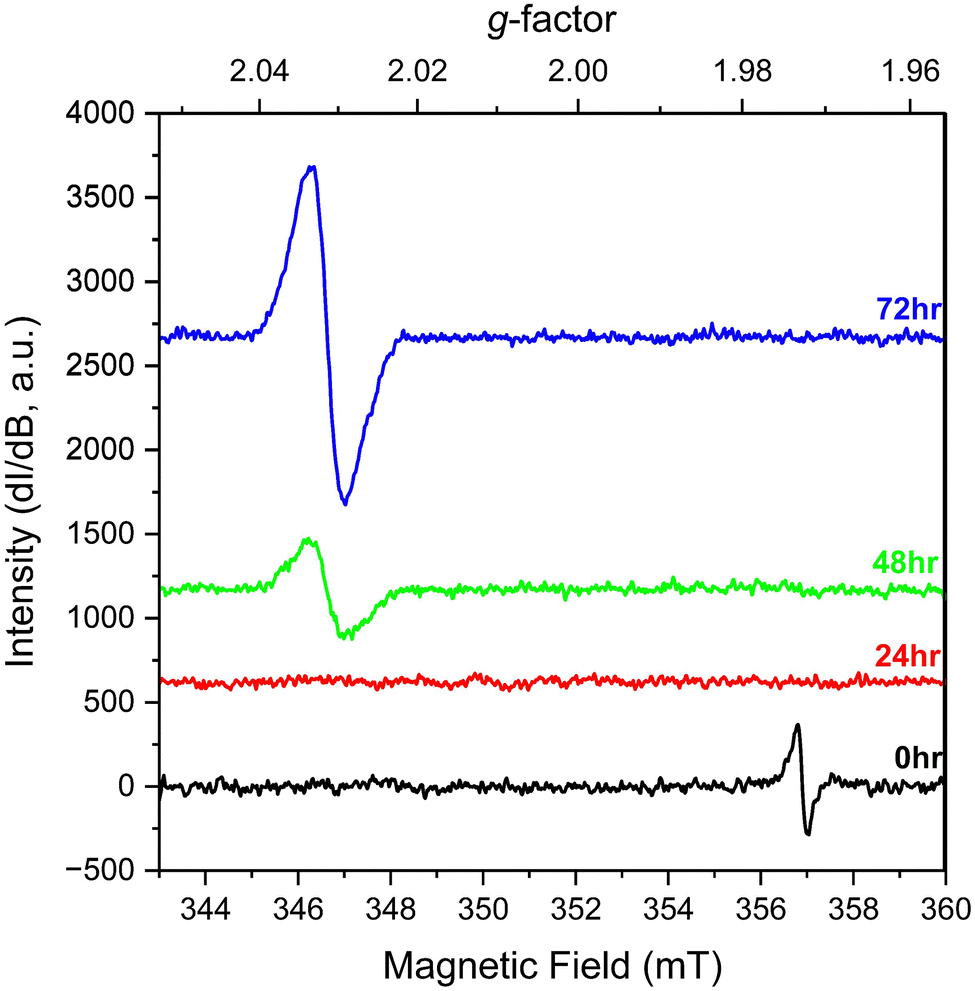 | ||
| Fig. 3 Solution EPR spectra of fresh, purified KUT-1 dissolved in water (pH = 12) at 0, 24, 48 and 72 h. | ||
Identifying the presence of O2˙− in solution by EPR spectroscopy without the use of a molecular spin-trap, such as 5,5-dimethyl-1-pyrroline N-oxide (DMPO), is very unusual because the half-life of the superoxide anion is ∼1.25 seconds at pH 10–12.22,23 Control experiments of solutions containing potassium hydroxide (pH 12) and H2O2 demonstrate this issue, as no EPR signatures for superoxide were observed for the entire 72 h (Fig S11, ESI†) without the addition of DMPO (Fig. S12 and S13, ESI†). To the best of our knowledge, the U(VI)–O2˙− signature is the only instance of superoxide resonance being observed in aqueous solution at room temperature without the addition of a spin-trap molecule. This highlights that the U(VI) cation stabilizes the superoxide radical longer than a free O2˙− anion in solution to enable detection by EPR spectroscopy at room temperature.
The results demonstrate that the O22− anion can oxidize to O2˙− even after binding to the U(VI) cation and formation can be enhanced by the presence of trace metals in the system. This has important implications in considering the long-term stability of uranyl peroxide materials as Kravchuk et al. demonstrated that U(VI) materials containing superoxide radicals engage in direct-air carbon capture and formation of peroxocarbonate phases.14,24 Within alkaline solutions, the oxidation of peroxide occurs more readily to form superoxide radicals that enhance carbonation reactions25 and our results suggest that this can be further influenced by the presence of U(VI) and Cr(VI/V) in solution. In addition, high radiation fields may promote the formation of superoxide from U(VI) peroxide complexes that can lead to additional reactivity over time.13
Overall, the current study demonstrates that U(VI) triperoxide solids and related soluble molecular species will change over time to form the superoxide species and may be further influenced by trace metals (i.e. Cr, Fe) that are commonly found in nuclear materials. Evidence of superoxide in solution without the addition of spin-traps demonstrates the stability of O2˙− under these conditions for several days. This level of stability has yet to be demonstrated for any other metal cation and has implications for the carbonation process within alkaline conditions and the transformation of U(VI) peroxide solid phases in high radiation fields.
This work was supported by the U. S. Department of Energy Basic Energy Sciences (DE-14037600). We thank Dr. Gary Buettner and Brett Wagner at the University of Iowa ESR Facility and Drs. Benjamin Stein and Samuel Greer at Los Alamos National Laboratory for their expert guidance, training, and support on the EPR spectroscopy. We acknowledge the University of Iowa Materials Analysis, Testing, and Fabrication Facility and Dr. Daniel Unruh for support associated with the X-ray diffractometer and Raman spectrometer. Computational efforts were additionally supported by the Theory and Computation facility of the Center for Functional Nanomaterials (CFN), which is a U.S. Department of Energy Office of Science User Facility, at Brookhaven National Laboratory under Contract No. DE-SC0012704.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- C. R. Armstrong, M. Nyman, T. Shvareva, G. E. Sigmon, P. C. Burns and A. Navrotsky, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 1874–1877 CrossRef CAS PubMed.
- B. E. Burakov, E. E. Strykanova and E. B. Anderson, Mater. Res. Soc. Symp. Proc., 1997, 465, 1309–1311 CrossRef CAS.
- B. Hanson, B. McNamara, E. Buck, J. Friese, E. Jenson, K. Krupka and B. Arey, Radiochim. Acta, 2005, 93, 159–168 CrossRef CAS.
- B. McNamara, E. Buck and B. Hanson, Mater. Res. Soc. Symp. Proc., 2002, 757, 401–406 CrossRef.
- B. J. Mincher, J. Radioanal. Nucl. Chem., 2018, 316, 799–804 CrossRef CAS.
- C. Ferradini and J. P. Jay-Gerin, Can. J. Chem., 1999, 77, 1542–1575 CrossRef CAS.
- B. J. Mincher, G. Elias, L. R. Martin and S. P. Mezyk, J. Radioanal. Nucl. Chem., 2009, 282, 645–649 CrossRef CAS.
- B. J. Mincher and S. P. Mezyk, Radiochim. Acta, 2009, 97, 519–534 CAS.
- N. Rodríguez-Villagra, L. J. Bonales and J. Cobos, MRS Adv., 2020, 5, 539–547 CrossRef.
- N. Rodríguez-Villagra, L. J. Bonales, A. Milena-Pérez and H. Galán, MRS Adv., 2023, 8, 207–213 CrossRef.
- P. C. Burns and K. A. Hughes, Am. Mineral., 2003, 88, 1165–1168 CrossRef CAS.
- K. A. H. Kubatko, K. B. Helean, A. Navrotsky and P. C. Burns, Science, 2003, 302, 1191–1193 CrossRef PubMed.
- S. K. Scherrer, C. Gates, H. Rajapaksha, S. M. Greer, B. W. Stein and T. Z. Forbes, Angew. Chem., Int. Ed., 2024, 63, e202400379 CrossRef CAS PubMed.
- D. V. Kravchuk, N. N. Dahlen, S. J. Kruse, C. D. Malliakas, P. M. Shand and T. Z. Forbes, Angew. Chem., Int. Ed., 2021, 60, 15041–15048 CrossRef CAS PubMed.
- P. D. C. Dietzel, R. K. Kremer and M. Jansen, J. Am. Chem. Soc., 2004, 126, 4689–4696 CrossRef CAS PubMed.
- D. M. Lindsay, D. R. Herschbach and A. L. Kwiram, Chem. Phys. Lett., 1974, 25, 175–181 CrossRef CAS.
- L. Zhang and P. A. Lay, Inorg. Chem., 1998, 37, 1729–1733 CrossRef CAS.
- A. G. Baldwin, N. J. Bridges and J. C. Braley, Ind. Eng. Chem. Res., 2016, 55, 13114–13119 CrossRef CAS.
- M. Dembowski, V. Bernales, J. Qiu, S. Hickam, G. Gaspar, L. Gagliardi and P. C. Burns, Inorg. Chem., 2017, 56, 1574–1580 CrossRef CAS PubMed.
- M. Orhanovic and R. G. Wilkins, J. Am. Chem. Soc., 1967, 89, 278–282 CrossRef CAS.
- T. L. Spano, A. Simonetti, L. Corcoran, P. A. Smith, S. R. Lewis and P. C. Burns, J. Nucl. Mater., 2019, 518, 149–161 CrossRef CAS.
- S. Scheinok, P. Leveque, P. Sonveaux, B. Driesschaert and B. Gallez, Free Radical Res., 2018, 52, 1182–1196 CrossRef CAS PubMed.
- L. Y. Zang and H. P. Misra, J. Biol. Chem., 1992, 267, 23601–23608 CrossRef CAS PubMed.
- D. V. Kravchuk and T. Z. Forbes, Acs Mater. Au, 2022, 2, 33–44 CrossRef CAS PubMed.
- U. Stoin, Z. Barnea and Y. Sasson, RSC Adv., 2014, 4, 36544–36552 RSC.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental methods, PXRD of solid phases, Raman spectroscopy, details of DFT study, EPR data. See DOI: https://doi.org/10.1039/d4cc03194f |
| This journal is © The Royal Society of Chemistry 2024 |