
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMerging electrocatalytic alcohol oxidation with C–N bond formation by electrifying metal–ligand cooperative catalysts†
Sitthichok
Kasemthaveechok
 a,
Patrice
Gérardo
b and
Niklas
von Wolff
*a
a,
Patrice
Gérardo
b and
Niklas
von Wolff
*a
aLaboratoire d'Électrochimie Moléculaire, Université Paris Cité/CNRS UMR7591, F-75013 Paris, France. E-mail: niklas.von-wolff@u-paris.fr
bLaboratoire de Chimie et Biochimie, Pharmacologiques et Toxicologiques, Université Paris Cité/CNRS UMR8601, F-75006 Paris, France
First published on 20th October 2023
Abstract
Electrification of thermal chemical processes could play an important role in creating a more energy efficient chemical sector. Here we demonstrate that a range of MLC catalysts can be successfully electrified and used for imine formation from alcohol precursors, thus demonstrating the first example of molecular electrocatalytic C–N bond formation.This novel concept allowed energy efficiency to be increased by an order of magnitude compared to thermal catalysis. Molecular EAO and the electrification of homogeneous catalysts can thus contribute to current efforts for the electrocatalytic generation of C–N bonds from simple building blocks.
Introduction
According to the International Energy Agency (IEA), the chemical sector needs to reduce its emissions by 17% from the current 925 Mt CO2 footprint by 2030 in order to align with a net-zero scenario by 2050.1 Achieving these ambitious goals heavily relies on increasing energy efficiency. Electrochemistry presents an opportunity to directly control the energy input of a reaction through the applied potential.2 By electrifying existing thermal processes, significant advancements have been made in recent years. This approach has unlocked novel reactivity3 and leveraged the inherent advantages of electrochemistry, including safety, scalability, and the utilization of affordable and environmentally friendly redox agents (electrons) in existing processes.4–8 A prime example of this is the electrochemical Birch reduction, which elegantly demonstrates these benefits.9 Electrocatalytic alcohol oxidation, which combines potentially renewable feedstocks with energy efficiency (see also the ESI†), holds particular interest for various applications such as low-voltage electrolyzers, fuel cells, and organic redox chemistry (Scheme 1A).10–14 Molecular systems utilizing hydrogen atom transfer (HAT) mediators, such as N-oxyl systems,15–20 or high-valent ruthenium and iron oxo species,21–24 have shown intriguing activity for electrocatalytic alcohol oxidation (EAO). However, their application is mostly limited to activated substrates and high oxidation potentials (Scheme 1B). Grützmacher and colleagues conducted seminal work demonstrating the possibility of electrifying thermal (transfer)-hydrogenation (TH) catalysts for molecular EAO under heterogeneous conditions.25,26 This work inspired the development of homogeneous systems based on similar (de)hydrogenation catalysts (Scheme 1C),27–33 including acceptor-less alcohol dehydrogenation (AAD) complexes.34 Despite the notable advancements achieved thus far, the realm of molecular electrocatalytic alcohol oxidation (EAO) using (de)hydrogenation catalysts remains confined to a limited array of substrates, primarily isopropanol and benzyl alcohol.29,31–34 We were thus interested if the scope of such systems could be extended to carbon–heteroatom bond formations. From a fundamental point of view, we were intrigued to know if the thermodynamic and kinetic challenges related to anodic processes tolerating amine substrates could be overcome and thus targeted C–N bond formation as a case study (Scheme 1D and ESI Section 7†). In addition, C–N bond formation has been identified as a pivotal objective in current electrocatalysis,35–44 with a specific emphasis on alcoholic substrates.45,46 We hypothesized that the unique attributes of so-called metal–ligand cooperative (MLC) catalysts,47 such as their low oxidation potential, high activity, and potential for substrate differentiation, make them ideal candidates for exploring the challenging realm of C–N bond formation under electrocatalytic conditions. In particular, we envisioned that MLC systems could potentially overcome some of the limitations posed by commonly employed hydrogen atom transfer (HAT) mediators, whose oxidation potential closely aligns with that of many amines.48 Imines are of particular interest and have found application both at the laboratory scale and in industrial synthesis, making them a suitable target.49 In this exploratory work, we present the first example of efficient molecular electrocatalytic C–N bond formation from alcohols to form imines (Scheme 1E).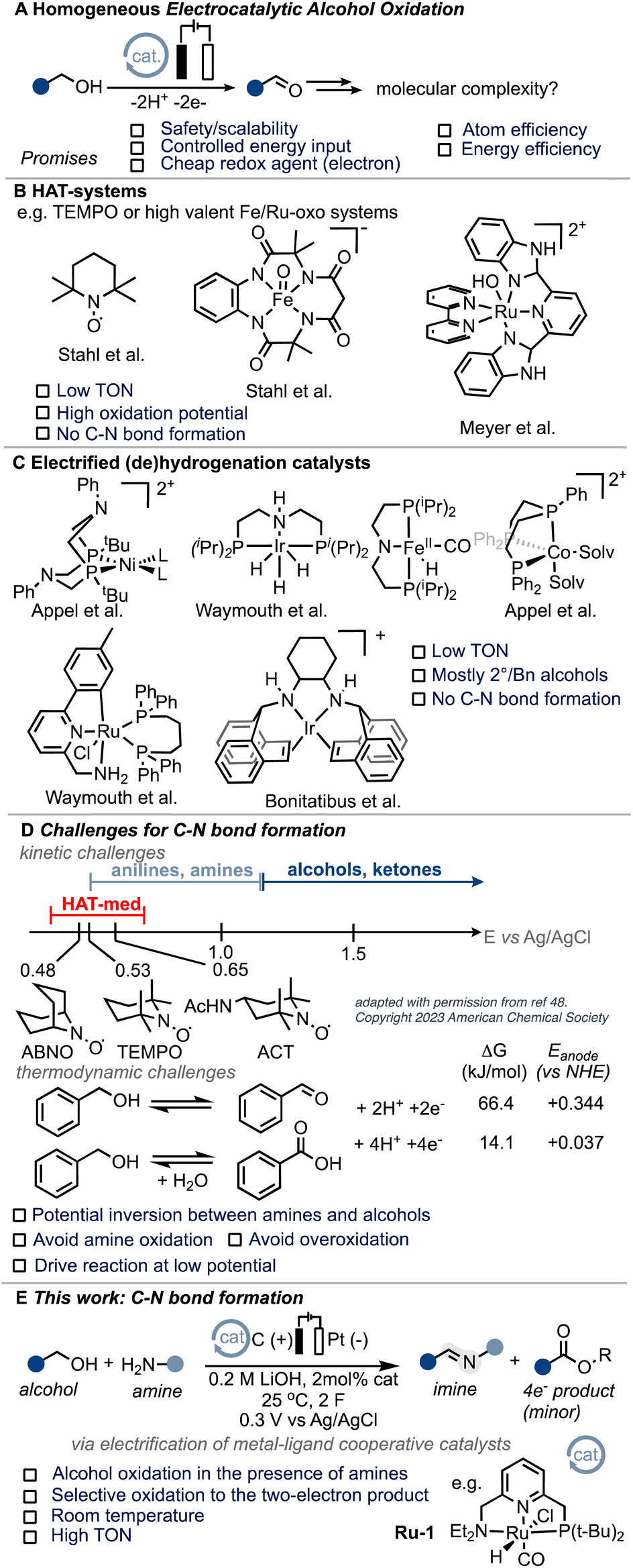 | ||
| Scheme 1 Homogeneous electrocatalytic alcohol oxidation and C–N bond formation. (A) Electrocatalytic alcohol oxidation. (B) and (C) HAT- and (de)hydrogenation systems. (D) Challenges for C–N bond formation. (E) This work. | ||
Results and discussion
In our endeavour to tackle the challenge of electrifying thermal dehydrogenation systems, we recently successfully employed the renowned Milstein catalyst Ru-1 (Scheme 1E) under electrochemical conditions, showcasing its versatility and potential for transformative electrocatalysis. Significantly, we unravelled the beneficial role of electricity in promoting turnover at significantly lower temperatures, a breakthrough that holds promise to increase the energy-efficiency of such systems in the future.50 Building upon these foundations, we sought to harness the benefits of MLC catalysts under electrochemical conditions, aiming to overcome the hurdles associated with alcohol oxidation in the presence of amines while mitigating the undesirable outcomes of alcohol overoxidation, undesired amine oxidation or competitive amine dehydrogenation.After screening optimal reaction conditions (see ESI Section 4†), we were pleased to find that Ru-1 is capable of promoting C–N bond formation in neat benzyl alcohol in the presence of n-hexyl amine (H2N-Hex) and 0.2 M LiOH to form the corresponding imine 1 in excellent yield (99%) and faradaic efficiency (F.E.) close to 100% (Table 1, entry 1). To confirm the successful electrification of Ru-1 for C–N bond formation, we performed corresponding blank experiments (see Table 1, note that the reactions were stopped after 6 h to compare electrochemical and thermal reactions). Notably, we eliminated any significant contributions from thermal activation of Ru-1 (ref. 51) by running the experiment without an applied potential (open-circuit potential, OCP), which resulted in considerably lower amine conversions (Table 1, entry 2). The presence of Ru-1 proved crucial for the successful formation of the C–N bond, as evidenced by the limited yield and faradaic efficiency obtained with a blank carbon electrode52 (Table 1, entry 3). This underscores the indispensability of Ru-1 as a catalyst for achieving the desired outcome. Motivated by our hypothesis that metal–ligand cooperative (MLC) complexes, exemplified by Ru-1, could offer advantages over the conventional hydrogen atom transfer (HAT) mediators employed in alcohol oxidation, we investigated the activity of (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO). However, under our reaction conditions, TEMPO displayed limited efficacy in promoting oxidative C–N bond formation (Table 1, entry 5). This lack of catalytic activity of TEMPO underscores the challenges associated with merging EAO with C–N bond formation. Notably, catalysts for this transformation must be capable of tolerating low working potentials and highly coordinating reaction media to enable the selective conversion of easily oxidizable substrates,53 aligning with the inherent difficulties in identifying suitable catalysts for this intricate transformation (Scheme 1D).
|
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | Deviation from standardsa | Yield 1b (%) | Product distribution (mmol) | Charge passed (mmol) | F.E. (%)b | ||
| 1b | Aldehyde | Ester | (Total F.E.)c | ||||
| a Conditions: 0.2 M LiOH in 6 mL solvent (neat BnOH), 50 μL hexylamine (65 mM), 1.3 mM Ru-1 (2 mol%), 0.3 V vs. Ag/AgCl, 25 °C, stopped at 6 h. b GC-MS yield with mesitylene as an internal standard. The amount of imine and ester formation from benzyl alcohol is subtracted from the product distribution and the yield (and F.E.) is corrected for the initial presence of benzaldehyde (see ESI Section 3 also for the calculations of F.E.). c The total F.E. includes electrochemically formed aldehyde and ester. | |||||||
| 1 | None | >99 | 0.257 | 0.142 | — | 0.788 | 65 (101) |
| 2 | No applied potential | 7 | 0.029 | 0.009 | — | n.d. | — |
| 3 | No catalyst | 11 | 0.022 | — | — | 0.41 | 10 (10) |
| 4 | No applied potential and no catalyst | 0 | — | — | — | — | — |
| 5 | TEMPO instead of Ru-1 | 2.5 | 0.009 | 0.006 | — | 0.041 | 44 (73) |
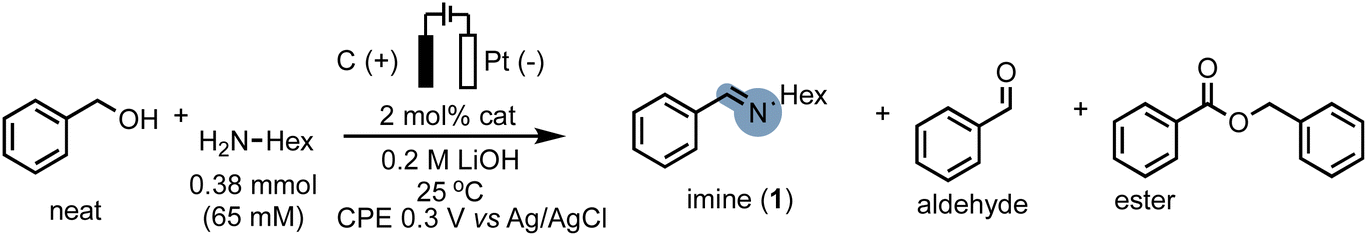
In order to demonstrate the generality of the concept of electrifying (de)hydrogenation catalysts for C–N bond formation, we conducted a comprehensive screening of common ruthenium-based systems (Scheme 2). Notably, both the pyridine-based PNNH and PNP pincer complexes Ru-2 (ref. 54) and Ru-3 exhibited higher activity towards C–N bond formation, achieving quantitative conversion of the amine. However, the high reactivity came at the cost of reduced faradaic efficiency (F.E). On the other hand, the MACHO-type aliphatic pincer complexes Ru-4, Ru-5, Ru-6, Ru-7, Ru-9, and Ru-10 (ref. 55) demonstrated decreased selectivity, with F.E. values dropping to around 60%. The increased flexibility in the ligand backbone of these systems could lead to reduced stability under electrocatalytic conditions, which aligns with the limited turnover numbers observed for similar iron-MACHO systems in the context of electrocatalytic alcohol oxidation.34 Furthermore, the presence of steric bulk around the metal center seemed to hinder the catalytic activity of the aliphatic systems (e.g., Ru-4), while the inherent configurational stability of aromatic pincer complexes enabled the tolerance of bulky tert-butyl groups (Ru-2, Ru-3). Notably, the utilization of Gusev's air-stable thio-MACHO complex Ru-8 (ref. 56) yielded results similar to those of the other aliphatic systems, albeit with a slightly higher amount of over-oxidation to the ester product, accompanied by a modest overall F.E. of approximately 65%. Nevertheless, this observation underscores the potential for fine-tuning the ligand environment to modulate the selectivity between two- and four-electron oxidation pathways within these systems. Having established the generality of our concept, we aimed to investigate the scope of the reaction by exploring its applicability to other benzyl alcohols. Since most commercial benzyl alcohols are solid under standard conditions, we employed optimized diluted conditions to assess the effectiveness of our system in promoting C–N bond formation with benzylic alcohols exhibiting varying electronic demands (Scheme 3 and ESI Section 4†). We observed the highest conversion rates for sterically accessible electron-rich alcohols, such as those bearing hydrogen, methoxy, and methyl groups in the para-position. These substrates successfully underwent conversion to the corresponding imine products (1, 1a, and 1b) with good yields and nearly complete F.E. In contrast, benzyl alcohols carrying electron-withdrawing groups in the para-position, such as trifluoromethyl, bromo, or nitro groups, exhibited reduced reactivity, resulting in lower yields and faradaic efficiency.
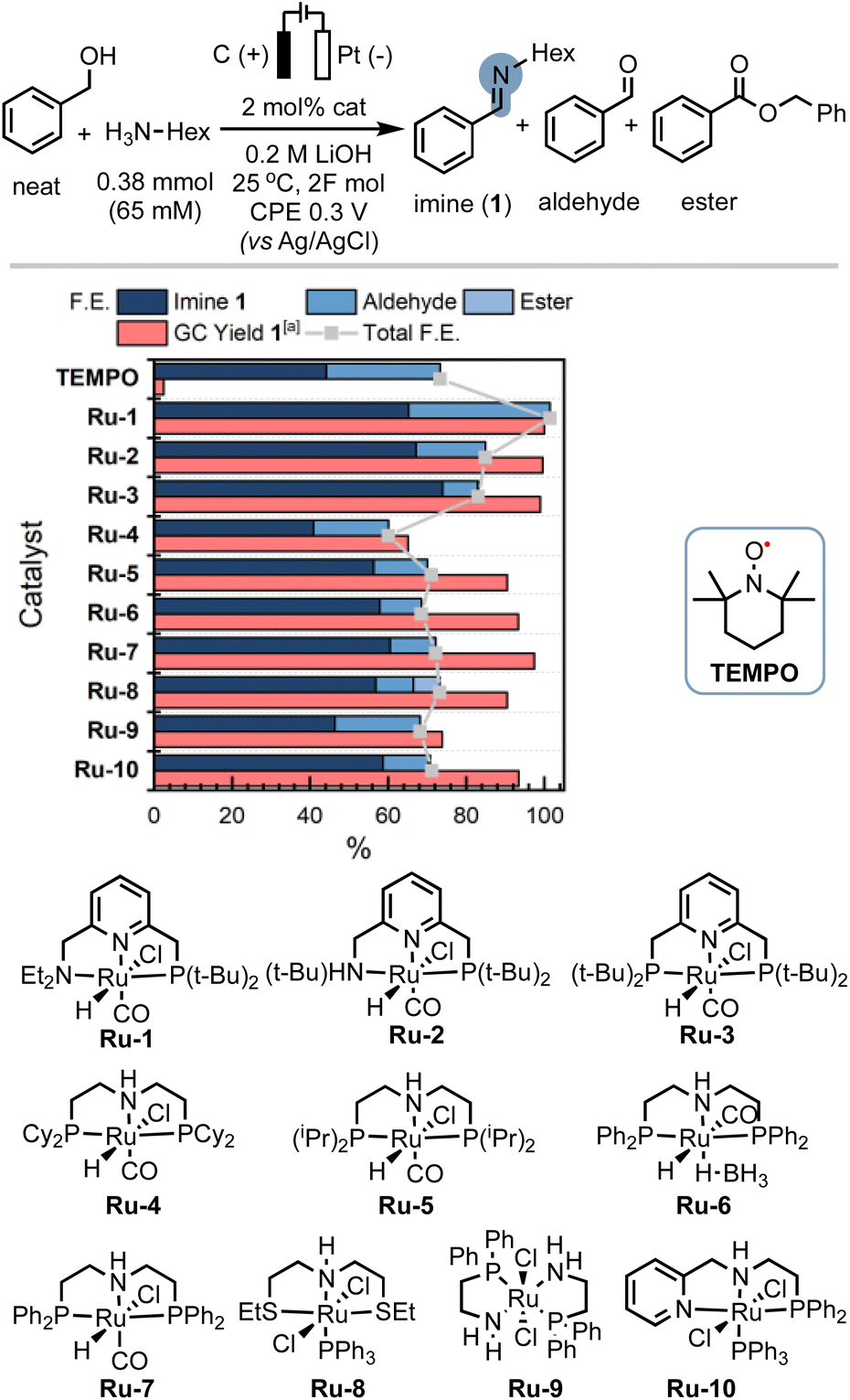 | ||
| Scheme 2 Ligand effects for the electrocatalytic alcohol oxidation with common metal–ligand cooperative catalysts. [a]GC-MS yield with mesitylene as an internal standard. The amount of imine formation from benzyl alcohol auto-oxidation is subtracted from the product distribution and the yield is corrected for the initial presence of benzaldehyde (see ESI Section 3†). | ||
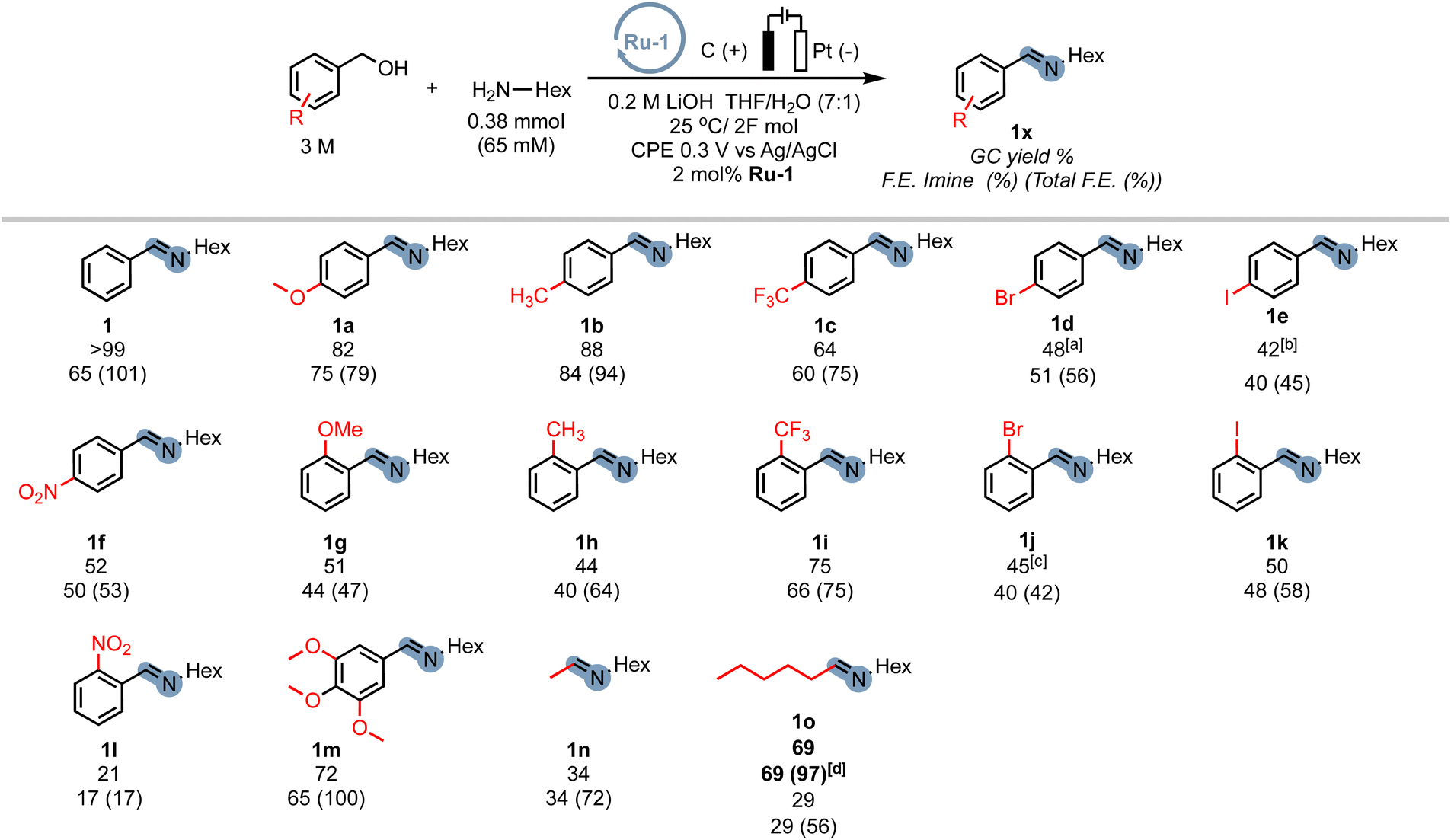 | ||
| Scheme 3 Scope of benzyl alcohols used for the electrocatalytic imine formation. Reactions are stopped after 2F per amine (to account for differences in electrode/cell dimensions), which is in general well below 6 hours. The initial amine concentration is adjusted for benzyl alcohol auto-oxidation (see ESI Section 3†). [a]The total F.E. includes electrochemically formed aldehyde and ester. [b]NMR yield. [c]Isolated yield. [d]Run at 0.1 V instead of 0.3 V vs. Ag/AgCl. | ||
The extent of the decrease in reactivity followed the order of increasing inductive effects of the substituents (1c–1f). The introduction of electron-donating groups in the ortho-position (1g, 1h) or electron-withdrawing groups (1i–1k) led to decreased yields, with o-CF3 (75%) and o-NO2 (21%) being outliers. However, electron-donating groups in the meta-position were well-tolerated (1m), resulting in a good yield of 72% and an excellent faradaic efficiency of 100%. Alkyl alcohols are a particular challenge for most (de)hydrogenation-based systems in EAO, where mostly secondary aliphatic alcohols are reported as substrates.30 Indeed, under standard conditions, primary aliphatic alcohols proved to be less competent coupling partners (1n, 1o), yielding modest yields and faradaic efficiency. Notably, the presence of hydroxylated amine side-products (mono and bis) was detected by GC-MS, constituting up to 19% of the reaction mixture. We hypothesized that the lower acidity of alkyl alcohols in organic media, as compared to benzyl alcohols, hinders sufficient deprotonation, leading to parasitic oxidation of the amine at the applied potential. Encouragingly, when the potential was adjusted to 0.1 V, the corresponding alkyl amine (1o) was recovered with a good yield of 69% and an excellent faradaic efficiency of 97%, while the accumulation of hydroxylation side-products was significantly reduced to under 4%. Thus, adjusting the working potential provides a straightforward approach to expand the scope of our procedure to include alkyl alcohols. While we did not seek to provide an extensive scope in this exploratory study, we were pleased to demonstrate the inherent adaptability of this procedure offering interesting avenues to expand the scope of electrocatalytic C–N bond formation.
Our procedure also demonstrated excellent tolerance towards various aliphatic amines (Scheme 4, 1-4), delivering moderate to good yields and high faradaic efficiencies (>64%). Even sterically hindered substrates (2, 3, and 9) were successfully converted, showcasing the versatility of our approach. Amines bearing electron-rich heterocyclic substituents such as furanyl or thiophenyl groups exhibited outstanding performance (5 and 6), with yields exceeding 75% and faradaic efficiencies exceeding 80%. Aniline proved less suitable due to parasitic polymerization, which led to high conversion of the starting material and low product yield (23%, 7, see ESI Table S8† for other non-successful substrates). Benzylic amines emerged as excellent coupling partners, as both electron-donating and electron-withdrawing groups in the para position were well-tolerated, leading to quantitative yields and faradaic efficiencies (8–14). Remarkably, amines bearing substituents in the ortho position of the phenyl group proved to be potent coupling partners (15–18), affording nearly quantitative yields and faradaic efficiencies. To further demonstrate the utility of our molecular EAO with C–N bond formation, we explored the synthesis of diimine motifs. Starting from the corresponding diamine, we successfully obtained 1,2-diimine (19), 1,3-diimine (20), and 1,4-diimine (21) with good yields (>53%) and high faradaic efficiencies (>64%). Additionally, we investigated the involvement of radical/HAT pathways by employing a radical-clock substrate. Notably, the exclusive formation of cyclopropyl imine (4) without detection of the ring-opened species indicated the absence of nitrogen-centred radicals/HAT pathways during electrolysis, underscoring the significance of catalytic systems operating at low potentials (see ESI Section 7†). These results collectively highlight the broad applicability and exciting potential of our procedure in facilitating efficient electrocatalytic C–N bond formation. Under our standard conditions, the Ru-1 catalyst demonstrates full conversion of the 65 mM amine starting material, as indicated by the accumulation of unreacted benzaldehyde (see Scheme 2). This suggests that the catalyst retains its activity even after the consumption of the limiting amine substrate. To investigate the longevity of Ru-1 under these conditions, we introduced additional equivalents of the amine substrate after passing 2F of charge. Interestingly, the production of the imine product continued, indicating that the catalyst remained active. After passing 4F of charge, catalyst deactivation became apparent (Scheme 5A). Despite this deactivation, we were able to achieve the formation of over 70 mmol of imine and a turnover number close to 90, demonstrating the reasonable stability of Ru-1. These results are particularly noteworthy when compared to those of other molecular electrocatalytic alcohol oxidation (EAO) systems based on transition metal catalysts, such as a diamine iridium complex (TON = 32),31 a cobalt–triphos complex (TON = 20)33 or a tan iridium PNP pincer complex (TON = up to 8).29 Our findings highlight the potential of our electrocatalytic approach for synthetic applications in alcohol oxidation. Electrifying thermal catalysis offers several advantages, including increased energy efficiency by controlling the energy input and providing excellent temporal and spatial control over reaction progress. To showcase these benefits, we conducted a standard controlled-potential electrolysis (CPE) run, where we analyzed the formation of the imine product at hourly intervals, followed by a 30-minute switch to open circuit potential (OCP) (Scheme 5B). Remarkably, imine formation ceased upon stopping the applied potential and was restored upon switching back to the working potential. This demonstrates the exceptional temporal control of the reaction under electrochemical conditions. Furthermore, we observed that interrupting the run did not affect the selectivity, as the reactivity stalled at room temperature in the absence of applied potential. This precise control over the reaction progression under electrochemical conditions opens up exciting possibilities for selective tandem reactions in the future. Building upon the demonstrated stability of Ru-1 during imine formation, we investigated the possible resting states and the fate of the catalyst after CPE. By extracting the crude reaction mixture with CDCl3, we obtained NMR resonances that closely matched those of independently synthesized samples of the benzyloxo complex Ru-OBn (ref. 57) and the hydroxo complex Ru-OH (ref. 58) (Scheme 5C). This suggests that under the employed conditions, Ru-OBn serves as the resting state of the catalyst. This finding aligns with previous experimental (ref. 59) observations and theoretical60 investigations of similar systems for thermal alcohol activation and dehydrogenation in basic media.
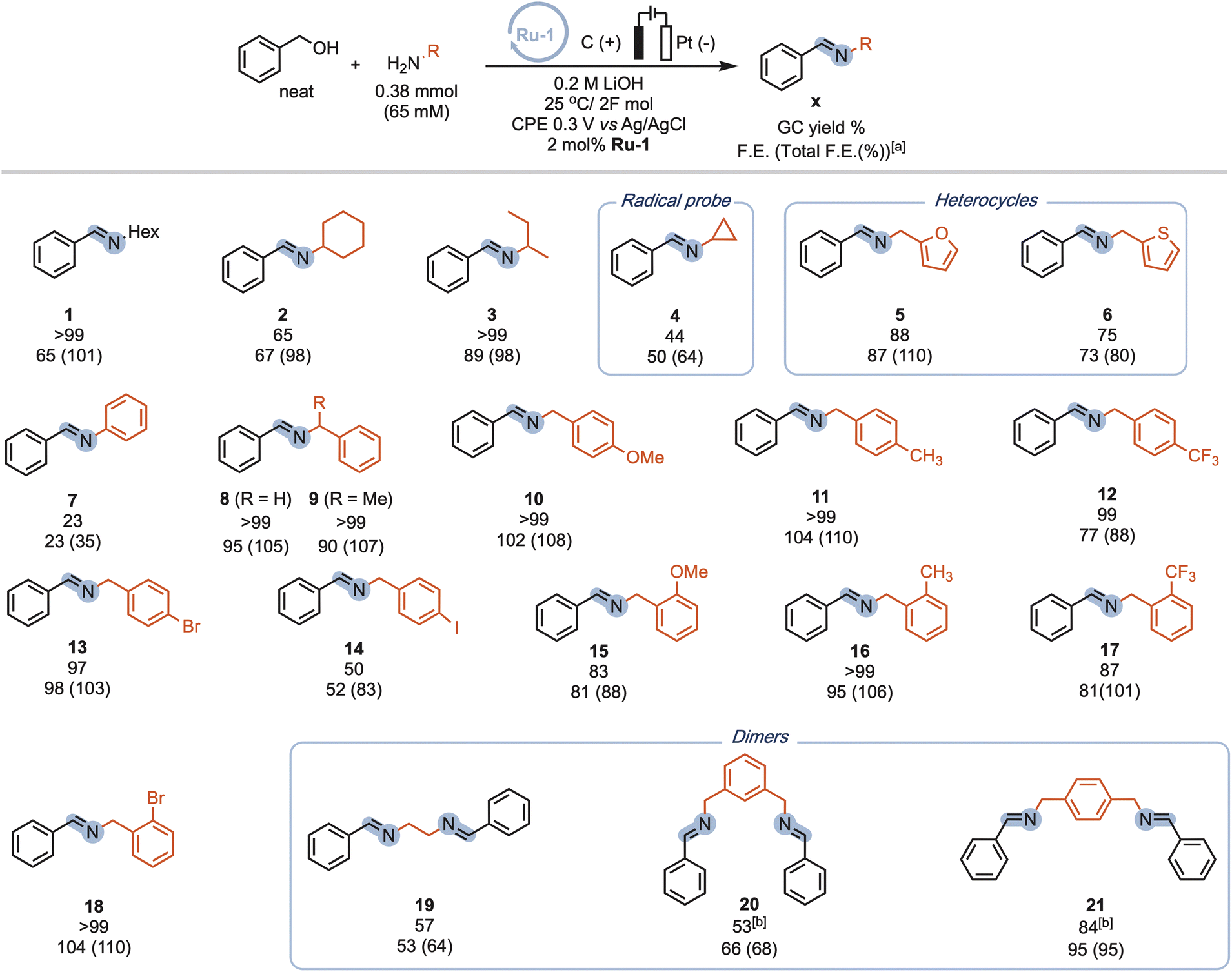 | ||
| Scheme 4 Scope of amine coupling partners used for the electrocatalytic imine formation. Reactions are stopped after 2F per amine (to account for differences in electrode/cell dimensions), which is in general well below 6 hours. The initial amine concentration is adjusted for benzyl alcohol auto-oxidation (see ESI Section 3†). [a]The total F.E. includes electrochemically formed aldehyde and ester. [b] NMR yield. | ||
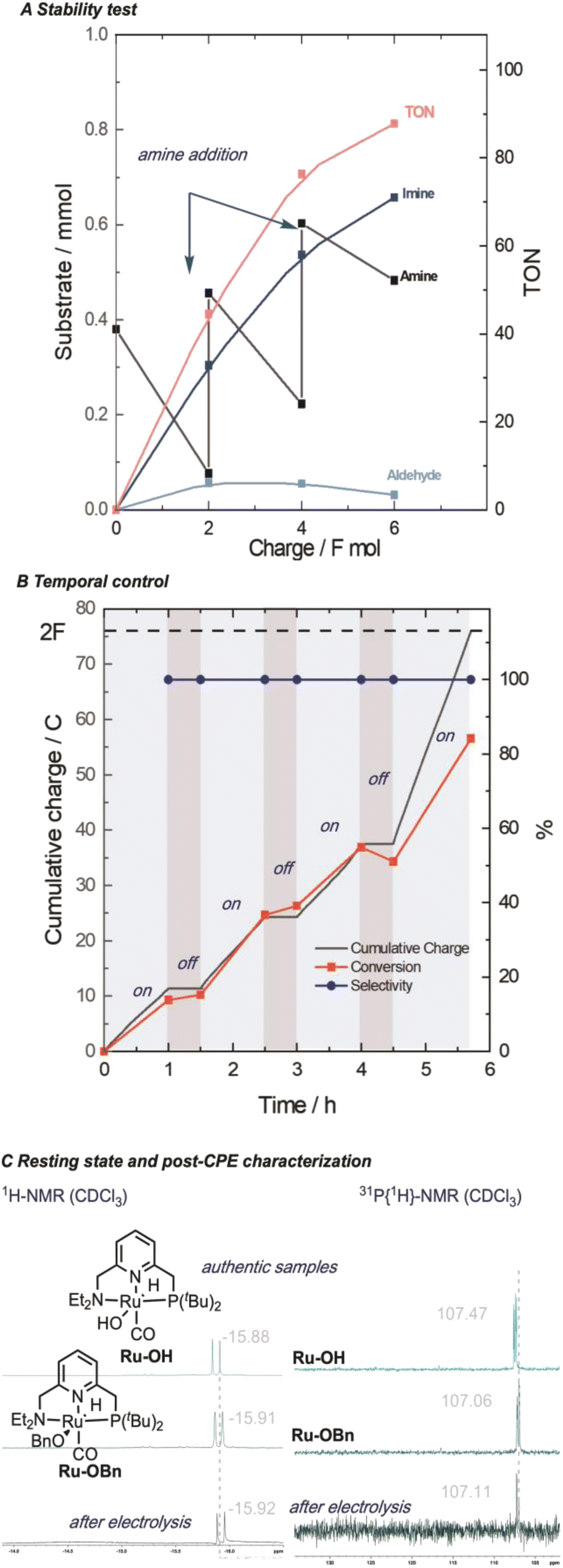 | ||
| Scheme 5 (A) Stability and (B) potential control experiments for electrocatalytic alcohol oxidation to imines by Ru-1. (C) Resting state/post-electrolysis characterization (1H and 31P) of the catalytic species. | ||
While our proof-of-concept study demonstrates the broad applicability of electrocatalytic alcohol oxidation (EAO) for C–N bond formation, the use of a large excess of alcohol substrate can be a limiting factor for practical applications. However, we are confident that by optimizing the reactor design, specifically the electrode surface-to-volume ratio, the excess alcohol can be significantly reduced. We nevertheless sought to prove that even with a simple home-made cell, excess alcohol can be significantly reduced. By repeatedly adding the amine substrate and catalyst after passing 2F of charge, we were able to isolate the imine product 1 on a sub-gram scale, resulting in a reduced excess alcohol ratio of approximately 4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 (300 mg, 33% isolated yield). Furthermore, we successfully recovered over 74% of the excess alcohol for reuse (Scheme 6A). These results indicate that even with our current experimental setup, the excess alcohol can be effectively reduced. Additionally, we demonstrated that under the standard conditions with a large excess of alcohol, the imine product can be efficiently separated from the excess alcohol and the alcohol can be recovered in an excellent yield of 92% (Scheme 6B). These preliminary findings highlight the potential scalability of EAO approaches, which would greatly benefit from the optimization of cell designs in future studies.
:1 (300 mg, 33% isolated yield). Furthermore, we successfully recovered over 74% of the excess alcohol for reuse (Scheme 6A). These results indicate that even with our current experimental setup, the excess alcohol can be effectively reduced. Additionally, we demonstrated that under the standard conditions with a large excess of alcohol, the imine product can be efficiently separated from the excess alcohol and the alcohol can be recovered in an excellent yield of 92% (Scheme 6B). These preliminary findings highlight the potential scalability of EAO approaches, which would greatly benefit from the optimization of cell designs in future studies.
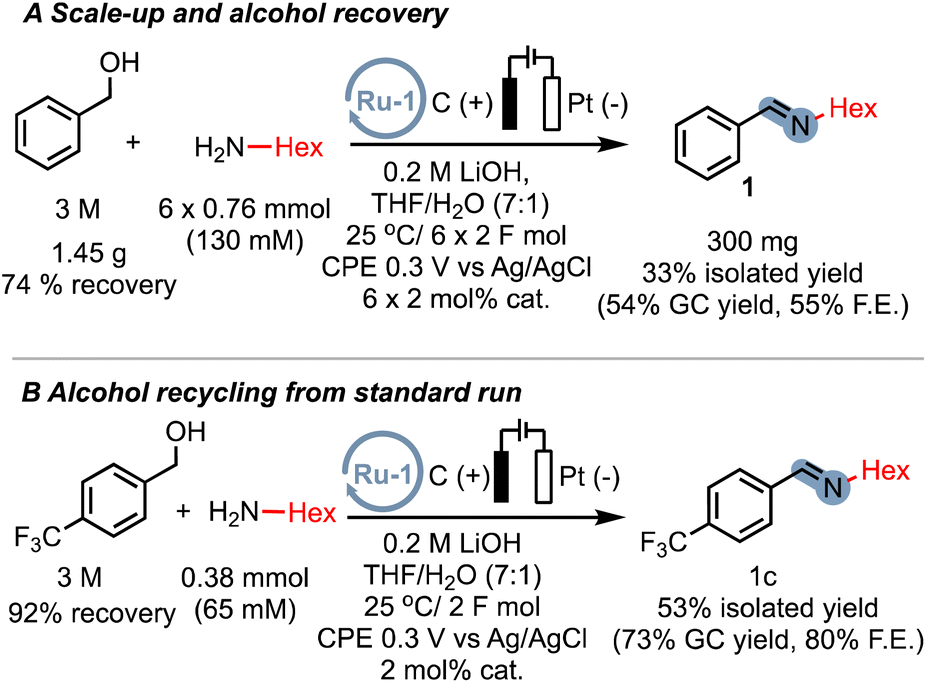 | ||
| Scheme 6 (A) Successive addition of both amine and catalyst equivalents for multi-mmol scale synthesis with starting material recovery. (B) Alcohol recovery experiment from a standard run. | ||
To provide a rational explanation for the observed reactivity of the ruthenium (de)hydrogenation catalysts employed in our study, we have developed putative catalytic cycles based on several key observations. These observations are as follows: (1) we propose that the initial step in the catalytic cycle involves the deprotonation of the alcohol substrate. This step is crucial for achieving selectivity in favour of amine oxidation, as alkyl alcohols with lower acidity exhibit increased levels of amine oxidation side-products. Deprotonation of the alcohol is consistent with our experimental findings regarding the reactivity of alkyl alcohols and provides a thermodynamically and kinetically favourable pathway for subsequent reactions. (2) Resting states: post-electrolysis NMR studies have indicated that hydroxo- and alkoxo–ruthenium complexes could be potential resting states of the catalysts. (3) Hydride transfer: we propose that hydride transfer from the alcohol to the catalyst proceeds through the alcoholate species. Alcoholates have been shown to be significantly better hydride donors compared to their corresponding alcohols.61 This hypothesis is also supported by theoretical studies on thermal activated alcohol dehydrogenation, where ion-pairing transition states are often proposed.60,62,63 (4) Oxidation of hydride intermediate: the oxidation of the hydride intermediate is expected to be readily feasible at the applied potential as shown in our previous study50 and could serve as thermodynamic driving force to regenerate active species.64 These observations can be rationalized by the putative catalytic cycle proposed in Scheme 7 (see also ESI Section 8†).
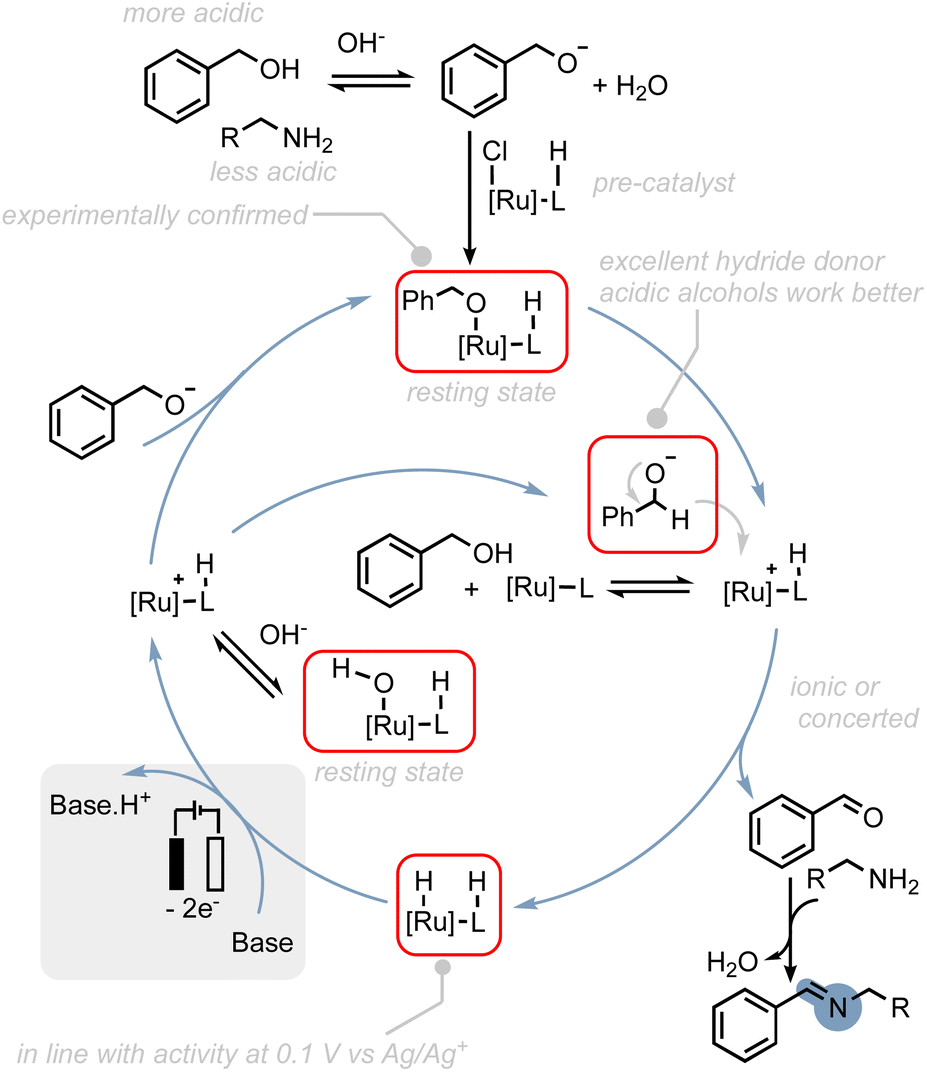 | ||
| Scheme 7 Possible reaction mechanism for the electrocatalytic alcohol oxidation with common ruthenium-based MLC catalysts. | ||
We believe that the electrification of thermal processes holds significant potential and can offer important benefits in the future. One of the key advantages of electrochemical approaches is their ability to achieve the desired transformations with significantly reduced time and temperature compared to traditional thermal conditions. In the case of imine bond formation, the conventional thermal process typically requires prolonged reaction times (>24 hours) and high temperatures (>110 °C).51 By contrast, our electrochemical approach allows for faster reaction kinetics and can be conducted at lower temperatures. We believe that energy efficiency of electrochemical approaches could be a crucial advantage. A rough estimation suggests that the energy efficiency of electrochemical methods for C–N bond formation can be an order of magnitude higher than that of traditional thermal methods (see ESI Section 9†). Electrifying thermal processes could thus help achieve higher energy efficiency in the chemical sector. While this study represents an initial step towards this goal, further research and development are needed to overcome the current limitations and expand its applicability on a larger scale. The optimization of reactor designs, catalyst systems, and process conditions will be crucial in harnessing the full potential of electrochemical approaches for C–N bond formation and other transformations.
Conclusions
In summary, this study represents the first example of molecular EAO for C–N bond formation. Smooth reaction conditions (room temperature, below 6 h) compare well to thermal conditions (>110 °C, >24 h), demonstrating the usefulness of this electrochemical approach. Over 30 imine products were obtained and long-term stability tests showed that TON close to 90 can be obtained for this molecular system. Furthermore, we demonstrated the excellent temporal control of the system by switching between working and open circuit potential (OCP), illustrating the unique opportunities presented by electrified organic redox chemistry. This temporal control opens up avenues for selective tandem reactions and the design of more complex reaction sequences. We demonstrate the scalability of the process, as well as the possibility of recycling excess alcohol. Building upon the success of this study, we are actively expanding the scope of molecular EAO, exploring new catalyst systems, and investigating the potential for other types of bond formations. We hope that this work will serve as a foundation for further developments at the interface of homogeneous catalysis and electrochemistry, driving innovation in sustainable synthetic methodologies.Author contributions
SK was responsible for the experimental investigation, writing of the original draft and optimizing the methodology. NVW was responsible for funding acquisition, conceptualization, supervision, and writing and reviewing the original draft. PG performed the HR-MS analysis.Conflicts of interest
A patent on the presented chemistry was deposited.Acknowledgements
The authors are grateful for a generous donation of the PNNH-ligand from the Milstein group. The IdEx of the Université Paris Cité is acknowledged for funding (ANR-18-IDEX-0001), as well as technology transfer agency ERGANEO for a maturation grant.References
- Chemicals – Analysis, https://www.iea.org/reports/chemicals, accessed November 24, 2022.
- J. E. Nutting, J. B. Gerken, A. G. Stamoulis, D. L. Bruns and S. S. Stahl, J. Org. Chem., 2021, 86, 15875–15885 CrossRef CAS PubMed.
- X. Dong, J. L. Roeckl, S. R. Waldvogel and B. Morandi, Science, 2021, 371, 507 CrossRef CAS PubMed.
- Y. Yuan and A. Lei, Nat. Commun., 2020, 11, 802 CrossRef CAS PubMed.
- M. Yan, Y. Kawamata and P. S. Baran, Chem. Rev., 2017, 117, 13230–13319 CrossRef CAS PubMed.
- C. Kingston, M. D. Palkowitz, Y. Takahira, J. C. Vantourout, B. K. Peters, Y. Kawamata and P. S. Baran, Acc. Chem. Res., 2020, 53, 72–83 CrossRef CAS PubMed.
- T. H. Meyer, I. Choi, C. Tian and L. Ackermann, Chem, 2020, 6, 2484–2496 CAS.
- B. A. Frontana-Uribe, R. D. Little, J. G. Ibanez, A. Palma and R. Vasquez-Medrano, Green Chem., 2010, 12, 2099–2119 RSC.
- B. K. Peters, K. X. Rodriguez, S. H. Reisberg, S. B. Beil, D. P. Hickey, Y. Kawamata, M. Collins, J. Starr, L. Chen, S. Udyavara, K. Klunder, T. J. Gorey, S. L. Anderson, M. Neurock, S. D. Minteer and P. S. Baran, Science, 2019, 363, 838 CrossRef CAS PubMed.
- J. Na, B. Seo, J. Kim, C. W. Lee, H. Lee, Y. J. Hwang, B. K. Min, D. K. Lee, H.-S. Oh and U. Lee, Nat. Commun., 2019, 10, 5193 CrossRef PubMed.
- S. Verma, S. Lu and P. J. A. Kenis, Nat. Energy, 2019, 4, 466–474 CrossRef CAS.
- A. W. Cook and K. M. Waldie, ACS Appl. Energy Mater., 2020, 3, 38–46 CrossRef CAS.
- N. von Wolff, O. Rivada-Wheelaghan and D. Tocqueville, ChemElectroChem, 2021, 8, 4019–4027 CrossRef CAS.
- M. Bellini, M. Bevilacqua, A. Marchionni, H. A. Miller, J. Filippi, H. Grützmacher and F. Vizza, Eur. J. Inorg. Chem., 2018, 2018, 4393–4412 CrossRef CAS.
- J. E. Nutting, M. Rafiee and S. S. Stahl, Chem. Rev., 2018, 118, 4834–4885 CrossRef CAS PubMed.
- M. Rafiee, M. Alherech, S. D. Karlen and S. S. Stahl, J. Am. Chem. Soc., 2019, 141, 15266–15276 CrossRef CAS PubMed.
- M. Rafiee, Z. M. Konz, M. D. Graaf, H. F. Koolman and S. S. Stahl, ACS Catal., 2018, 8, 6738–6744 CrossRef CAS.
- M. Rafiee, K. C. Miles and S. S. Stahl, J. Am. Chem. Soc., 2015, 137, 14751–14757 CrossRef CAS PubMed.
- M. C. Ryan, L. D. Whitmire, S. D. McCann and S. S. Stahl, Inorg. Chem., 2019, 58, 10194–10200 CrossRef CAS PubMed.
- A. C. Cardiel, B. J. Taitt and K.-S. Choi, ACS Sustainable Chem. Eng., 2019, 7, 11138–11149 CrossRef CAS.
- A. Paul, J. F. Hull, M. R. Norris, Z. Chen, D. H. Ess, J. J. Concepcion and T. J. Meyer, Inorg. Chem., 2011, 50, 1167–1169 CrossRef CAS PubMed.
- A. K. Vannucci, J. F. Hull, Z. Chen, R. A. Binstead, J. J. Concepcion and T. J. Meyer, J. Am. Chem. Soc., 2012, 134, 3972–3975 CrossRef CAS PubMed.
- H. Kuiry, D. Das, S. Das, S. Chakraborty, B. Chandra and S. S. Gupta, Faraday Discuss., 2022, 234, 42–57 RSC.
- A. Das, J. E. Nutting and S. S. Stahl, Chem. Sci., 2019, 10, 7542–7548 RSC.
- S. P. Annen, V. Bambagioni, M. Bevilacqua, J. Filippi, A. Marchionni, W. Oberhauser, H. Schönberg, F. Vizza, C. Bianchini and H. Grützmacher, Angew. Chem., Int. Ed., 2010, 49, 7229–7233 CrossRef CAS PubMed.
- M. Bellini, M. Bevilacqua, J. Filippi, A. Lavacchi, A. Marchionni, H. A. Miller, W. Oberhauser, F. Vizza, S. P. Annen and H. Grützmacher, ChemSusChem, 2014, 7, 2432–2435 CrossRef CAS PubMed.
- J. Lybaert, S. Trashin, B. U. W. Maes, K. De Wael and K. Abbaspour Tehrani, Adv. Synth. Catal., 2017, 359, 919–925 CrossRef CAS.
- K. R. Brownell, C. C. L. McCrory, C. E. D. Chidsey, R. H. Perry, R. N. Zare and R. M. Waymouth, J. Am. Chem. Soc., 2013, 135, 14299–14305 CrossRef CAS PubMed.
- C. M. Galvin and R. M. Waymouth, J. Am. Chem. Soc., 2020, 142, 19368–19378 CrossRef CAS PubMed.
- K. M. Waldie, K. R. Flajslik, E. McLoughlin, C. E. D. Chidsey and R. M. Waymouth, J. Am. Chem. Soc., 2017, 139, 738–748 CrossRef CAS PubMed.
- P. J. Bonitatibus, M. P. Rainka, A. J. Peters, D. L. Simone and M. D. Doherty, Chem. Commun., 2013, 49, 10581–10583 RSC.
- C. J. Weiss, E. S. Wiedner, J. A. S. Roberts and A. M. Appel, Chem. Commun., 2015, 51, 6172–6174 RSC.
- S. P. Heins, P. E. Schneider, A. L. Speelman, S. Hammes-Schiffer and A. M. Appel, ACS Catal., 2021, 11, 6384–6389 CrossRef CAS.
- E. A. McLoughlin, B. D. Matson, R. Sarangi and R. M. Waymouth, Inorg. Chem., 2020, 59, 1453–1460 CrossRef CAS PubMed.
- J. E. Kim, S. Choi, M. Balamurugan, J. H. Jang and K. T. Nam, Trends Chem., 2020, 2, 1004–1019 CrossRef CAS.
- J. Li, Y. Zhang, K. Kuruvinashetti and N. Kornienko, Nat. Rev. Chem, 2022, 6, 303–319 CrossRef CAS PubMed.
- C. Guo, W. Zhou, X. Lan, Y. Wang, T. Li, S. Han, Y. Yu and B. Zhang, J. Am. Chem. Soc., 2022, 144, 16006–16011 CrossRef CAS PubMed.
- Z. Mei, Y. Zhou, W. Lv, S. Tong, X. Yang, L. Chen and N. Zhang, ACS Sustainable Chem. Eng., 2022, 10, 12477–12496 CrossRef CAS.
- J. Li and N. Kornienko, Chem. Sci., 2022, 13, 3957–3964 RSC.
- Z. Tao, C. L. Rooney, Y. Liang and H. Wang, J. Am. Chem. Soc., 2021, 143, 19630–19642 CrossRef CAS PubMed.
- C. L. Rooney, Y. Wu, Z. Tao and H. Wang, J. Am. Chem. Soc., 2021, 143, 19983–19991 CrossRef CAS PubMed.
- C. Lv, J. Liu, C. Lee, Q. Zhu, J. Xu, H. Pan, C. Xue and Q. Yan, ACS Nano, 2022, 16, 15512–15527 CrossRef CAS PubMed.
- G.-L. Yang, C.-T. Hsieh, Y.-S. Ho, T.-C. Kuo, Y. Kwon, Q. Lu and M.-J. Cheng, ACS Catal., 2022, 12, 11494–11504 CrossRef CAS.
- L. Chen, C. Tang, Y. Zheng, K. K. Ostrikov and Y. Jiao, Energy Fuels, 2022, 36, 7213–7218 CrossRef CAS.
- J. Shao, N. Meng, Y. Wang, B. Zhang, K. Yang, C. Liu, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2022, 134, e202213009 CrossRef.
- N. Meng, J. Shao, H. Li, Y. Wang, X. Fu, C. Liu, Y. Yu and B. Zhang, Nat. Commun., 2022, 13, 5452 CrossRef CAS PubMed.
- J. R. Khusnutdinova and D. Milstein, Angew. Chem., Int. Ed., 2015, 54, 12236–12273 CrossRef CAS PubMed.
- F. Wang and S. S. Stahl, Acc. Chem. Res., 2020, 53, 561–574 CrossRef CAS PubMed.
- J. P. Adams, J. Chem. Soc., Perkin Trans., 2000, 1, 125–139 RSC.
- D. Tocqueville, F. Crisanti, J. Guerrero, E. Nubret, M. Robert, D. Milstein and N. von Wolff, Chem. Sci., 2022, 13, 13220–13224 RSC.
- B. Gnanaprakasam, J. Zhang and D. Milstein, Angew. Chem., Int. Ed., 2010, 49, 1468–1471 CrossRef CAS PubMed.
- D. A. Stevens and J. R. Dahn, J. Electrochem. Soc., 2001, 148, A803 CrossRef CAS.
- H. Roth, N. Romero and D. Nicewicz, Synlett, 2015, 27, 714–723 CrossRef.
- E. Fogler, J. A. Garg, P. Hu, G. Leitus, L. J. W. Shimon and D. Milstein, Chem.—Eur. J., 2014, 20, 15727–15731 CrossRef CAS PubMed.
- D. G. Gusev, ACS Catal., 2016, 6, 6967–6981 CrossRef CAS.
- D. Spasyuk, S. Smith and D. G. Gusev, Angew. Chem., Int. Ed., 2013, 52, 2538–2542 CrossRef CAS PubMed.
- C. A. Huff, J. W. Kampf and M. S. Sanford, Chem. Commun., 2013, 49, 7147–7149 RSC.
- S. W. Kohl, L. Weiner, L. Schwartsburd, L. Konstantinovski, L. J. W. Shimon, Y. Ben-David, M. A. Iron and D. Milstein, Science, 2009, 324, 74–77 CrossRef CAS PubMed.
- Y.-Q. Zou, N. von Wolff, M. Rauch, M. Feller, Q.-Q. Zhou, A. Anaby, Y. Diskin-Posner, L. J. W. Shimon, L. Avram, Y. Ben-David and D. Milstein, Chem. – Eur. J., 2021, 27, 4715–4722 CrossRef CAS PubMed.
- P. A. Dub and J. C. Gordon, ACS Catal., 2017, 7, 6635–6655 CrossRef CAS.
- C. Amatore, J. Badoz-Lambling, C. Bonnel-Huyghes, J. Pinson, J. M. Saveant and A. Thiebault, J. Am. Chem. Soc., 1982, 104, 1979–1986 CrossRef CAS.
- N. Govindarajan, V. Sinha, M. Trincado, H. Grützmacher, E. J. Meijer and B. de Bruin, ChemCatChem, 2020, 12, 2610–2621 CrossRef CAS.
- V. Sinha, N. Govindarajan, B. de Bruin and E. J. Meijer, ACS Catal., 2018, 8, 6908–6913 CrossRef CAS PubMed.
- C. L. Mathis, J. Geary, Y. Ardon, M. S. Reese, M. A. Philliber, R. T. VanderLinden and C. T. Saouma, J. Am. Chem. Soc., 2019, 141, 14317–14328 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Calculation of F.E., reaction optimization, thermochemical calculations, CV studies, and GC-MS and NMR data. See DOI: https://doi.org/10.1039/d3sc03408a |
| This journal is © The Royal Society of Chemistry 2023 |