
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceModel reactions for the evaluation of poly- and multifunctional molecules as potential interfacial agents for the compatibilization of polyethylene/poly(ethylene-co-vinyl alcohol) blends†
Elisa
Gitzhofer
a,
Bertrand
Vileno
b,
Michel
Bouquey
a and
Delphine
Chan-Seng
 *a
*a
aUniversité de Strasbourg, CNRS, Institut Charles Sadron UPR 22, F-67000 Strasbourg, France. E-mail: delphine.chan-seng@ics-cnrs.unistra.fr
bUniversité de Strasbourg, CNRS, Institut de Chimie de Strasbourg UMR 7177, F-67000 Strasbourg, France
First published on 23rd January 2023
Abstract
In aiming to identify an interfacial agent for polyethylene (PE)/poly(ethylene-co-vinyl alcohol) (EVOH) blends, which are the main components found in flexible food packaging, triallyl isocyanurate and a molecule bearing two (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) groups were initially investigated with model molecules of targeted polymers, but the results were only moderate. The design of a suitable interfacial agent led to 4-glycidyl-2,2,6,6-tetramethylpiperidin-1-oxyl, which demonstrated covalent coupling of the TEMPO group with the model molecule of PE and the glycidyl group with the hydroxyl group of the model molecule of EVOH. This approach with model molecules could be a way to screen molecules that could have potential as interfacial agents to compatibilize polymers from materials that need to be upcycled.
Introduction
The protection of the environment and the need to save planetary resources have become major issues for our society in recent years.1 Plastics do not appear in this context as materials of the future and have been blamed as very polluting due to their poor biodegradability and, in most cases, their exhaustive resources. However, their production continues to increase doubling between 2000 and 20192 and exceeding in 2022 the threshold of 450 Mt per year. Their properties such as, light weight, thermal and electrical insulation, impermeability to gases and liquids, and ease of transformation into different shapes and volumes, designate them as materials with unequalled levels of performance in all fields rendering unrealistic the consideration of stopping their production. A more adaptable solution would be to develop strategies to better collect plastics after use (22 Mt being disposed of in the environment), facilitate their sorting, and validate them by different recycling approaches to satisfy the new model of a circular economy of production as it is currently estimated that only 9% of plastic waste is recycled worldwide.3The packaging sector is by far the largest consumer of plastics in the world with a need close to 160 Mt annually with the manufacture mainly of single-use packaging. Many packages are produced for the food industry because of their barrier properties, which guarantee an extended shelf life for products to be consumed. To preserve fresh products, it is necessary to design multi-layered flexible packaging and therefore this comprises multiple materials to satisfy all the constraints of the functional specifications, in particular those concerning the oxygen and water tightness of the packaging. The superimposition of different plastic films of a few microns in thickness, each one linked together by a layer of polymer adhesive, constitutes an indissociable complex that is a major obstacle to the recycling of this packaging. Chemical recycling, currently very popular,4 is not sufficiently selective to separate the different polymers present, while mechanical recycling,5 the most obvious to develop (economically and technologically) for thermoplastic polymeric materials, generates heterophased materials with non-stabilized interfaces leading to, for example, a decrease in macroscopic mechanical properties.6 These products are, therefore, generally incinerated when possible or buried more conventionally. To meet this challenge and thus be able to offer a credible recycling alternative to these plastics, the use of an agent able to stabilize the interface between the phases of the polymer blends has been considered using copolymers (block, graft, and random copolymers), fillers (e.g. carbon black, glass fibers, carbon nanotubes, and inorganic particles), and Janus particles.7 A new method of recycling polymeric materials in a mixture has been developed to be complementary to the existing traditional processes (mechanical,5,8 chemical,9,10 or thermal11 recycling). This concept called mechanical–chemical recycling (MCR) consists of synthesizing in situ, i.e. in the molten state of the polymer mixture, compatibilizing agents to stabilize the different phases that are present. This method has been developed since the late 1980s under the impulse of plastics producers who wished to complete their range of materials by the elaboration of polymer blends necessitating its deployment on a large scale. The first examples have involved branched- and ideally block-copolymer compatibilizing agents.12–14 Not too many reactions are suitable for the formation of the copolymer because they have to meet strict requirements, such as fast kinetics, temperature resistance without degradation or side reactions, and irreversibility. In this context, Orr et al. have compared the kinetics of different coupling reactions between targeted chemical functions.15 It appears from this work that each polymer blend requires a specific adaptation of the reactive system to satisfy the compatibility criteria, often with a prior synthesis of monofunctional macromolecular chains. Their approach consisted of exploiting the only macromolecular chains available in the polymer blend to develop the compatibilization copolymers. For that, the concept foresaw a preliminary synthesis of a universal agent called the recombination agent whose role was to be able to associate two macromolecular chains of different chemical natures and which could not carry reactive chemical functions. Peroxides16–18 have been commonly investigated as recombination agents for a wide range of polymer blends to improve interfacial adhesion. Upon heating, the peroxide will produce primary radicals able to abstract hydrogens on polymer chains. This radical formed on a polymer chain could react with a radical present on another polymer chain to create bridges between polymer chains. The use of maleic anhydride19,20 in the presence of a thermal initiator has been also considered to graft in situ maleic anhydrides on polymer chains, while the succinic anhydride formed could react with functional groups present on the polymer of the blend. Existing maleic anhydride-grafted polymers (e.g. polyolefins, aliphatic polyesters, copolymers) have also been used as compatibilizing agents by adding them to a polymer blend.21–24 Other strategies in reactive compatibilization have been reported and include the use of silane-, isocyanate-, oxazoline-, epoxide-, and phosphite-based coupling agents to induce interfacial stability in polymer blends.7
The cohesion of the stacking into multilayered films of a few micrometers in thickness is ensured by an adhesive deposited between each layer such as OREVAC®, a unique range of functionalized polymers with maleic anhydride functions. These functions can be advantageously used to react with the alcohol functions of EVOH25,26 during the melt blending of the multilayer flexible food packaging, thus allowing the in situ development of copolymers adapted to the compatibility of the blend.27 As most food packaging does not benefit from these functionalized copolymers,28 it is essential to develop an alternative method to the one described previously with the use of a recombination agent. In order to evaluate the relevance of this method, we chose to work with the same grades of polymeric materials used for the concoction of films intended for flexible packaging. With the aim to treat flexible plastic food packaging structured in alternating layers of apolar non-functionalized polyolefins (polyethylene, PE or polypropylene, PP) and vinyl alcohol-based copolymers (EVOH),29 we decided in this study to investigate model molecules of PE and EVOH to identify suitable compatibilizing agents for this polymer blend. The reactions were conducted at high temperature in the presence of a radical initiator to induce proton abstractions on the polymer chains. These macroradicals were expected to react with molecules that would link two polymer chains, preferably of different natures. Various molecules were investigated from commercially available triallyl isocyanurate (TAIC) to designed molecules bearing one (2,2,6,6-tetramethylpiperidin-1-yl)oxyl (TEMPO) group and one glycidyl function to promote simultaneously specific reactions with each polymer. The formed products were investigated by 1H NMR spectroscopy, mass spectrometry, electron paramagnetic resonance (EPR), and capillary viscometry to evaluate the potential of these molecules to act as compatibilizing agents for PE/EVOH blends.
Results and discussion
Triallyl isocyanurate as a coupling agent
Triallyl isocyanurate (TAIC) is commonly used upon irradiation or thermal treatment to improve the heat resistance and mechanical properties of polymers by crosslinking polymers,30–32 to compensate for the degradation of aliphatic polyesters during sterilization processes,33,34 and to promote in situ reactive compatibilization of polymer blends. In the latter case, only a few polymer systems have been reported, such as polyolefin/styrenic (co)polymers35,36 and polyesters.37 TAIC in the presence of a source of radicals was investigated here as a promoter of compatibilization by thermal treatment for a blend of PE and EVOH. Octadecane (C18) and 2-undecanol (C11-OH) were selected as model molecules of PE and EVOH, respectively, in order to overcome the solubility issues associated with the use of the corresponding polymers (Fig. 1a) as C18, C11-OH, and the resulting products were fully soluble in chloroform and dichloromethane at room temperature permitting the characterizations to be performed at room temperature and the identification of the most promising compatibilizing agent to be tested with polymers. Dicumyl peroxide (DCP) was chosen as the source of radicals upon heating (i.e. 1 h half-life of 135 °C). DCP heated above its 1 h half-life temperature will abstract a proton from the model molecules forming a radical on the chain that could react with one of the allyl groups present in TAIC. The investigation started with C18 using 4 mol% of TAIC and 2 mol% of DCP over a period of 15 min at 150 °C leading to the formation of a white solid that was insoluble in CDCl3 indicating that the extent of crosslinking was too high for the targeted application as the flexibility of the material would be lost. The amount of TAIC was decreased to 0.5 and 1 mol% while keeping the TAIC/DCP molar ratio identical. The reaction mixtures were cloudy, but the products were soluble in CDCl3 permitting their characterization by 1H NMR spectroscopy. The characteristic peaks of the allyl groups of TAIC at 4.49, 5.28, and 5.87 ppm disappeared indicating their complete consumption (Fig. 1b, full spectra available in ESI, Fig. S1†). The same phenomenon was observed when conducting the reaction solely with C11-OH or with an equimolar mixture of C18 and C11-OH. The latter product was further characterized by electrospray ionization mass spectrometry (ESI-MS) showing the formation of different species corresponding to the coupling of one or multiple molecules of C18 and C11-OH with TAIC (Fig. 1c, zoom available in ESI, Fig. S2†). To estimate the extent of this coupling reaction, capillary viscometry was performed using a micro-Ostwald viscometer in a thermostated bath at 25 °C by dissolving the raw product in CH2Cl2 at a concentration of 400 mg mL−1. The difference in kinematic viscosity between the reaction mixture before the reaction and after 30 min of reaction (Δη) for the equimolar mixture C18/C11-OH was 0.05 ± 0.01 mm2 s−1, suggesting an increase in molecular weight of at least a fraction of the C18 and C11-OH introduced and thus confirming the results observed by ESI-MS. The decrease of the amount of TAIC by half, while keeping the TAIC/DCP ratio constant, led to a decrease of Δη, which was expected as the number of coupling reactions will be lower. The effect of the TAIC/DCP ratio was also investigated by keeping the amount of TAIC introduced to 1 mol% and varying the amount of DCP (0.5, 1.0, and 1.5 mol%). No trend was observed, but the highest Δη was obtained for the reaction conducted with the lowest amount of DCP suggesting that the production of a higher concentration of the primary radical may not be suitable in favoring the coupling reaction between TAIC and the model molecules, and may induce a larger amount of β-hydrogen abstraction reactions, which is unavoidable under these conditions.38 However, a better understanding of the extent of this reaction was not possible due to the insolubility of longer alkyl chains and some of the inconsistencies observed in the capillary viscosity measurements.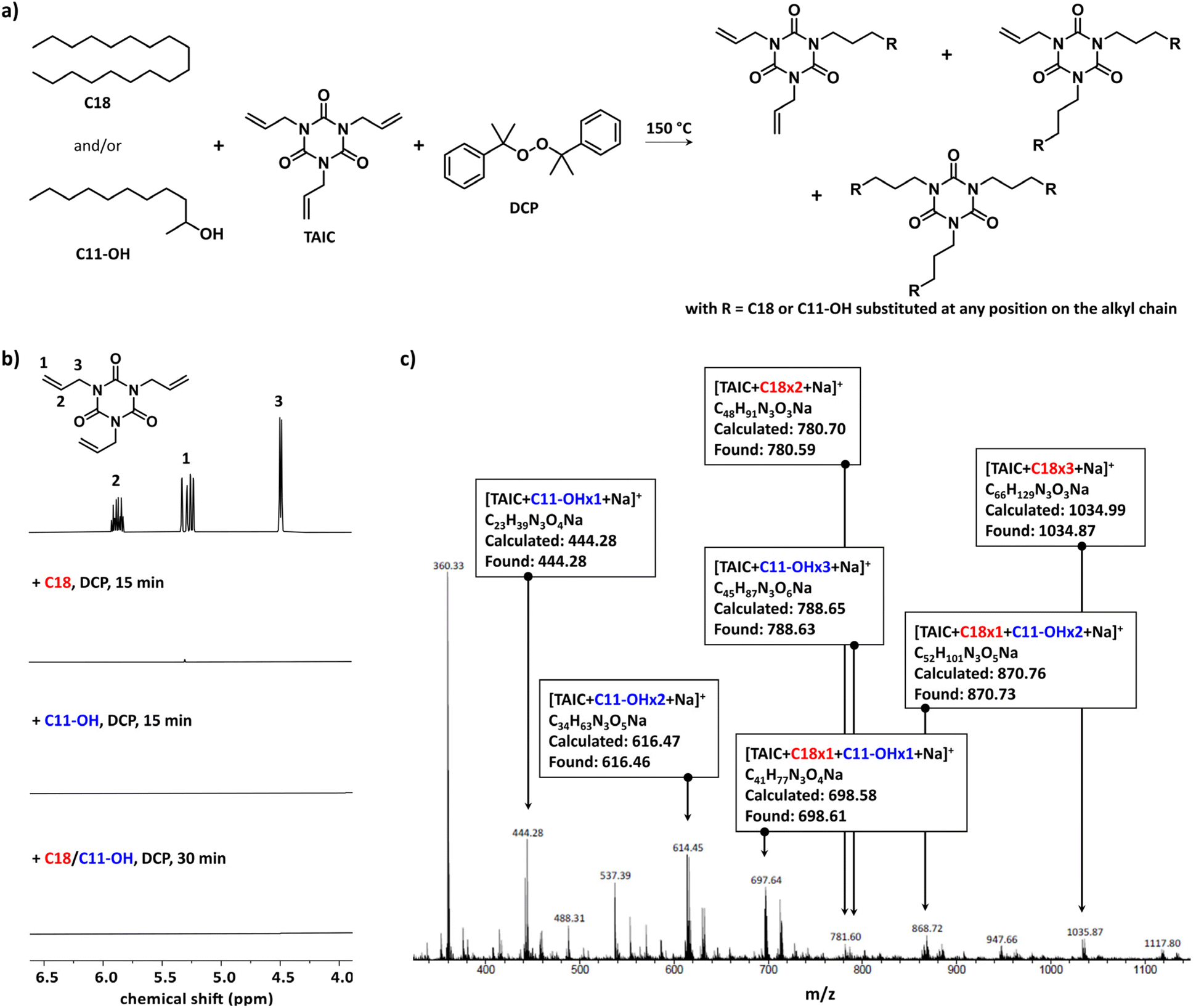 | ||
| Fig. 1 Coupling of TAIC (1 mol%) on C18 and/or C11-OH in the presence of DCP (0.5 mol%) at 150 °C: (a) reaction scheme, (b) monitoring of the consumption of the allyl group using 1H NMR spectroscopy in CDCl3 (full spectra in ESI, Fig. S1†), and (c) ESI-MS spectrum of the raw product obtained from the reaction performed on the mixture of C18 and C11-OH (1/1) (zoom on the molecular peaks and their simulation in ESI, Fig. S2†). | ||
TEMPO-based molecules as coupling agents
(2,2,6,6-Tetramethylpiperidin-1-yl)oxyl (TEMPO) is a persistent nitroxide radical that does not induce initiation, but is able to react with other radical species. This property has been exploited in the field of polymer science when using TEMPO, for example, as a controlling agent in radical polymerization,39 as a reversible redox-active moiety on polymer chains for battery applications,40 and as a scavenger of reactive oxygen species for the development of polymers with therapeutic activities.41 Nitroxide radical coupling is an attractive post-polymerization modification strategy for polymer functionalization and crosslinking.42 The main approach involves the coupling of the nitroxide with a carbon-centered macroradical formed by hydrogen abstraction on a polymer chain affording an alkoxyamine group that has been employed in peroxide-initiated functionalization of polyolefins. TEMPO has been explored by Nakatani et al. to create in situ a polystyrene-g-TEMPO that upon thermal treatment led to the formation of polystyrene-g-polypropylene acting as a compatibilizer of the polystyrene/polypropylene blends.43 Another approach has been described by Park et al. involving first, the insertion of TEMPO on PE, which was then used as a macroinitiator to polymerize styrene with a small amount of maleic anhydride affording PE-g-poly(styrene-co-maleic anhydride), with the latter being used as a compatibilizer of PE and starch blends.44 Herein, we propose developing TEMPO-based molecules as interfacial agents between PE and EVOH using model molecules.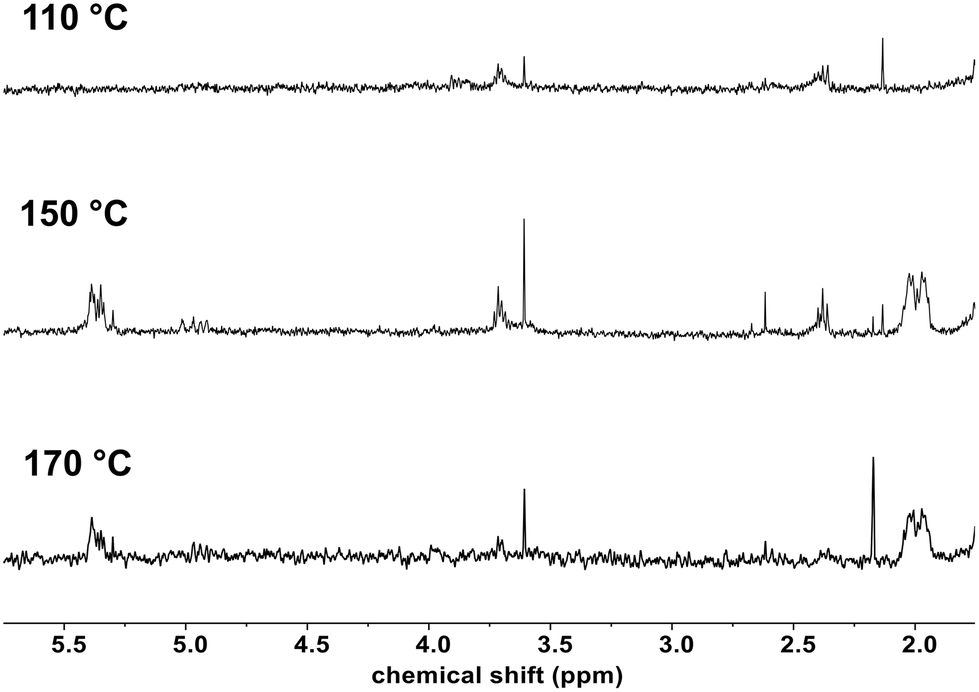 | ||
| Fig. 2 1H NMR spectra (CDCl3) of the reaction mixture of C18 with 4 mol% of TEMPO in the presence of 2 mol% of DCP at different temperatures. | ||
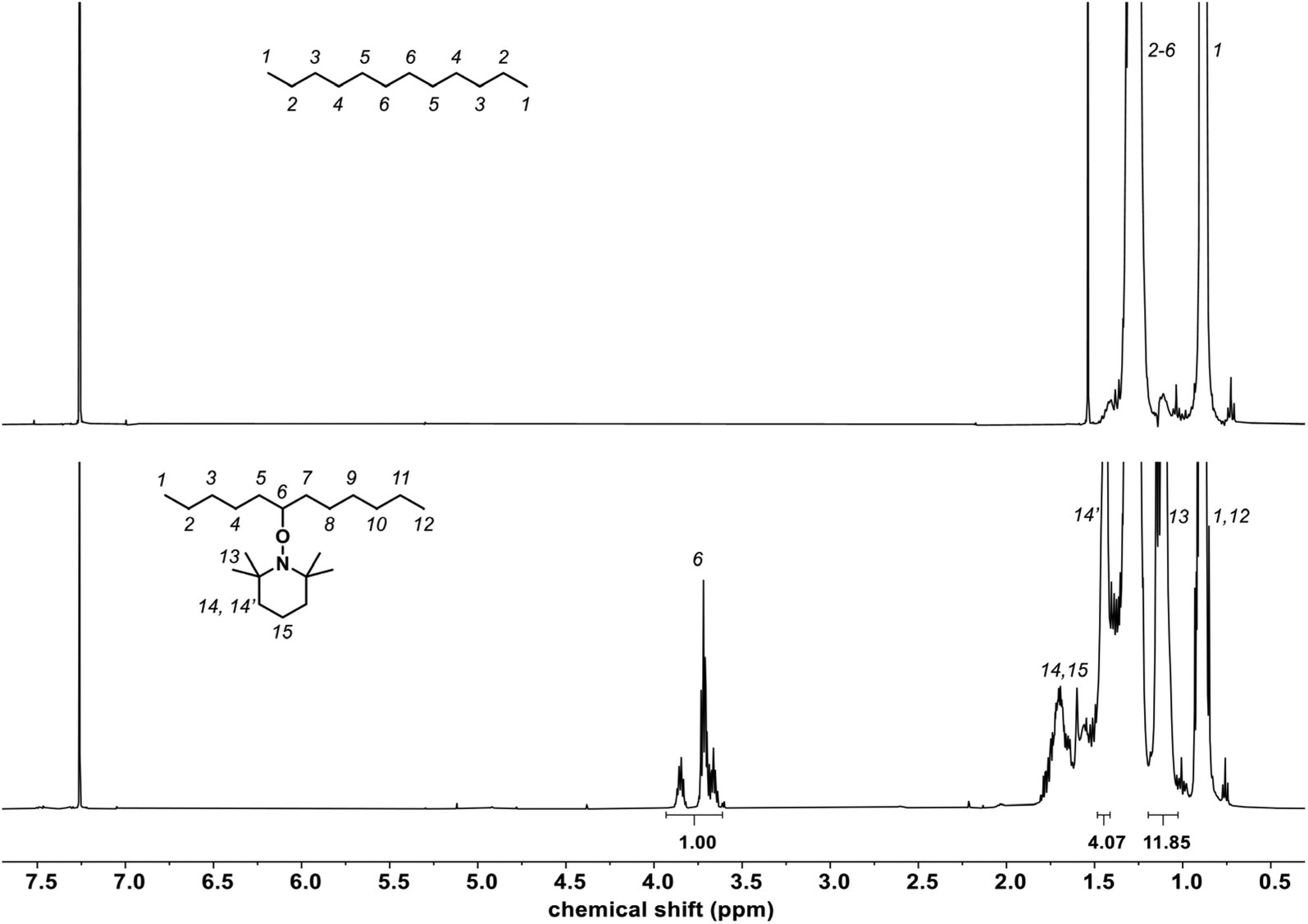 | ||
| Fig. 3 1H NMR spectra (CDCl3) of C12 (top) and TEMPO-grafted C12 (bottom) formed by the reaction of C12 with TEMPO (4 mol%) and DCP (2 mol%) at 150 °C after 19 h and concentrated by distillation. The NMR assignment of TEMPO-grafted C12 is illustrated with the TEMPO grafted in the center, but it could be on any carbon of the C12 chain. | ||
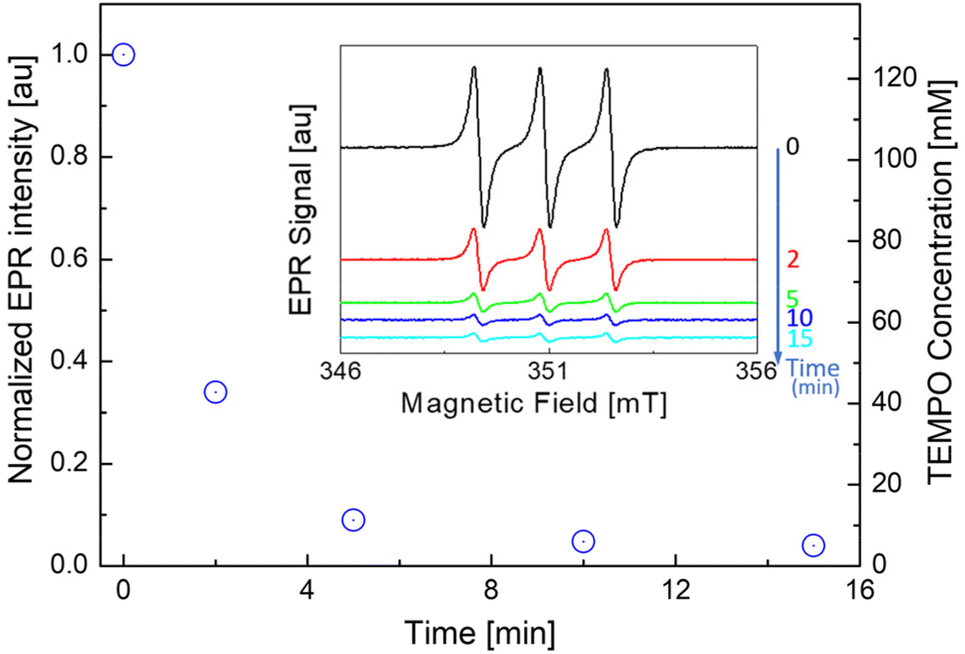 | ||
| Fig. 4 EPR monitoring of the TEMPO concentration for the reaction of C18 with 4 mol% TEMPO and 2 mol% DCP at 150 °C: TEMPO-normalized EPR intensities (I/I0 = I(t)/I(t = 0)) were obtained at t = 0, 2, 5, 10 and 15 min with corresponding EPR signals (inset). | ||
The grafting of TEMPO on C11-OH (the model molecule of EVOH) was also studied using 4 mol% TEMPO and 2 mol% DCP at 150 °C. After 1 h of reaction, the reaction medium was partially discolored. Unfortunately, the methine on which the alcohol was attached had a chemical shift at 7.8 ppm (ESI, Fig. S6†), superimposing on the area within which was expected the characteristic peak of TEMPO-grafted C11-OH to appear, rendering the monitoring of the reaction difficult. However, the formation of TEMPO-grafted C11-OH was observed by mass spectrometry (ESI, Fig. S7†).
 | ||
| Scheme 1 Synthesis route of diTEMPO (a molecule with two TEMPO groups). | ||
The grafting of diTEMPO was studied with C18 using 4 mol% diTEMPO and 4 mol% DCP at 150 °C for 30 min leading to a partial discoloration of the reaction medium. The monitoring of the reaction by 1H NMR spectroscopy showed the grafting of diTEMPO on C18 by the appearance of the peak characteristic of the proton on the carbon on which a TEMPO group was grafted at 3.7 ppm (Fig. 5). The grafting was fast occurring within the first 5 min of the reaction without significant increase of the grafting at longer reaction times. The grafting was estimated to be the same as the number of molecules of diTEMPO corresponding to half of the amount of TEMPO groups as observed previously. However, 1H NMR spectroscopy did not allow any distinguishing between the contribution of one or both TEMPO units of the diTEMPO in the grafting reaction. Capillary viscometry was used under similar conditions as previously described to determine the kinematic viscosity of the reaction mixture after the reaction, which was compared to the viscosity before the reaction. The kinematic viscosity increased from 0.595 mm2 s−1 (before the reaction) to 0.628 mm2 s−1 after 30 min of reaction, representing a small difference in kinematic viscosity (Δη = 0.033 mm2 s−1). By increasing the quantity of diTEMPO to 8 mol% (DCP 8 mol%), the difference in kinematic viscosity increased to 0.058 mm2 s−1. The same experiment was conducted (8 mol% diTEMPO, 8 mol% DCP) by replacing C18 with C11-OH, which led to no difference in kinematic viscosity (Δη = 0 mm2 s−1) suggesting that diTEMPO was able to covalently connect two C18 chains together, which was not the case for C11-OH, thus preventing the ability of diTEMPO to act as a compatibilizing agent for blends of PE and EVOH.
 | ||
| Fig. 5 Monitoring of the grafting of diTEMPO (4 mol%) on C18 in the presence of DCP (4 mol%) at 150 °C. The peak at 0.9 ppm was taken as a reference for all the terminal methyl groups of modified and unmodified C18, while the peak at 3.7 ppm corresponded to the proton on the carbon onto which a TEMPO group was grafted. | ||
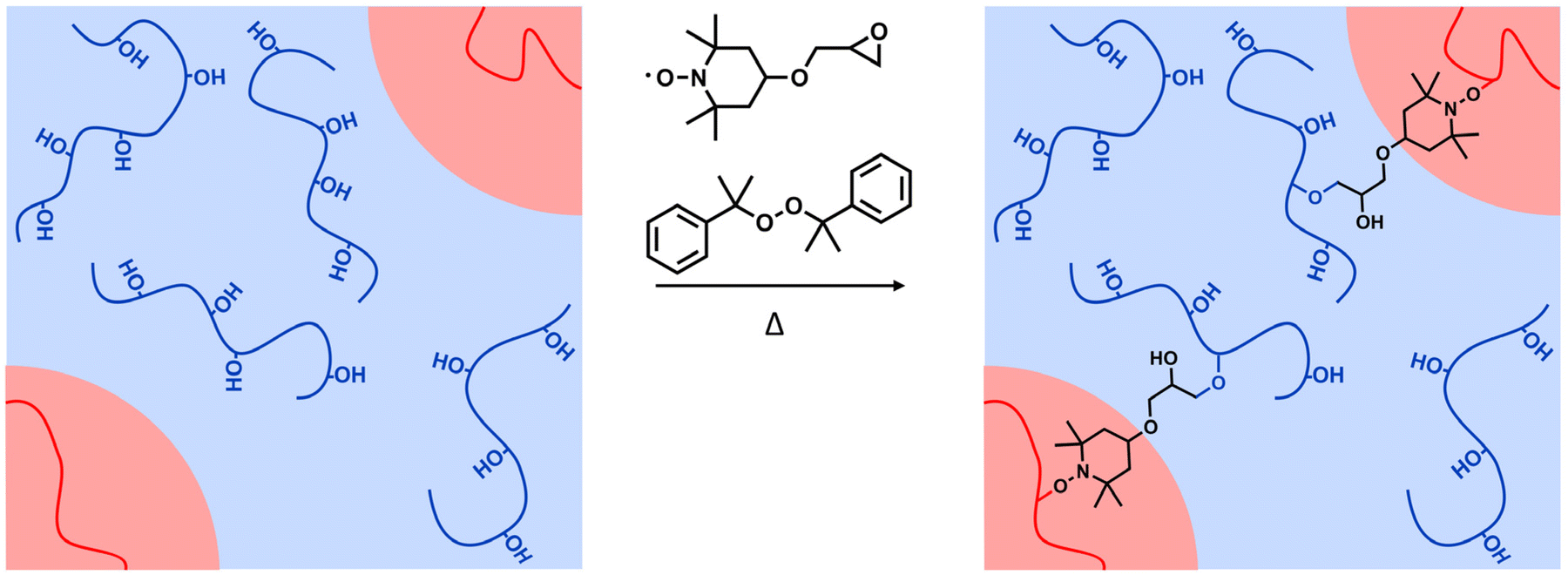 | ||
| Fig. 6 TEMPO-epoxy as a proposed compatibilizing agent to stabilize the interface between PE (in red) and EVOH (in blue) phases through the grafting of the TEMPO group of TEMPO-epoxy onto PE and the coupling reaction of the epoxy group of TEMPO-epoxy with the hydroxyl groups of EVOH. | ||
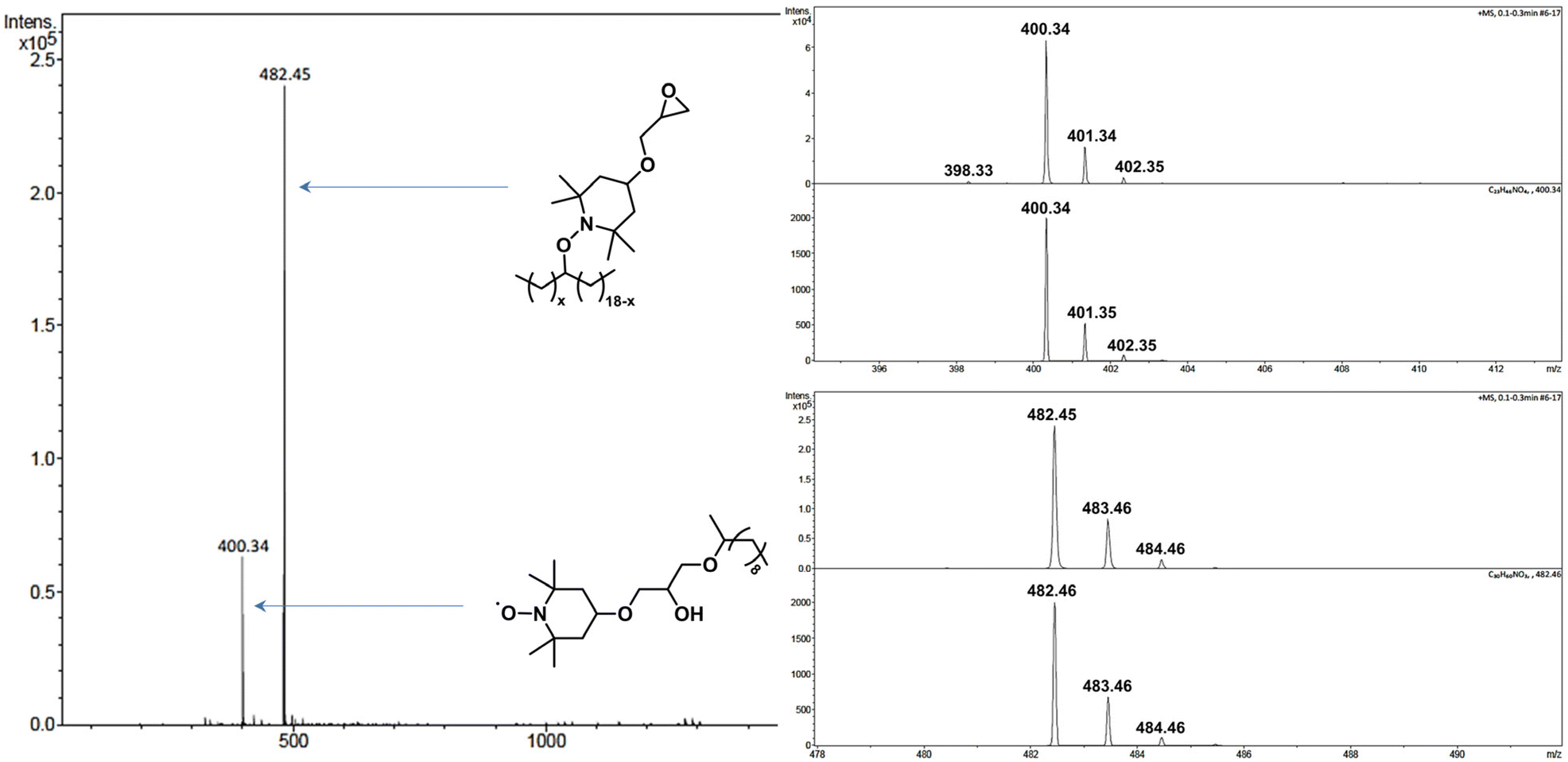 | ||
Fig. 7 Mass spectrum of the crude product from the reaction of C18 and C11-OH (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) with TEMPO-epoxy in the presence of DCP (2 mol%) at 150 °C for 15 min. For each molecular peak in the inset, the spectrum on top corresponds to the experimental results, and that on the bottom is the simulated pattern according to the molecular formula. :1) with TEMPO-epoxy in the presence of DCP (2 mol%) at 150 °C for 15 min. For each molecular peak in the inset, the spectrum on top corresponds to the experimental results, and that on the bottom is the simulated pattern according to the molecular formula. | ||
Summary
The design of suitable and efficient interfacial agents to promote the compatibilization of polymer blends is still a challenge and is worth pursuing specially to provide solutions to the upcycling of plastic waste. The identification of a universal molecule that could be used for any type of polymer blend remains the Holy Grail in this field. TAIC and diTEMPO were demonstrated here as inappropriate to compatibilize PE/EVOH blends through the use of model molecules. This work showed a reasoned approach to conceive a suitable interfacial agent for this polymer blend. A molecule bearing a TEMPO and a glycidyl group has been demonstrated as a promising candidate to add to a blend of PE and EVOH. The design of recombination agents is a unique and attractive approach for researchers in this field to identify compatibilizing agents for specific blends of polymers. These results pave the way to investigations aimed at developing adequate interfacial agents for other polymer blends in existing waste composed of different incompatible polymers.Experimental
Materials
Adipoyl chloride (TCI, >98.0%), benzoyl peroxide (BPO, Aldrich, 75%, remainder water), dicumyl peroxide (DCP, Aldrich, 98%), epichlorohydrin (99%, Alfa Aesar), 4-hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl free radical (4-hydroxy-TEMPO, TCI, >98.0%), n-dodecane (Alfa Aesar, 99%), n-octadecane (C18, Alfa Aesar, 99%), phenylhydrazine (Alfa Aesar, 97%), terephthaloyl chloride (TCI, >99.0%), tetrabutylammonium hydrogen sulfate (TBABS, TCI, >98%,), 2,2,6,6-tetramethylpiperidine-1-oxyl radical (TEMPO, Alfa Aesar, 98+%), triallyl isocyanurate (TAIC, TCI), triethylamine (NEt3, Acros Organics, >97%), 2-undecanol (C11-OH, TCI, >98%), potassium carbonate (K2CO3, Fisher Chemical, 99%), silica gel (Millipore Sigma, high purity grade, pore size 60 Å, 230–400 mesh), sodium sulfate (Na2SO4, Fisher Chemical, anhydrous 99.6%), cyclohexane (Carlo Erba, RE), anhydrous dichloromethane (Sigma, ≥99.8%, contains amylene as stabilizer), ethyl acetate (Carlo Erba, pure), and dichloromethane (Carlo Erba, pure-stabilized with amylene, RE) were used as received, except if noted otherwise. Anhydrous tetrahydrofuran (THF) was obtained on a GT S100 purification station. 4-Glycidyl-2,2,6,6-tetramethylpiperidin-1-oyl (TEMPO-epoxy) was synthesized as described in the literature, except the eluent used for the column chromatography (gradient of cyclohexane and ethyl acetate 70:30 to 50:50).49
Characterization
1H and 13C NMR spectra were recorded on a 400 MHz Bruker Avance III HD spectrometer equipped with a BBO-type probe at 25 °C.Mass measurements were carried out on a Bruker Daltonics microTOF spectrometer (Bruker Daltonik GmbH, Bremen, Germany) equipped with an orthogonal electrospray (ESI) interface. Calibration was performed using Tuning mix (Agilent Technologies). Sample solutions were introduced into the spectrometer source with a syringe pump (Harvard type 55 1111: Harvard Apparatus Inc., South Natick, MA, USA) with a flow rate of 5 μL min−1.
Capillary viscosity measurements were conducted using a micro-Ostwald capillary (2 mL, k = 0.01098 mm2 s−2 or k = 0.01064 mm2 s−2) in dichloromethane with a concentration of 400 mg mL−1, in a thermostated bath at 25 °C.
Resonance paramagnetic electronic (RPE) analyses were carried out on a continuous-wave EPR X-band spectrometer (EMXplus from Bruker Biospin GmbH, Germany) equipped with a high sensitivity resonator (4119HS-W1). This setup was used to record field-swept spectra from ca. 2 mg of C18-TEMPO resuspended in 1 mL of dichloromethane. The main experimental parameters were: microwave power 1.8 mW, modulation amplitude 1 G, time constant of ca. 40 ms, and 10 ms conversion time, 12 mT were swept in 30 s per scan and 2 scans were accumulated. All experiments were performed at room temperature (T = 295 K ± 1 K). EPR intensity (I(t)) values were then obtained through careful double integration of the EPR signal after baseline subtraction (pure dichloromethane solution). TEMPO concentrations were then evaluated using the calculated TEMPO concentration at t = 0 as reference.
Model reaction of C18 and/or C11-OH in the presence of TAIC
C18 (3.00 g, 11.8 mmol) or C11-OH (2.00 mL, 9.63 mmol) was placed in a round-bottom flask. DCP and TAIC in the required amounts were added. The round-bottom flask was heated at 40 °C to melt the reagents and the solution was degassed by bubbling argon for 15 min. The reaction mixture was stirred in a bath thermostated at 150 °C for 15 min under argon. For the reactions conducted with a mixture of C18 and C11-OH, 1.00 g of C18 (3.93 mmol) and 0.82 mL of C11-OH (3.93 mmol) were used.Model reactions of PE and EVOH model molecules in the presence of TEMPO
C18 (1.00 g, 3.93 mmol), C12 (1.00 mL, 4.40 mmol) or C11-OH (3.00 mL, 14.50 mmol) was placed in a round-bottom flask. DCP (or BPO) and TEMPO in the required amounts were added. For C18 the round-bottom flask was heated at 50 °C to melt the reagents, while for C12 and C11-OH the solution was stirred at room temperature. The solution was degassed by bubbling argon for 15 min. The reaction mixture was stirred in a bath thermostated at the required temperature under argon. For the experiment with a purification step by distillation, the reaction mixture was washed with acetonitrile followed by distillation on a Kugelrohr apparatus (160 °C, 12 mbar).Synthesis of diTEMPO
4-Hydroxy-TEMPO (2.09 g, 12.10 mmol) and NEt3 (2.00 mL, 14.30 mmol) were placed in a round-bottom flask and dissolved in 17 mL of anhydrous CH2Cl2 under argon. The reaction mixture was cooled to 0 °C. A solution of terephthaloyl chloride (1.11 g, 5.49 mmol) in anhydrous THF (2 mL) was added dropwise into the round-bottom flask. The reaction mixture was stirred at room temperature for 20 h. The reaction mixture was then washed with an aqueous solution of K2CO3 (10% w/v, 20 mL), dried over Na2SO4, and concentrated by rotary evaporation. The crude product was purified by column chromatography on silica gel using a gradient of solvents, namely dichloromethane and ethyl acetate (95:5 to 90:10), as eluent affording a light orange solid (1.48 g). The integrals were not indicated on the 1H NMR spectrum (ESI, Fig. S8†) as the product was not pure (presence of residual NEt3, 4-hydroxy-TEMPO, and monofunctionalized terephthaloyl chloride). The structural characteristics reported below are only for the terephthaloyl with two TEMPO groups.
1H NMR: δ (CDCl3 + 1 drop of phenylhydrazine, 7.26 ppm) 7.97 (s, 4H), 5.23 (m, 2H), 1.98 (m, 4H), 1.74 (m, 4H), 1.21 (s, 12H), 1.19 (s, 12H). ESI (m/z): calculated from C26H38N2O6 474.27; found 474.27.
Reaction of diTEMPO with C18 or C11-OH
C18 (3.00 g, 12 mmol) or C11-OH (2.5 mL, 12 mmol) was placed in a round-bottom flask. DCP (0.13 g, 0.48 mmol) and diTEMPO (0.23 g, 0.48 mmol) were added to the round-bottom flask, which was then heated to 50 °C and degassed by bubbling argon for 15 min. The reaction mixture was stirred in a bath thermostated at 150 °C for 30 min under argon.Reaction of TEMPO-epoxy with C18 and/or C11-OH
C18 (1.00 g, 3.93 mmol) or C11-OH (0.82 mL, 3.93 mmol) was placed in a round-bottom flask. DCP (22 mg, 0.08 mmol) and TEMPO-epoxy (36 mg, 0.16 mmol) were added to the round-bottom flask, which was heated at 50 °C to melt the reagents and degassed by bubbling argon for 15 min. The reaction mixture was stirred in a thermostated bath at 150 °C for 15 min under argon. For the reactions conducted with a mixture of C18 and C11-OH, 1.00 g of C18 (3.93 mmol) and 0.82 mL of C11-OH (3.93 mmol) were used in the presence of the required amount of DCP and TEMPO-epoxy.Author contributions
Elisa Gitzhofer: methodology, investigation, visualization. Bertrand Vileno: methodology, investigation, visualization. Michel Bouquey: supervision, funding acquisition, writing. Delphine Chan-Seng: conceptualization, methodology, supervision, visualization, writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the institute Carnot MICA (VARIfORM) that supported the research assistant position of EG. The authors thank the polymer characterization facilities at the Institut Charles Sadron, the mass spectrometry facilities of the University of Strasbourg, and the Cronenbourg NMR core facilities.References
- M. Norton, Chem. – Eur. J., 2020, 26, 7737–7739 CrossRef CAS PubMed.
- Y. X. Jiang and M. Jiang, Adv. Mater. Res., 2013, 750–752, 811–815 Search PubMed.
- C. Zhao, M. Liu, H. Du and Y. Gong, Sustainability, 2021, 13, 3662 CrossRef.
- P. Quicker, M. Seitz and J. Vogel, Waste Manage. Res., 2022, 40, 1494–1504 CrossRef CAS PubMed.
- Z. O. G. Schyns and M. P. Shaver, Macromol. Rapid Commun., 2021, 42, 2000415 CrossRef CAS PubMed.
- Mixing in polymer processing, ed. C. Rauwendaal, Marcel Dekker, 1991 Search PubMed.
- Compatibilization of Polymer Blends: Micro and Nano Scale Phase Morphologies, Interphase Characterization and Properties, ed. A. R. Ajitha and S. Thomas, Wiley, 2020 Search PubMed.
- J. Maris, S. Bourdon, J.-M. Brossard, L. Cauret, L. Fontaine and V. Montembault, Polym. Degrad. Stab., 2018, 147, 245–266 CrossRef CAS.
- T. Thiounn and R. C. Smith, J. Polym. Sci., 2020, 58, 1347–1364 CrossRef CAS.
- A. Rahimi and J. M. García, Nat. Rev. Chem., 2017, 1, 0046 CrossRef.
- W. Kaminsky, J. Anal. Appl. Pyrolysis, 1985, 8, 439–448 CrossRef CAS.
- T. Sadik, V. Massardier, F. Becquart and M. Taha, J. Appl. Polym. Sci., 2013, 127, 1148–1156 CrossRef CAS.
- S. Touhtouh, F. Becquart, C. Pillon and M. Taha, Polymers, 2011, 3, 1734–1749 CrossRef CAS.
- C. Chevallier, F. Becquart and M. Taha, Mater. Chem. Phys., 2013, 139, 616–622 CrossRef CAS.
- C. A. Orr, J. J. Cernohous, P. Guegan, A. Hirao, H. K. Jeon and C. W. Macosko, Polymer, 2001, 42, 8171–8178 CrossRef CAS.
- J. M. Ferri, D. Garcia-Garcia, E. Rayón, M. D. Samper and R. Balart, Polymers, 2020, 12, 1344 CrossRef CAS PubMed.
- A. Hejna, M. Klein, M. R. Saeb and K. Formela, Polym. Test., 2019, 73, 143–151 CrossRef CAS.
- T. Vivier and M. Xanthos, J. Appl. Polym. Sci., 1994, 54, 569–575 CrossRef CAS.
- J. B. Olivato, M. V. E. Grossmann, F. Yamashita, D. Eiras and L. A. Pessan, Carbohydr. Polym., 2012, 87, 2614–2618 CrossRef CAS.
- S. Park and S. Choe, Macromol. Res., 2005, 13, 297–305 CrossRef CAS.
- S. Horiuchi, N. Matchariyakul, K. Yase, T. Kitano, H. K. Choi and Y. M. Lee, Polymer, 1997, 38, 59–78 CrossRef CAS.
- I. P. Mahendra, B. Wirjosentono, Tamrin, H. Ismail, J. A. Mendez and V. Causin, J. Polym. Res., 2019, 26, 215 CrossRef CAS.
- C. Jiang, S. Filippi and P. Magagnini, Polymer, 2003, 44, 2411–2422 CrossRef CAS.
- H. Jang, S. Kwon, S. J. Kim and S.-I. Park, Int. J. Mol. Sci., 2022, 23, 7166 CrossRef CAS PubMed.
- P. Cassagnau, V. Bounor-Legaré and F. Fenouillot, Int. Polym. Process., 2007, 22, 218–258 CrossRef CAS.
- Y. Zhuang, N. Saadatkhah, M. S. Morgani, T. Xu, C. Martin, G. S. Patience and A. Ajji, Can. J. Chem. Eng., 2022, 101, 59–77 CrossRef.
- Y. Nasri, M. T. Benaniba and M. Bouquey, Mater. Today: Proc., 2022, 53, 91–95 CAS.
- I. Chodak, Polym.-Plast. Technol. Eng., 2004, 43, 1769–1777 CrossRef CAS.
- W. Du, W. Zhong, Y. Lin, L. Shen and Q. Du, Eur. Polym. J., 2004, 40, 1987–1995 CrossRef CAS.
- H. Zhao, J. Chen, H. Zhang, Y. Shang, X. Wang, B. Han and Z. Li, RSC Adv., 2017, 7, 37095–37104 RSC.
- M. D. Stelescu, E. Manaila and N. Zuga, Polym. J., 2011, 43, 792–800 CrossRef CAS.
- J. S. Forsythe, D. J. T. Hill, A. L. Logothetis, P. J. Pomery and A. K. Whittaker, J. Appl. Polym. Sci., 1999, 73, 169–175 CrossRef CAS.
- F. Jin, S.-H. Hyon, H. Iwata and S. Tsutsumi, Macromol. Rapid Commun., 2002, 23, 909–912 CrossRef CAS.
- S.-L. Yang, Z.-H. Wu, W. Yang and M.-B. Yang, Polym. Test., 2008, 27, 957–963 CrossRef CAS.
- P. van Ballegooie and A. Rudin, Polym. Eng. Sci., 1988, 28, 1434–1442 CrossRef CAS.
- L. Yang, J. Huang, X. Lu, S. Jia, H. Zhang, G. Jin and J. Qu, J. Appl. Polym. Sci., 2015, 132, 41543 Search PubMed.
- P. Zytner, F. Wu, M. Misra and A. K. Mohanty, ACS Omega, 2020, 5, 14900–14910 CrossRef CAS PubMed.
- L. D. Loan, Pure Appl. Chem., 1972, 30, 173–180 CrossRef CAS.
- J. Nicolas, Y. Guillaneuf, C. Lefay, D. Bertin, D. Gigmes and B. Charleux, Prog. Polym. Sci., 2013, 38, 63–235 CrossRef CAS.
- T. Janoschka, M. D. Hager and U. S. Schubert, Adv. Mater., 2012, 24, 6397–6409 CrossRef CAS PubMed.
- Q. Xu, C. He, C. Xiao and X. Chen, Macromol. Biosci., 2016, 16, 635–646 CrossRef CAS PubMed.
- S. Coiai, E. Passaglia and F. Cicogna, Polym. Int., 2019, 68, 27–63 CrossRef CAS.
- H. Nakatani, H. Hayashi and S. Motokucho, React. Funct. Polym., 2021, 169, 105090 CrossRef CAS.
- E.-S. Park and J.-S. Yoon, J. Appl. Polym. Sci., 2003, 88, 2434–2438 CrossRef CAS.
- D. Belekian, E. Beyou, P. Chaumont, P. Cassagnau, J. J. Flat, S. Quinebèche, Y. Guillaneuf and D. Gigmes, Eur. Polym. J., 2015, 66, 342–351 CrossRef CAS.
- G. E. Garrett, E. Mueller, D. A. Pratt and J. S. Parent, Macromolecules, 2014, 47, 544–551 CrossRef CAS.
- S. Akbar, E. Beyou, P. Chaumont and F. Melis, Macromol. Chem. Phys., 2010, 211, 2396–2406 CrossRef CAS.
- T. D. Lee and J. F. W. Keana, J. Org. Chem., 1975, 40, 3145–3147 CrossRef CAS.
- Z. Jia, C. A. Bell and M. J. Monteiro, Macromolecules, 2011, 44, 1747–1751 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d2py01495e |
| This journal is © The Royal Society of Chemistry 2023 |